Introduction
Neuroendocrine tumors (NETs) arise from
neuroendocrine cells, located in different tissues throughout the
body. Gastroenteropancreatic NETs derive from neuroendocrine cells
that are disseminated throughout the gastrointestinal (GI) tract or
form neuroendocrine islets within the exocrine pancreatic tissue.
Given its length, the GI tract is the largest neuroendocrine organ,
enclosing more neuroendocrine cells than any other part of the
human body (1). This could explain
the fact that Digestive system (DS) is the most common site of NETs
development, followed by the bronchopulmonary tree. Distribution of
DS NETs at each specific site/organ is as follows: esophagus
<1%, stomach 7%, small intestine 17%, appendix 5%, colon 5%,
rectum 15%, pancreas 45%, liver 1% (2).
Grade (G), based on Ki67 proliferation index and
mitotic count, has proven to be a powerful prognostic indicator
(3–6). Low grade DS NETs, are considered in
general of good prognosis with a survival rate ranging between 38%
(pancreas) and 88% (rectum). Lymph nodes and liver are the most
common sites of metastases (20–50 and 60% respectively) (2), while, to the best of our knowledge,
peritoneal carcinomatosis has been reported only once in G1
gastrointestinal NETs (7).
Case report
Written informed consent was obtained from the
patient. A 60-year-old, female, Caucasian patient, with an
unremarkable past medical history, was admitted in a provincial
hospital, with a 20-day history of atypical, crampy abdominal pain,
vomiting and diarrhea. Initial diagnostic work-up included an upper
and lower gastrointestinal (GI) endoscopy, both of which were
normal, and an abdominal CT scan, which revealed mild dilatation of
jejunal loops. Patient's basic laboratory values and tumor markers
(CEA, Ca19-9) were within normal range. She was discharged with a
diagnosis of recurrent partial bowel obstruction and was referred
to the GI Department of a tertiary hospital (Agios Savvas
Anticancer Hospital, Athens, Greece), for further
investigation.
Due to worsening clinical condition, patient was
transferred to the Department of Surgical Oncology with a working
diagnosis of complete small bowel obstruction. An exploratory
laparotomy was performed, which revealed multiple tumoral lesions
in the wall of the jejunum, causing complete intestinal
obstruction. Due to extensive regional lymphadenopathy up to the
root of the associated mesentery, an extensive resection of jejunum
was performed, with primary anastomosis. Inspection of the
abdominal cavity did not reveal hepatic or other splanchnic
metastases. However, a suspicious peritoneal nodule measuring 2 cm
in the pelvic cul-de-sac was excised for biopsy. Postoperative
course was uneventful and patient was discharged on the 8th
postoperative day.
The surgical specimen consisted of a 76-cm-long
small intestine segment obstructed at a distance of 20 cm from the
distal resection margin and dilated proximal to the obstruction.
Obstruction was caused by 4 whitish neoplastic foci measuring 7, 8,
12 and 20 mm in maximum diameter located in the jejunal wall
causing mucosal erosion. Mesenteric fat seemed to be
macroscopically involved by the neoplasm. Forty nine lymph nodes
were retrieved measuring 2–10 mm, 2 of which from the mesentery
root. Twenty five tissue cassettes were prepared including 1 from
each neoplastic focus (4 cassettes), 1 from the peritoneal nodule,
1from nonlesional mucosa, 2 from the intestinal resection margins
and 17 containing the dissected lymph nodes. Tissue cassettes were
processed according to standard protocol. H&E sections were
examined by 2 specialist pathologists (AF and SS) with interest in
neuroendocrine tumors.
Sections showed variable sized solid nests composed
of uniform neoplastic cells with bland nuclei and salt and pepper
chromatin. Tumoral cells invaded widely the intestinal wall and
perivisceral fat, abating but not penetrating the serosa (Fig. 1A and B). Mitotic activity assessed in
60 tumoral HPFs was very low resulting in a mitotic count <2 per
10 HPF in the hot spots. Moreover, plenty of blood/lymph vessel
emboli and foci of perineural infiltration were recognized
(Fig. 1C). Of the 47 lymph nodes
dissected from the perivisceral fat, 6 were positive for metastatic
disease (Fig. 1D). The 2 additional
lymph nodes excised from the root of the mesentery were also
positive for disease. Finally, the nodule that was found in the
pelvic cul-de-sac was proven a metastatic implant.
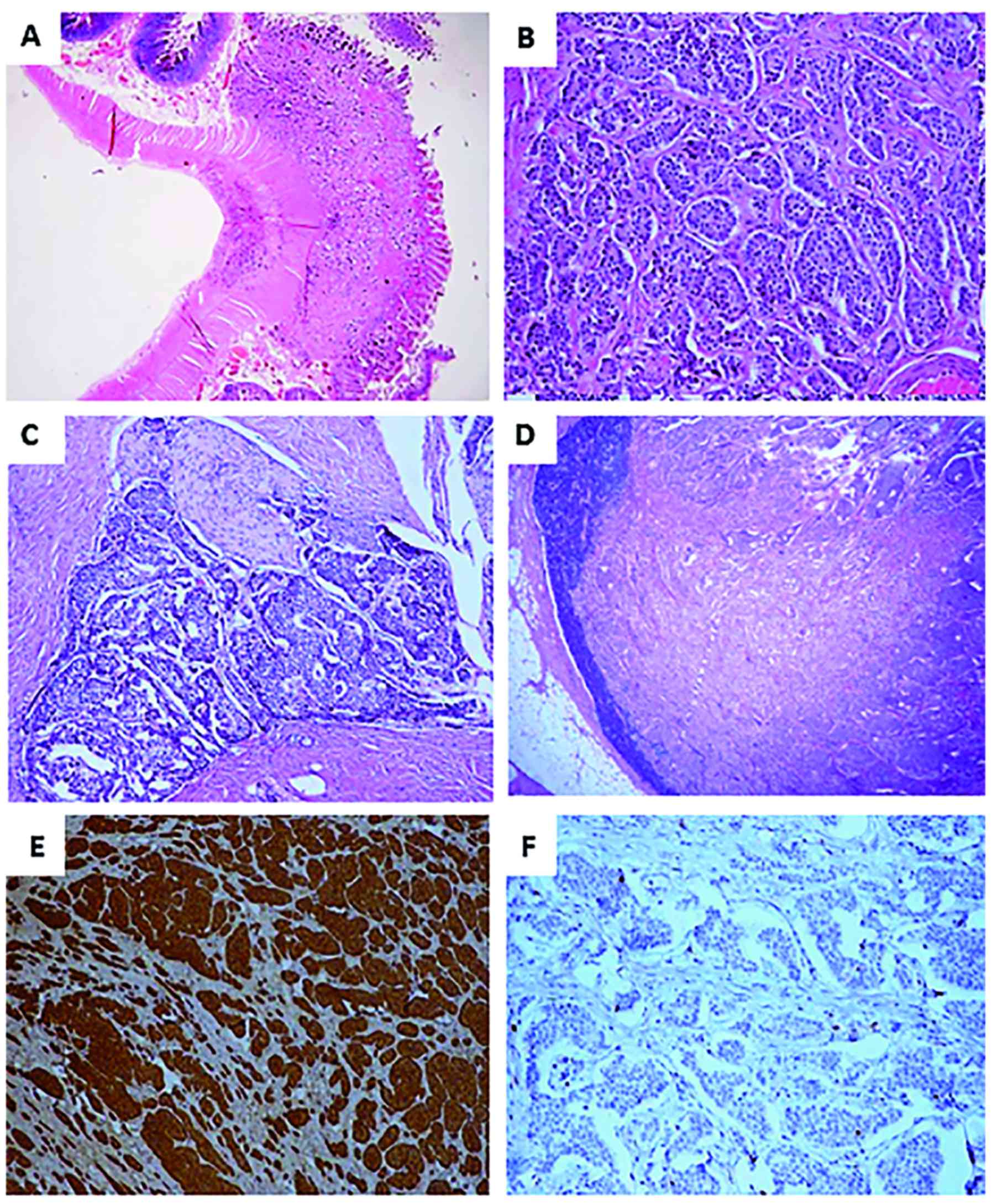 | Figure 1.Tumor histological features. (A)
Jejunal wall infiltrated by the tumor (H&E; magnification,
×10). (B) Tumor composed of uniform neoplastic cells arranged in
solid nests (HE; magnification, ×200). (C) Tumor perineural
infiltration (H&E; magnification, ×100). (D) Lymph node
metastasis (H&E; magnification, ×40). (E) Diffuse tumor
positivity for synaptophysin (synaptophysin immunostaining, DAB
chromogen; magnification, ×100). (F) Ki67 tumor proliferation index
(Ki67/MIB1 immunostaining, DAB chromogen; magnification, ×100).
H&E, hematoxylin and eosin; DAB, 3,3′-diaminobenzidine. |
Tumor immunohistochemical analysis was performed in
3 tissue blocks (from the 2 largest neoplastic foci and the
peritoneal nodule) using antibodies against chromogranin A,
synaptophisin, serotonin and Ki67. The EnVision Staining System
(Dako, Glostrup, Denmark) with 3,3′ diaminobenzidine (DAB) as a
chromogen, and a PT Link, Pre-Treatment Module was applied
according to standard protocols. Table
I shows details on antibodies and methodology. Diffuse
positivity was evident for chromogranin A (Fig. 1E), synaptophysin and serotonin. The
percentage of Ki67 positivity in 2,000 neoplastic cells was
estimated in areas of strongest nuclear labeling by the 2
pathologists independently. Both of them concluded that Ki67
proliferation index was <2% (Fig.
1F). Histological diagnosis was NET Grade1, pT3N1M1 (TNM 7th
edition, 2007), stage IV, according to the 2010 American Joint
Committee on Cancer (AJCC). The patient received m-TOR inhibitor
(Everolimus) and is free of disease 18 months post surgery.
 | Table I.Antibodies used for histological
diagnosis. |
Table I.
Antibodies used for histological
diagnosis.
| Antibody type | Antibody
dilution |
|---|
| Synaptophysin (mouse
monoclonal AB, clone DAK-SYNAP) | 1:50 |
| Chromogranin A (mouse
monoclonal AB, clone DAK-A3) | 1:100 |
| Serotonin (mouse
monoclonal AB, clone 5HT-H209) | 1:50 |
| Ki67 (mouse
monoclonal AB, clone MIB-1) | 1:50 |
Discussion
Natural history of NETs is poorly understood. They
arise from enterochromaffin cells, which are multipotent stem cells
that migrate from the neural crest to the gut ectoderm (8–10).
According to the embryological origin, NETs are classified as
foregut, midgut or hindgut. Foregut NETs refer to tumors arising in
the respiratory tract, thymus, thyroid, stomach, duodenum and
pancreas. Midgut NETs develop in the small bowel, appendix and
ascending colon (11), while hindgut
tumors appear in the transverse, descending colon and rectum
(12,13).
NETs can be functional (40%) or non functional (60%)
depending on the excess of hormones (serotonin, substance P) and/or
peptides (chromogranin, synaptophysin) secretion. Functional NETs
can cause symptoms such as flushing (95%), secretory diarrhea (78%)
and abdominal crumps (50%) (14–16). Non
functional NETs can grow undetected for years, causing symptoms in
later stages due to mass effect, such as intestinal blockage or
bleeding. The majority of NETs are sporadic and only 10% are
familial, arising in the context of autosomal dominant inherited
syndromes (MEN1-2, VHL, NF) (17,18).
NET's are the second most prevalent group of tumors
in the GI tract. Through the years, WHO has applied various
classifications to GI NETs. In 1980 GI NETs were categorized as
follows: I. Carcinoid, II. Mucocarcinoid, III. Mixed forms
(Carcinoid-Adenocarcinoma) and IV. Pseudotumor lesions. In 2000,
WHO revised the previous classification in the following
categories: I. Well differentiated endocrine carcinoma (WDEC), II.
Poorly differentiated/small cell carcinoma (PDEC), III. Mixed
endocrine-exocrine carcinoma (MEEC) and III. Tumor - like
lesions.
Over the years, it was shown that proliferation
rate, described as the number of mitoses per 10 HPF and the
percentage of Ki67 positive neoplastic cells, provides significant
prognostic information for NETs, such as recurrence or metastatic
potential (3). In 2010 a new
classification was established, grading GI NETs based on mitotic
count and Ki67 index (percentage of Ki67 positive cells in
500–2,000 cells counted in areas of strongest nuclear label), as
follows: I. NET Grade 1: Ki67 1–2% and/or up to 1 mitosis/10 HPF,
II. NET Grade 2: Ki67 3–20% and/or 2–20 mitoses/10 HPF, II.
Neuroendocrine carcinomas (NECs): Ki67 >20% and/or >20
mitoses/10 HPF. III. Mixed adeno-neuroendocrine carcinoma (MANEC):
≥30% of tumor cells with neuroendocrine phenotype. Tables II and III, show the latest grading GI NETs
classification and Table IV
presents the transition from previous classifications to the new
grading categories (4).
| Table II.Grading system of gastrointestinal
neuroendocrine neoplasms (World Health Organization 2010). |
Table II.
Grading system of gastrointestinal
neuroendocrine neoplasms (World Health Organization 2010).
| Grade | Mitotic count (/10
HPF) | Ki-67 index (%) |
|---|
| G1 | <2 | <2 |
| G2 | 2–20 | 3–20 |
| G3 | >20 | >20 |
| Table III.World Health Organization 2010
classification of gastrointestinal neuroendocrine neoplasms. |
Table III.
World Health Organization 2010
classification of gastrointestinal neuroendocrine neoplasms.
| Type of
neuroendocrine neoplasm | Description |
|---|
| NET | Low to intermediate
grade (G1-G2), well to moderately differentiated neoplasms |
| NEC (small cell to
large cell type) | High grade (G3),
moderate to poorly differentiated neoplasms |
| MANEC | Neoplasms with ≥30%
of tumor cells that have NE characteristics |
| Table IV.WHO classifications of
gastrointestinal neuroendocrine neoplasms. |
Table IV.
WHO classifications of
gastrointestinal neuroendocrine neoplasms.
| Classification
no. | WHO (1980) | WHO (2000) | WHO (2010) |
|---|
| 1 | Carcinoid islet cell
tumor | Well differentiated
endocrine tumor | NET G1 |
| 2 | Muco-carcinoid | Well differentiated
endocrine carcinoma | NET G2 |
| 3 | Mixed forms carcinoid
-adenocarcinoma | Poorly differentiated
endocrine carcinoma | Neuroendocrine
carcinoma |
| 4 | Pseudotumor
lesions | Mixed
endocrine-exocrine cell neoplasm | Mixed adeno-endocrine
carcinoma |
| 5 | – | Tumor-like
lesions | – |
Difficulties arise when applying NET WHO 2010
classification in practice. First of all, categorization of NETs
with a Κi67 index between 2 and 3% is unclear. To address this
issue, Yamaguchi et al (5)
studied retrospectively 45 NET G1/G2 cases and showed that the
cutoff value for predicting metastases or recurrence was 2.8%. They
concluded that the categorization of NETs into G1 or G2 based on
Ki67 index of 3% can predict metastases or recurrences (5).
Apart from grade, stage, referring to tumor size,
extent of invasion and metastatic status is an indispensable tool
for therapeutic intervention and prognosis estimation and should
always be taken into consideration (19). According to some epidemiological data
from a 6 year surveillance study in USA during the period of
1988–2004, medium survival was 203 months for localized tumors, 114
months for tumors with regional extension and 39 months for distant
metastatic tumors (20). The TNM
classification of Malignant Tumours, the most widely used
organ/site specific cancer staging system, is also applied to GI
NETs. The recently published 8th edition of TNM classification
acknowledges the importance of the number of lymph nodes metastasis
and the presence of mesenteric mass, incorporating for the first
time this information in the N category of the TNM system (21). According to the new classification,
presence of mesenteric neoplastic mass measuring more than 2cm in
maximum diameter corresponds to N2 category, even in the absence of
lymph node metastasis. Another novelty is that the new M1 category
(distant metastasis) includes 3 sub-categories, namely hepatic
metastasis only (M1a), extrahepatic metastasis only (M1b) and
hepatic and extrahepatic metastase (M1c). Concerning the presented
case, the neoplastic mass found adhered to the peritoneum of the
rectouterine pouch (cul-de-sac) does not qualify for a mesenteric
mass. On the other hand and despite the improvements in the new TNM
classification, it remains unclear whether it should be considered
an extrahepatic metastasis.
Recent data suggest that useful information
concerning NET clinical outcome could be derived from circulating
tumor cells (CTCs) expressing epithelial cell adhesion molecule
(EpCAM), possibly with more predictive power than WHO grading
system (22). However, CTCs as
prognostic biomarkers cannot be widely used at present time.
The therapeutic options for NETs are the following:
i) Surgery: Curative (rarely), ablative (very often); ii)
debulking: Radiofrequency ablation (RFA)/embolization,
chemoembolization/radioembolization; irradiation, external (bone,
brain metastasis)/tumor targeted, radioactive therapy; iii) medical
therapy: Chemotherapy, biological treatment (somatostatin analogs,
a-Interferon, m-TOR inhibitors, VEGF R inhibitors, Other TKI's
(23).
The presented case is of special interest regarding
its prognosticators. On one hand, tumor grade, according to
proliferation rate, is low (G1), suggesting an indolent clinical
course. On the other hand, many histological features, namely,
multiplicity and size of tumor, depth of invasion, lymphatic and
vascular emboli, nodal metastases and peritoneal implant, point
towards aggressive tumor behavior. According to English literature,
small intestinal NETs have a tendency tο multiplicity (30%) and
those multiple tumors have been associated with a worse clinical
outcome (24,25). Microscopic tumors, as small as 3 mm,
can give rise to nodal and distant metastases. The current protocol
of the College of American Pathologists (CAP), cites a 12%
frequency of lymph node metastasis for small intestinal low grade
NETs measuring <1 cm (13). It is
not clear why low grade tumors in this location present with
aggressive histological features and it seems possible that their
behavior is underestimated.
As far as peritoneal carcinomatosis is concerned, it
represents a complication encountered in high grade tumors
(7). To the best of our knowledge,
only one additional to the present case NET G1, Stage IV with
peritoneal carcinomatosis has been previously reported (7). Prognosis of GI NETs depends both on
stage and grade. The 5-year survival rate for stages I–III is
70–80% and for stage IV, 35–80%. Patients with G1 NETs have a 94%
5-year survival, with G2, 83% and with G3, 50% (26). Prognosis of a well differentiated NET
with peritoneal carcinomatosis cannot be estimated, since neither
the WHO 2010 classification nor the TNM system, even in the
recently published 8th edition, can be useful for prognostic
stratification. On occasion of the present case, it becomes evident
that criteria for metastatic disease should be reconsidered to
include peritoneal metastases, in order to provide patients with
the most suitable therapeutic scheme (27).
We reported a case of a jejunal G1, stage IV NET,
with peritoneal carcinomatosis. Although multiplicity and nodal
metastases is not unusual for low grade NETs in this part of the GI
tract, peritoneal carcinomatosis is an extremely rare finding.
Surgeons and histopathologists should be familiar with such
eventualities and tumor boards need to conclude whether aggressive
therapeutic interventions may have any impact on patients' long
term survival.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
FA conceived the study and drafted the manuscript.
DK interpreted the data and revised the manuscript critically for
important intellectual content. NK designed the study, acquired the
data and drafted the manuscript. DM analyzed the data and revised
the manuscript. SS interpreted the data and revised the manuscript
critically for important intellectual content. All authors approved
the final version of the manuscript for publication and have agreed
to be accountable for all aspects of the work.
Ethics approval and consent to
participate
Written informed consent was obtained from the
patient.
Patient consent for publication
Written informed consent was obtained from the
patient.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AJCC
|
American Joint Committee on Cancer
|
|
CAP
|
College of American pathologists
|
|
CT
|
computed tomography
|
|
CTCs
|
circulating tumor cells
|
|
DS
|
digestive system
|
|
G
|
grade
|
|
HPF
|
high power field
|
|
GI
|
gastrointestinal
|
|
NEC
|
neuroendocrine carcinoma
|
|
NET
|
neuroendocrine tumor
|
References
|
1
|
Rehfeld JF: The new biology of
gastrointestinal hormones. Physiol Rev. 78:1087–108. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hauso O, Gustafsson B, Kidd M, Waldum H,
Drozdov I, Chan A and Modlin I: Neuroendocrine tumour epidemiology.
Cancer. 113:2655–2664. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rindi G, Petrone G and Inzani F: The 2010
WHO classification of digestive neuroendocrine neoplasms: a
critical appraisal four years after its introduction. Endocr
Pathol. 25:186–192. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rindi G, Arnoid R, Bosman FT, Capella C,
Klimstra DS, Kloppel G, Komminorth P and Solcia E: Nomeclature and
classification of the neuroendocrine neoplasms of the digestive
system. In: WHO Classification of Tumours of the Digestive System.
4th. IARC Press; Lyon, France: pp. 13–14. 2010
|
|
5
|
Yamaguchi T, Fujimori T, Tomita S,
Ichikawa K, Mitomi H, Ohno K, Shida Y and Kato H: Clinical
validation of the gastrointestinal NET grading system: Ki67 index
criteria of the WHO 2010 classification is appropriate to predict
metastasis or recurrence. Diagn Pathol. 8(65)2013.
|
|
6
|
Karakuş E, Helvacı A, Ekinci O and and
Dursun A: Comparison of WHO 2000 and WHO 2010 classifications of
gastroenteropancreatic neuroendocrine tumours. Turk J
Gastroenterol. 25:81–87. 2014. View Article : Google Scholar
|
|
7
|
Celotti A, Pulcini G, Schieppati M,
Ministrini S, Berruti A and and Ronconi M: An unususal case of a
well-differentiated neuroendocrine tumor of the ileum with
peritoneal carcinomatosis : A case report. World J Surg Oncol.
13:1353–1361. 2015. View Article : Google Scholar
|
|
8
|
Langley K: The neuroendocrine consept
today. Ann NY Acad Sci. 733:1–17. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Warner RR: Enteroendocrine tumors other
than carcinoid. A review of clinically significant advances.
Gastroenterology. 128:1668–1684. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Uberg K: Carcinoid tumors: Current
concepts in diagnosis and treatment. The Oncologist. 3:339–45.
1998.PubMed/NCBI
|
|
11
|
Griniatsos J and Michail O: Appendiceal
neuroendocrine tumors. Resent insights and clinical implications.
World J Gastrointest Oncol. 2:192–96. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ni S and Sheng W, Du X and Sheng W:
Pathologic research update of colorectal neuroendocrine tumors.
World J Gastroenterol. 16:1713–19. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Konishi T, Watahabe T, Nagawa H, Oya H,
Ueno M, Kuroyanagi H, Fujimoto Y, Akiyoshi T, Yamaguchi T and Muto
T: Treatment of colorectal carcinoids. A new paradigm. World J
Gastrointest Surg. 2:153–56. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Begum N, Maasberg S, Plockinger U, Anlauf
M, Rinke A, Popperl G, Lehnert H, Izbicki HR, Krausch M, Vashist
YK, et al: Zentralbl Chir. 139:276–283. 2014.PubMed/NCBI
|
|
15
|
Modlin I. M, Shapiro MD and Kidd M:
Signified Oberdorfer. Origins and perspectives of carcinoid tumors.
Human Pathology. 35:1440–51. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Krols LK: Carcinoid tumors and the
carcinoid syndrome. What is new in therapeutic pipeline. The
carcinoid cancer foundation: carcinoid symposium 2002. http://www.carcinoid.orgJuly 8–2017
|
|
17
|
Oberg K: The genetics of neuroendocrine
tumors. Semin Oncol. 40:37–44. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kaltsas G, Besser M and Grossman A: The
diagnosis and medical management of advanced neuroendocrine tumors.
Endocr Rev. 25:458–511. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jamalli M and Chetty R: Predicting
prognosis on gastroentero-pancreatic neuroendocrine tumors. An
overview and the value of Ki 67 immunostaining. Endocr Pathol.
19:282–288. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yao JC, Hassan M, Phan A, Dagohoy C, Leary
C, Mares JE, Abdalla EK, Fleming JB, Vauthey JN, Rashid A, et al:
One hundred years after ‘carcinoid’. Epidemiology of and prognostic
factors for neuroendocrine tumors in 38,525 cases in the United
States. J Clin Oncol. 26:3063–3072. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Well-Differentiated Neuroendocrine Tumours
of the Gastrointestinal Tract. Union for International Cancer
Control (UICC). TNM Classification of Malignant Tumours. 8th.
Wiley-Blackwell; Hoboken, NJ: pp. 96–99. 2017
|
|
22
|
Khan MS, Kirkwood A, Tsigani T,
Garcia-Hernnandez I, Hartley JA, Caplin ME and Meyer T: Circulating
tumor cells as prognostic markers in neuroendocrine tumors. J Clin
Oncol. 31:365–372. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mei D, Alexandria T and James C: New
strategies for advanced neuroendocrine tumors. CCR. 18:1830–37.
2012.
|
|
24
|
Burke AP, Thomas RM, Elsayed AM and Sobin
H: Carcinoids of jejunum and ileum. Cancer. 79:1086–93. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yantiss RK, Odze RD, Farraye FA and
Rosenberg AE: Solitary versus multiple carcinoid tumors. A clinical
and pathologic review of 68 cases. Am J Surg Pathol. 27:811–817.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jann H, Roll S, Couverald A, Hentic O,
Pavel M, Muller-Nordhorn J, Koch M, Rocken C, Rindi G, Ruszniewski
P, et al: Neuroendocrine tumors of midgut- hindgut origin.
Tumor-node-metastasis classification determines clinical outcome.
Cancer. 117:3332–3341. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shi Ch, Adsay V, Bergsland EM, Berlin J,
Branton PA, Fitzgibbons PL, Frankel WL, Kakar S, Klepeis V,
Klimstra DS, et al: Protocol for the Examination of Specimens from
Patients with Neuroendocrine Tumors (Carcinoid Tumors) of the
Jejunum and Ileum. Version: Jejunum/Ileum NET 1.0.0.0. Cancer
protocol templates. College of American Pathologists (CAP), 2017.
http://www.cap.org/ShowProperty?nodePath=/UCMCon/Contribution%20Folders/WebContent/pdf/cp-jejunal-Ileal-net-17protocol-1000.pdfJuly
8–2017
|














