1. Introduction
The proto-oncogene c-SRC (SRC) is a
non-receptor tyrosine kinase, its expression and activity is
enhanced in various human cancers and correlates with malignancy
progression and development of distant metastasis (1-3).
Since there is increasing evidence of its crucial role in tumor
progression (4,5) c-SRC has emerged as a promising
target for anticancer therapy. Consequently, SRC inhibitors have
been evaluated in the development of clinical therapies (6,7).
However, the exact mechanisms of action of c-SRC and the
critical respective pathway involved in malignancy are not fully
elucidated.
c-SRC is involved in the maintenance of normal cell
homeostasis regulating a wide range of cellular events, including
cell growth, differentiation, proliferation, survival, adhesion,
migration and motility (8,9). In normal cells, the expression levels
and activity of c-SRC are strictly regulated by several mechanisms.
The kinase activity of c-SRC is controlled by C-terminal SRC kinase
(CSK), which phosphorylates a conserved tyrosine residue in the
c-SRC carboxy-terminal domain (Tyr530). This is reversed by
phosphatases such as protein tyrosine phosphatase 1B (PTP1B),
resulting in c-SRC activation. Additionally, activation of
growth-factor receptors leads to their association with the c-SRC
homology 2 (SH2) domain, which disrupts inhibitory intramolecular
interactions to promote c-SRC activation. Other proteins, such as
CRK-associated substrate (CAS) and FAK, bind to the c-SRC SH2 and
SH3 domains to stimulate c-SRC activation by a similar mechanism.
Moreover, c-SRC is also negatively regulated via the
ubiquitin-proteasome pathway, which is mediated by E3
ubiquitin-ligase Cbl and Cullin-5 (10-12).
Hence, c-SRC is regulated at both transcriptional and
post-translational levels by a variety of mechanisms (10-12).
The disruption of any of the c-SRC regulatory mechanisms may
trigger cancer phenotypes through uncontrolled proliferation,
enhanced survival, and invasiveness, in cooperation with other
oncogenic signals (2). Once
activated, as by growth factors or integrins, c-SRC triggers
downstream signaling pathways, including the RAS/MAPK,
phosphatidylinositol 3-kinase (PI3K)/AKT, and STAT pathways,
leading to malignant phenotypic changes (13).
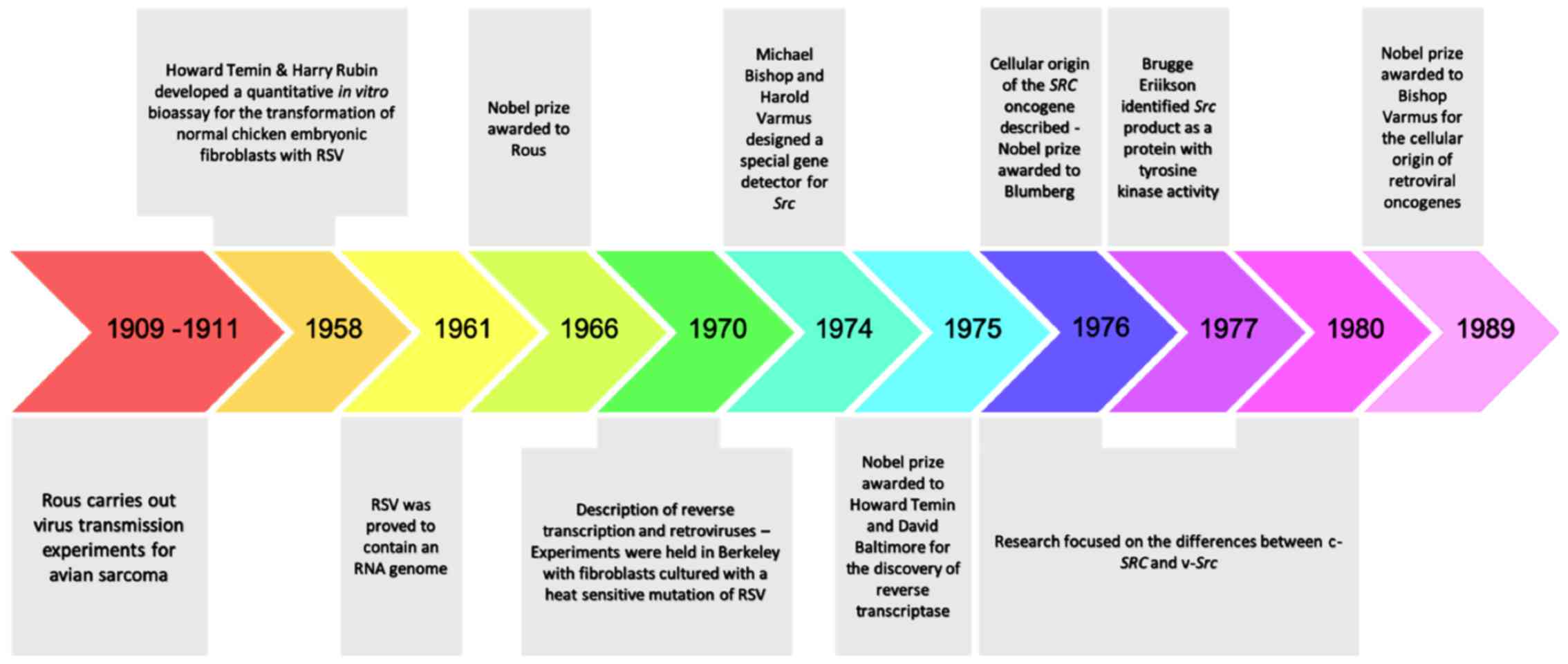
2. Discovery of Rous sarcoma virus
In 1909, at the Rockefeller Institute, Peyton Rous
started his studies on a sarcoma that had been developed in the
breast muscle of a hen. In his original experiments, Rous managed
to transmit the tumor to other birds of the same species, by
implanting fragments of the initial tumor. In his subsequent
experiments, he developed a short protocol for the induction of
tumors in chickens. He used a chicken with sarcoma of breast
muscle, removed the mass and broke it up into small chunks of
tissue. Subsequently he ground up sarcoma with sand and filtrated
it through a fine pore filter. Finally, he injected the filtrate
into a young chicken, and observed the growth of sarcomas. He then
hypothesized that the tumor-inducing agent should be an oncogenic
virus, later becoming known as Rous sarcoma virus (RSV), since this
agent was possible to pass through a filter too fine to contain
bacteria or chicken cells and was capable of causing cancer with a
predictable pattern (14,15). This finding was of great importance
as it was the first proof of viral carcinogenesis and thus
triggered the discovery of many other types of tumor-inducing
viruses in non-human primates such as mice, cats, rabbits (16-19)
and later, of the first oncogenic human virus, Epstein Barr in
1964(20). Additionally, the
discovery of this pioneer oncogenic retrovirus (RSV) was the
hallmark of the onset of the development of research on the
molecular mechanisms of carcinogenesis (21).
For almost half a century the research interest was
focused on chemical carcinogenesis (22-27).
The revival of research regarding oncogenic retroviruses came in
1958 in the Laboratory of Renato Dulbecco. Temin and Rubin
developed a quantitative in vitro bioassay for the
transformation of normal chicken embryonic fibroblasts with RSV.
More specifically, in their experiment, they showed that when the
virus was introduced to Petri dishes where embryonic fibroblasts
where cultured, the RSV(+) cells obtained an evolutionary advantage
and were transformed, acquiring cancer morphology under the
microscope, i.e., they were less adherent and often rounded up,
with increased size and/or number of nucleoli (28). In 1966, the Nobel prize was finally
awarded to Peyton Rous for his discovery. The next question that
arose was whether the transformation of cellular phenotypes was due
to the constant influence of the RSV genome. In 1970, an experiment
in Berkeley confirmed the above hypothesis. In this experiment,
when fibroblasts where cultured with a heat-sensitive mutation of
RSV at permissible temperatures (37°C) the cells were
transformed. When the cultures containing these cells were
transferred to an impermissible temperature (41°C), the
fibroblasts regained their normal morphology and they re-acquired a
cancerous morphology when re-exposed to 37°C. It was
evident that the transforming phenotype was maintained from the
ongoing effects of this protein (29-31).
The Src oncogene of RSV became the prototype for dozens of
other transforming genes in oncogenic viruses. Its product was
identified by Brugge and Erikson in 1977, as a protein with
tyrosine-kinase activity.
3. Cellular origin of retroviral
oncogenes
In 1961, the RSV was proved to contain an RNA genome
(32), whose continuous presence
was necessary for maintaining cell transformation. However, the
mechanism by which the viral RNA genome was incorporated into the
infected cells remained undefined. In 1970, the simultaneous
research of Temin and Baltimore led to the discovery of reverse
transcriptase, an enzyme that catalyzes the transcription of the
retroviral RNA into DNA (33), and
that is also present in RSV. Through reverse transcriptase, the
monoclonal RNA of the virus is converted to a double-stranded DNA,
and the viral genome is then incorporated into the nuclear DNA via
another enzyme, called integrase (34). Initially, it was considered that a
copy of the src transforming gene exists only within
infected cells (35-38).
In 1974, the laboratory of Michael Bishop and Harold Varmus, taking
advantage of the reverse transcriptase, undertook the design of a
special gene detector for src, in order to understand its
properties and origin. To their surprise, they found that the
src detector could also be hybridized with the genetic
material of non-infected cells of chicken and other species (two
copies per genome of diploid cells) (35,37,38).
They also observed that the more distant the evolutionary affinity
with the chicken, the weaker the degree of hybridization. The data
supported the idea that the src sequences found in
non-infected cells, are actually part of their normal genome (the
cellular version of src=c-src) (35-39).
In 1975, the Nobel prize was awarded to Temin and Baltimore, for
the discovery of reverse transcriptase (33,34).
From 1976 to 1980 the research focused on the
differences between the c-src and, the v-src, which
is located within the RSV genome. The first one exhibited
physiological cellular behavior as opposed to the second, which
acts as a potent oncogene. The explanation was simple; the
src gene of RSV was not initially present in the primordial
RSV retrovirus. A pre-existing virus (ALV=src negative) was
detected that caused leukosis in birds and which, through genetic
modifications, incorporated sequences from the genome of infected
cells (RSV=src positive). Subsequent experiments showed that
the structure of the RSV genome is closely related to this common
infectious agent of birds, called ALV (35-40).
Both of them include three genes: Gag, pol and
env. The gag gene encodes for proteins that take part
in the formation of the nucleoprotein nucleus; the pol gene
encodes for integrase and reverse transcriptase; and the env
gene determines the glycoprotein precursors. The only difference
between the two genomes lies in the ability of the src gene
to cause cellular transformation (39,40).
Thus, for the first time, the concept of proto-oncogene was
introduced, implying that a normal gene can be altered by mutation
or by a pre-viral insertion, to become an oncogene, thereby
contributing to cancer development. Since 1980, retroviruses have
been used as probes, to detect the corresponding proto-oncogenes in
humans, and researchers have shifted the focus on chemical
carcinogenesis (41-43).
This second theory confirmed the cellular origin of retroviral
oncogenes and additionally contributed to the unravelling of
possible mechanisms for proto-oncogene activation, such as
amplification, pre-viral insertion, single nucleotide polymorphism
and translocation (41-45).
In 1989, the Nobel prize was awarded to Bishop and Varmus for the
discovery of the cellular origin of retroviral oncogenes (46,47).
The most important historic milestones on Src research are
presented in Fig. 1.
4. MicroRNAs as the fine tuners of SRC
oncogenic signaling
As mentioned above, c-SRC is the first
reported oncogene and its product is the first non-receptor
tyrosine kinase to be identified (48). In many human neoplasms, including
colorectal, breast, prostate, pancreatic, head and neck, and lung
carcinomas, gliomas and melanoma, SRC overexpression has
already been detected. In fact, its dysregulation could be
characterized as an oncogenic signature and as a key factor for
tumor progression (3,5,49).
However, the molecular mechanisms underlying c-SRC-mediated tumor
progression are not fully understood. Recent studies have
highlighted several microRNAs (miRNAs) as key molecules in
SRC-mediated tumor progression (50). miRNAs are a family of small,
endogenous and evolutionarily conserved non-coding RNAs (containing
about 22 nucleotides) involved in the regulation of essential
cellular and functional processes, including proliferation,
differentiation, survival and stress responses. The majority of
miRNAs are transcribed from DNA sequences into primary miRNAs
(pri-miRNAs) and processed into precursor miRNAs (pre-miRNA), and
finally mature miRNAs. Their functionality is bimodal, since they
locate complementary mRNAs and either regulate protein translation
or induce degradation of the target mRNA (51). Hence, miRNAs act either as oncogenes
or tumor suppressors and are important regulators of gene
expression at the post-transcriptional level (52). In subsequent experiments, microarray
profiling revealed that c-SRC regulates a set of miRNAs, which act
as tumor suppressors, when their expression is downregulated.
Generally, miRNAs are commonly silenced in human cancers by
mutation, methylation, loss of heterogeneity or by other
post-transcriptional modifications (53). Studies on the function of these
miRNAs uncovered miRNA-mediated c-SRC oncogenic signaling and
crosstalk between Src and other oncogenic signaling
pathways, such as the focal adhesion-mediated pathway and the
mammalian target of rapamycin, mTOR (50).
Recently, the mechanisms underlying SRC-mediated
activation of mTOR signaling, a major downstream effector of the
PI3K pathway, were found to be regulated by miRNA expression in
various cancer types (54,55). More precisely, functional analysis
showed that transcription of miR-99a, which is often downregulated
in various human cancers, and is regulated by SRC-related oncogenic
pathways, like the epidermal growth factor receptor (EGFR) pathway.
It was demonstrated that mir-99a targets mTOR and fibroblast growth
factor receptor 3 (FGFR3), both of which are strongly related with
human cancers (56,57). In conclusion, this study indicated
that miR-99a is the missing link between SRC and mTOR, which have
both been correlated with human cancer. Furthermore, miRNA-mediated
mTOR regulation has also been shown in studies of miR-100 and
miR-199-3p (58,59). Further studies suggested that miRNAs
also regulate focal adhesion and activation of downstream effectors
in SRC-activated cancer cells. More specifically, integrin-linked
kinase (ILK) is targeted by miR-542-3p, a downregulated miRNA in
SRC-transformed cells (60-63).
Apart from the fact that downregulation of miR-542-3p corresponds
with upregulation of c-SRC and ILK, there is also a correlation
between ILK upregulation and c-SRC activation in human colon cancer
tissues. Furthermore, it was found that miR-542-3p-mediated ILK
downregulation induces inactivation of c-SRC and FAK in human colon
cancer cells (feedback loop).
Last but not least, miRNA mediates regulation of
SRC expression itself, and this could also be a logical
explanation for the resistance that is observed when SRC-targeting
drugs are used. In detail, miR-23b functions as a tumor suppressor
and as a mediator of metastasis in different cell lines (64). miR-27b, which targets paxillin, a
platform for adaptor proteins and a critical component of the focal
adhesion complex, is under the control of the PI3K pathway
(65-68).
Taking into consideration that both of them are downregulated in
human castration-resistant prostate cancers (69), c-SRC could be regulated by the
miR-23b/27b 24-1 gene cluster via a dual mechanism: Regulation of
c-SRC kinase activity via either miR-27b or miR-23b mediated
regulation of paxillin. As a result, upregulation of c-SRC
expression may amplify the positive-feedback loop mediated by the
miR-23b/27b 24-2 gene cluster thus, inducing tumor progression
mediated by c-SRC activity (50).
5. miRNA-mediated SRC oncogenic signaling in
selected cancer types
As many miRNAs are down-regulated in human cancers
through various genetic and epigenetic alterations, such as
methylation and loss of heterogeneity, research was focused on the
role of down-regulated miRNAs in c-SRC transformation (53). Subsequent experiments highlighted
the key role of miR-137 in the development of SRC-mediated human
colon cancer (70). To elucidate
the role of miR-137 and its correlation with SRC signaling, the
HCT116 cell line, anti-sense miRNAs and also dasatinib (a specific
SRC kinase inhibitor) were used. It was finally concluded that
miR-137 is down-regulated in the early stages of cancer progression
(70). In another experiment, the
role of miR-129-1-3p in human colon cancer was evaluated by
assessing miR-129-1-3p expression in 10 pairs of primary colon
tumors and adjacent non-cancerous tissues using qRT-PCR and western
blot analysis to examine the activity of SFK (SRC pY418). It was
clarified that miR-129-1-3p was markedly downregulated and SFK
activity was greatly upregulated in colon cancer tissues (71). Additional studies demonstrated that
certain miRNAs induce SRC oncogenic signaling by targeting SRCIN1,
a specific SRC kinase signaling inhibitor. For example, miR-665
suppresses SRCIN1 expression, which normally acts as a negative
regulator of MAPK/ERK signaling in ovarian cancer cells (72). In ovarian cancer, sustained
activation of MAPK/ERK signaling is associated with strong cell
proliferation and metastatic potential (73). The western blotting results showed
that inhibition of miR-665 increased SRCIN1, at both the mRNA and
protein level, and inactivated MAPK/ERK pathway in ovarian cancer
(74). Similar findings were
reported in the case of miR-150. It was observed that miR-150
promotes the proliferation and migration of lung cancer cells by
targeting SRC kinase signaling inhibitor 1 (SRCIN1), therefore
acting as an oncogene (75).
Subsequent studies examined the role of miR-17-5p in the evolution
of osteosarcoma and revealed a component of the
miR-17-5p/SRCIN1/EMT signaling pathway. Furthermore, classic EMT
markers such as N-cadherin, E-cadherin and Snail were quantified by
western blot analysis. Finally, it was proven that SRCIN1 is a
direct target of miR-17-5p and silencing of this miRNA could change
the expression of EMT markers and arrest cell growth (76). SRCIN1 was found to be downregulated
in breast cancer in previous studies (77). Moreover, miR-374a was shown to
induce cell proliferation, invasion and migration of gastric cancer
cell via binding to SRCIN1(78). It
was also found to be involved in pancreatic cancer through the axis
miR-374a/SRCIN1/EMT (79). Finally,
a recent study focused on the identification of miR-373 levels in
metastatic neuroblastoma samples and its interaction with
SRCIN1(80).
In conclusion, it becomes evident that miRNA
dysfunction is involved in various human cancers and miRNAs can
function as both oncogenes and tumor suppressors (81,82).
Due to their implication in the regulation of sustained cell growth
signaling, miRNAs are considered as potential biomarkers and
therapeutic targets for cancer treatment (83).
6. Exosomes as the fine tuners of oncogenic
signaling
As mentioned above, SRC functions as a
molecular signaling switch and plays a central role in the
regulation of cell proliferation, differentiation, adhesion, and
migration in normal cells (8), and
is commonly upregulated in various human cancer cells. The
activation of src is strictly regulated by several molecular
mechanisms. For example, the kinase activity of SRC is negatively
regulated by the phosphorylation of a regulatory tyrosine at its
c-terminal tail, catalyzed by CSK (84,85).
On the other hand, SRC is positively regulated through several
extracellular signals, such as growth factors and extracellular
matrices, which lead to the interaction with certain adaptor
proteins, including FAK and Cas (49,86),
and consequently to the activation of downstream signaling
pathways. Furthermore, cellular localization of SRC, determines its
activity. Inactive SRC is located to the perinuclear region, and
once activated, it is translocated to the plasma membrane, under
the control of members of the Rho family (87).
Recent studies have shown that activated SRC is
downregulated through degradation by either lysosomes or
proteasomes, with the functional difference between them remaining
unclear (10,88-90).
More precisely, the E3 ubiquitin ligase Cbl mediates the
ubiquitination of SRC and induces its degradation via the
ubiquitin-proteasome pathway (89,90).
In a recent study, ubiquitination of activated SRC at Lys429 was
demonstrated to promote its secretion via small extracellular
vesicles (sEVs) (91). In this
experiment, MDCK cells expressing a modified SRC that can be
activated by hydroxytamoxifen were used in order to mimic SRC
upregulated cancer cells. When proteasome inhibition (MG132) was
performed, no accumulation of ubiquitinated SRC was noted,
suggesting that ubiquitination of SRC preferentially promotes its
secretion via sEVs to decrease the levels of activated SRC in these
cells. It was also identified that Lys 429 is a critical
ubiquitination site required for sEV-mediated secretion. In an
attempt to determine how the mutation at Lys429 on SRC (R429)
affects the cell, it was observed that it caused resistance to
ubiquitination and decreased its secretion via sEVs. Additionally,
since the cbl ablation caused a less potent suppression of the sEV
secretion, it was hypothesized that other E3 ligases might also be
required. In addition, activation of R429 mutant enhanced
SRC-induced invasive phenotypes, supporting the hypothesis of a
stronger activation of FAK at the early stages (86,91).
These findings have shed light on this missing link between SRC
ubiquitination and sEV secretion, and suggest a tumor suppressive
role for the secretion of SRC via sEVs. The fact that SRC is
detected in exosomes from various cancer cells, such as colorectal
(92), prostate (93), and breast (94) cancer cells, indicates that secretion
of SRC via exosomes may be a common mechanism used to regulate SRC
in a wide array of cell types and seems to constitute a novel
promising therapeutic target (95).
7. SRC inhibitors as anticancer agents in
clinical trials
The role of SRC in oncogenesis has prompted
the detection of other members of the SRC family of protein kinases
and the search for anticancer therapies. To this end, most of the
FDA-approved inhibitors of related protein kinases are directed
toward neoplastic diseases. However, since SRC is not a primary
driver of tumorigenesis, but rather a participant in pathways of
cell division, invasion, migration and survival, administration of
existing inhibitors of SRC as a monotherapy has not been proved
efficient in cancer treatment (96). Moreover, there are currently no
available prognostic biomarkers related to SRC activity that could
be used for patient selection in clinical trials.
Currently, four oral SRC/multi-kinase inhibitors
have been approved by the FDA for the treatment of various
malignancies. Bosutinib, a BCR-Abl, SRC, Lyn, Hck, Kit, and PDGFR
inhibitor approved for the treatment of Philadelphia-positive
chronic myeloid leukemia (Ph+CML) and acute
lymphoblastic leukemia (ALL), is currently evaluated in clinical
trials for the treatment of breast cancer, glioblastoma and other
solid tumors (97-99).
Dasatinib, an inhibitor of BCR-Abl, SRC, Lck, Fyn, Yes, PDGFR, and
other kinases, approved for the treatment of CML is currently
evaluated in clinical trials against various solid tumors (100). This inhibitor is also evaluated in
combination with insulin-like growth factor 1 Receptor (IGF-1R)
antibody AMG479 against embryonal or alveolar rhabdomyosarcoma.
Ponatinib, an inhibitor of BCR-Abl, PDGFR, VEGFR, members of the
SRC family and other kinases, approved for the treatment of CML and
ALL is currently evaluated in clinical trials against several
leukemias (101). Vandetanib is an
inhibitor of EGFR, VEGFR, RET, members of the SRC family and other
kinases, approved for the treatment of medullary thyroid carcinoma
and is currently evaluated in clinical trials against numerous
solid tumors (102-104).
Saracatinib (AZD0530) an SRC and BCR-Abl inhibitor is currently
evaluated in clinical trials against colorectal, gastric, ovarian,
small and non-small cell lung cancers and against metastatic
osteosarcoma in the lung (105-107).
A related drug (AZD0424) alone or in combination with other agents
is in Phase I clinical trials against various types of solid
tumors. KX2-391 is another orally administered small molecule SRC
kinase inhibitor with potential antineoplastic activity.
Interestingly, instead of binding to the ATP-binding site, like
other SRC inhibitors, KX2-391 specifically binds to the peptide
substrate binding site of SRC kinase; in this way, kinase activity
is eliminated, potentially resulting in the inhibition of primary
tumor growth and the suppression of metastasis. This inhibitor is
being evaluated in clinical trials against multiple cancer types,
either alone or in combination with paclitaxel (108).
At present, there is a critical number of clinical
trials that investigate the therapeutic value of putative specific
SRC or SRC-related inhibitors as anti-cancer agents, alone or in
combination with other agents (Table
I) (108). The clinical
efficacy of these agents against the above-mentioned cancer types
remains to be established.
 | Table ICombinatorial treatments of specific
SRC or SRC-related inhibitors and other anti-cancer agents in
clinical trials (Only studies with published results are
shown). |
Table I
Combinatorial treatments of specific
SRC or SRC-related inhibitors and other anti-cancer agents in
clinical trials (Only studies with published results are
shown).
| SRC inhibitor | Combinatorial
treatment | Additional
molecular target(s) | Cancer type | Clinical phase | ClinicalTrials.gov Identifier | Results/Major side
effects |
|---|
| Dasatinib | Afatinib | EGFR | NSCLC | Phase I |
NCT01999985(109) | The MTD of Afatinib
in combination with Dasatinib was set to 40 and 140 mg,
respectively. All subjects showed an objective response rate within
6 months. PFS rate in participants with acquired EGFR resistance
was measured to 5.5 (2.6 to 8.5) months. Mainly mild adverse
effects including anemia, diarrhea, nausea, vomiting, cough and
fatigue. |
| | Trastuzumab,
Paclitaxel | HER2,
Chemotherapeutic treatment | Metastatic breast
cancer | Phase I/II |
NCT01306942(110) | ORR was 79.3% (95%
CI 60.3-92), clinical benefit rate 82.8% (95% CI 64.2-94.2). Median
time to progression 23.9 months, median PFS 23.9 months. No grade 4
toxicity was seen. Grade 3 toxicities included: Ejection fraction
decrease, neutropenia, hyponatremia, fatigue and sensory neuropathy
and one left ventricular systolic dysfunction. Phosphorylated
(p)-SRC was reduced in peripheral blood mononuclear cells.
Phosphorylated SRC, ERK and AKT were also reduced in epidermal
keratinocytes. |
| | Ixabepilone | Chemotherapeutic
treatment | Metastatic breast
cancer | Phase I/II |
NCT00924352(111) | The MTD of
dasatinib (taken daily, continuously) when given in combination
with ixabepilone (administered on Days 1, 8, and 15 of a 28-day
cycle) was determined at 100 mg. Respectively, the MTD of
ixabepilone was 20 mg/m2. The PFS of the Combination of
Dasatinib and Ixabepilone (Phase II) was 6.01 (2.92 to 8.08)
months. 19.64% faced serious adverse effects, while many patients
had diarrhea, neutropenia, anemia, nausea and fatigue. |
| | Docetaxel | Chemotherapeutic
treatment | Metastatic Hormone
Refractory Prostate Cancer | Phase I/II |
NCT00439270(112) | Thirteen of 46
patients (28%) had a grade 3-4 toxicity. Durable 50% PSA declines
occurred in 26 of 46 patients (57%). 60% had a partial response.
30% had disappearance of a lesion on bone scan. In bone marker
assessments, 33 of 38 (87%) and 26 of 34 (76%) had decreases in
urinary N-telopeptide or bone-specific alkaline phosphatase levels,
respectively. 61% received single-agent dasatinib after docetaxel
discontinuation and had stabilization of disease for an additional
1 to 12 months. |
| | Erlotinib | EGFR | NSCLC | Phase I/II |
NCT00826449(113) | MTD was 150 mg of
erlotinib and 70 mg of dasatinib daily based on 12 patients treated
in the phase I portion. The 35 NSCLC patients treated in phase II
had an overall disease control rate of 59% at 6 weeks. Five
patients (15%) had partial responses; all had activating EGFR
mutations. Median PFS was 3.3 months. |
| Bosutinib | Exemestane | Hormonal
antineoplastic treatment | Metastatic hormone
receptor-positive/HER2-negative breast cancer (in post-menopausal
women) | Phase II |
NCT00793546(114) | 93% of the patients
experienced treatment-related adverse effects, including diarrhea
and hepatotoxicity; 10% faced serious treatment-related adverse
effects. One patient (300 mg/day) achieved confirmed partial
response; three (400 mg/day, n=2; 300 mg/day, n=1) maintained
stable disease for >24 weeks; a best response of progressive
disease occurred in 36% of the patients. Median PFS was 12.3 weeks
(80% confidence interval: 11.0-15.6). |
| | Letrozole | Hormonal
antineoplastic treatment | Breast cancer (in
post-menopausal women) | Phase II |
NCT00880009(115) | 69% of the subjects
experienced treatment-related adverse effects, most commonly
diarrhea. Treatment-related hepatotoxicity occurred in 38%. One
patient achieved confirmed partial response; one had stable disease
for >24 weeks. |
| | Capecitabine | Chemotherapeutic
treatment | Advanced solid
tumors | Phase I/II |
NCT00959946(116) | No dose-limiting
toxicities observed. 6% experienced dose limiting toxicities. Most
common treatment-related adverse events were diarrhea, nausea,
vomiting, palmar-plantar erythrodysesthesia (PPE), fatigue. Best
overall confirmed partial response or stable disease >24 weeks
(all tumor types) was observed in 6 and 13% of patients. |
| | Imatinib | BCR-ABL | Chronic Myelogenous
Leukemia | Phase III |
NCT02130557(117) | The MMR rate at 12
months was significantly higher with bosutinib vs. imatinib (47.2%
vs. 36.9%, respectively; P=.02), as was complete cytogenetic
response (CCyR) rate by 12 months (77.2% vs. 66.4%, respectively;
P=.0075). Cumulative incidence was favorable with bosutinib with
earlier response times. 1.6% receiving bosutinib and 2.5% receiving
imatinib experienced disease progression to accelerated/blast
phase. 22.0% of patients receiving bosutinib and 26.8% of patients
receiving imatinib discontinued treatment, most commonly for
drug-related toxicity (12.7 and 8.7%, respectively). Cardiac and
vascular toxicities were uncommon. |
| Saracatinib
(AZD0530) | Carboplatin,
Paclitaxel | Chemotherapeutic
treatment | Advanced ovarian
cancer | Phase II |
NCT00610714(118) | ORR for triple
combination patients was 53,4%. PFS for triple combination was 8.28
(0 to 11.04) months, over 7.79 (0.72 to 12.12) months for double
combination without Saracatinib. Serious adverse effects were
recorded in 43.81% of the patients, including mainly febrile
neutropenia. Non-serious adverse effects were observed in 97.14% of
the subjects. |
| | Paclitaxel | Chemotherapeutic
treatment | Ovarian Cancer,
Fallopian Tube Cancer, Primary Peritoneal Cancer | Phase II/III |
NCT01196741(119) | The 6-month PFS
rate was 29% (Pxl + S) vs. 34% (wPxl + P) (P=0.582). Median PFS was
4.7 vs. 5.3 months (hazard ratio 1.00, 95% confidence interval
0.65-1.54; P=0.99). Rate Response (complete + partial) was 29%
(wPxl + S) vs. 43% (wPxl + P), P-value=0.158. Grade 3/4 adverse
events were 36% vs. 31% (P=0.624); the most frequent G3/4
toxicities were vomiting, abdominal pain and diarrhea. Febrile
neutropenia was more common in the saracatinib arm (4.3%) than
placebo (0%). Response, PFS and Overall survival were all
significantly (P<0.05) better in patients with taxane interval
≥6 months/no prior taxane (n=85) than those <6 months (n=22),
regardless of randomisation. |
8. Conclusion
The discovery of the Src gene was the trigger
for the emergence of other oncogenes, as well as the understanding
of the genetic basis of cancer. Therefore, different molecular
mechanisms are involved in tumor progression, differentiation and
migration. Despite the fact that the src gene is now well
studied, the molecular pathways mediating cancer progression have
not yet been clarified. The contribution of miRNAs and exosomes in
the acquisition of malignant phenotype may contribute an emerging
therapeutic strategy of combinational therapies with dual pathway
inhibition, although further studies are needed. Finally, both
exosomes and miRNAs could be useful diagnostic, prognostic and
predictive biomarkers in SRC-induced carcinogenesis, thus
contributing to a more rational and effective classification and
treatment of these patients.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
AS, GS, MG, DAS, SB and VZ contributed to the
conception, reference selection and writing of this work, and read
and approvel the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
DAS is the Editor-in-Chief for the journal, but had
no personal involvement in the reviewing process, or any influence
in terms of adjudicating on the final decision, for this article.
The other authors declare that they have no competing
interests.
References
|
1
|
Frame MC: Src in cancer: Deregulation and
consequences for cell behaviour. Biochim Biophys Acta.
1602:114–130. 2002.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ishizawar R and Parsons SJ: c-Src and
cooperating partners in human cancer. Cancer Cell. 6:209–214.
2004.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Summy JM and Gallick GE: Src family
kinases in tumor progression and metastasis. Cancer Metastasis Rev.
22:337–358. 2003.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Irby RB and Yeatman TJ: Role of Src
expression and activation in human cancer. Oncogene. 19:5636–5642.
2000.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Yeatman TJ: A renaissance for SRC. Nat Rev
Cancer. 4:470–480. 2004.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Kim LC, Song L and Haura EB: Src kinases
as therapeutic targets for cancer. Nat Rev Clin Oncol. 6:587–595.
2009.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Krishnan H, Miller WT and Goldberg GS: SRC
points the way to biomarkers and chemotherapeutic targets. Genes
Cancer. 3:426–435. 2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Brown MT and Cooper JA: Regulation,
substrates and functions of src. Biochim Biophys Acta.
1287:121–149. 1996.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Playford MP and Schaller MD: The interplay
between Src and integrins in normal and tumor biology. Oncogene.
23:7928–7946. 2004.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Hakak Y and Martin GS: Ubiquitin-dependent
degradation of active Src. Curr Biol. 9:1039–1042. 1999.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Laszlo GS and Cooper JA: Restriction of
Src activity by Cullin-5. Curr Biol. 19:157–162. 2009.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Okada M, Nada S, Yamanashi Y, Yamamoto T
and Nakagawa H: CSK: A protein-tyrosine kinase involved in
regulation of src family kinases. J Biol Chem. 266:24249–24252.
1991.PubMed/NCBI
|
|
13
|
Ingley E: Src family kinases: Regulation
of their activities, levels and identification of new pathways.
Biochim Biophys Acta. 1784:56–65. 2008.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Rous P: Transmission of a malignant new
growth by means of a cell-free filtrate. J Amer Med Assoc. 56:198.
1911.
|
|
15
|
Rous P: A sarcoma of the fowl
transmissible by an agent from the tumor cells. J Exp Med.
13:397–411. 1911.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Bittner JJ: The Milk-influence of breast
tumors in mice. Science. 95:462–463. 1942.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Gross L: ‘Spontaneous’ leukemia developing
in C3H mice following inoculation in infancy, with AK-leukemic
extracts, or AK-embrvos. Proc Soc Exp Biol Med. 76:27–32.
1951.PubMed/NCBI
|
|
18
|
Shope RE and Hurst EW: Infectious
papillomatosis of rabbits: With a note on the histopathology. J Exp
Med. 58:607–624. 1933.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Sweet BH and Hilleman MR: The vacuolating
virus, S.V.40. Proc Soc Exp Biol Med. 105:420–427. 1960.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Epstein MA, Achong BG and Barr YM: Virus
particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet.
1:702–703. 1964.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kurth R and Bannert N: Beneficial and
detrimental effects of human endogenous retroviruses. Int J Cancer.
126:306–314. 2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Boveri T: Zur Frage der Entstehung
maligner tumoren. Gustav Fischer, Jena, 1914 (In German).
|
|
23
|
Conney AH, Miller EC and Miller JA: The
metabolism of methylated aminoazo dyes. V. Evidence for induction
of enzyme synthesis in the rat by 3-methylcholanthrene. Cancer Res.
16:450–459. 1956.PubMed/NCBI
|
|
24
|
Cook JW, Hewett CL and Hieger I: 106. The
isolation of a cancer-producing hydrocarbon from coal tar. Parts I,
II, and III. J Chemical Soc (Resumed). 395–405. 1933.
|
|
25
|
Miller EC and Miller JA: The presence and
significance of bound aminoazo dyes in the livers of rats fed
para-dimethylaminoazobenzene. Cancer Res. 7:468–480. 1947.
|
|
26
|
Pott P: Chirurgical observations relative
to the cataract, the polypus of the nose, the cancer of the
scrotum, the different kinds of ruptures, and the mortification of
the toes and feet. Carnegy TJ for Hawes L, Clarke W and Collins R,
London, 1775.
|
|
27
|
Yamagiwa K and Ichikawa K: Experimental
study of the pathogenesis of carcinoma. CA Cancer J Clin.
27:174–181. 1977.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Temin HM and Rubin H: Characteristics of
an assay for rous sarcoma virus and rous sarcoma cells in tissue
culture. Virology. 6:669–688. 1958.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Duesberg PH and Vogt PK: Differences
between ribonucleic acids of transforming and nontransforming avian
tumor viruses. Proc Natl Acad Sci USA. 67:1673–1680.
1970.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Martin GS: Rous sarcoma virus: A function
required for maintenance of transformed state. Nature.
227:1021–1023. 1970.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Toyoshima K and Vogt PK: Temperature
sensitive mutants of an avian sarcoma virus. Virology. 39:930–931.
1969.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Crawford LV and Crawford EM: Properties of
rous sarcoma virus purified by density gradient centrifugation.
Virology. 13:227–232. 1961.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Baltimore D: Viral Rna-Dependent DNA
Polymerase-Rna-Dependent DNA polymerase in virions of rna tumour
viruses. Nature. 226:1209–1211. 1970.
|
|
34
|
Temin HM and Mizutani S: RNA-dependent DNA
polymerase in virions of Rous sarcoma virus. Nature. 226:1211–1213.
1970.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Ali M and Baluda MA: Synthesis of avian
oncornavirus DNA in infected chicken cells. J Virol. 13:1005–1013.
1974.PubMed/NCBI
|
|
36
|
Baluda MA: Widespread presence, in
chickens, of DNA complementary to the RNA genome of avian leukosis
viruses. Proc Natl Acad Sci USA. 69:576–580. 1972.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Hayward WS and Hanafusa H: Detection of
avian tumor virus RNA in uninfected chicken embryo cells. J Virol.
11:157–167. 1973.PubMed/NCBI
|
|
38
|
Stehelin D, Varmus HE, Bishop JM and Vogt
PK: DNA related to the transforming gene(s) of avian sarcoma
viruses is present in normal avian DNA. Nature. 260:170–173.
1976.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Baluda MA and Drohan WN: Distribution of
deoxyribonucleic acid complementary to the ribonucleic acid of
avian myeloblastosis virus in tissues of normal and tumor-bearing
chickens. J Virol. 10:1002–1009. 1972.PubMed/NCBI
|
|
40
|
Hanafusa T, Hanafusa H and Miyamoto T:
Recovery of a new virus from apparently normal chick cells by
infection with avian tumor viruses. Proc Natl Acad Sci USA.
67:1797–1803. 1970.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Perucho M, Goldfarb M, Shimizu K, Lama C,
Fogh J and Wigler M: Human-tumor-derived cell lines contain common
and different transforming genes. Cell. 27:467–476. 1981.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Shih C, Padhy LC, Murray M and Weinberg
RA: Transforming genes of carcinomas and neuroblastomas introduced
into mouse fibroblasts. Nature. 290:261–264. 1981.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Shih C, Shilo BZ, Goldfarb MP, Dannenberg
A and Weinberg RA: Passage of phenotypes of chemically transformed
cells via transfection of DNA and chromatin. Proc Natl Acad Sci
USA. 76:5714–5718. 1979.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Reddy EP, Reynolds RK, Santos E and
Barbacid M: A point mutation is responsible for the acquisition of
transforming properties by the T24 human bladder carcinoma
oncogene. Nature. 300:149–152. 1982.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Tabin CJ, Bradley SM, Bargmann CI,
Weinberg RA, Papageorge AG, Scolnick EM, Dhar R, Lowy DR and Chang
EH: Mechanism of activation of a human oncogene. Nature.
300:143–149. 1982.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Raju TN: The Nobel Chronicles. 1989: John
Michael Bishop (b 1936) and Harold Eliot Varmus (b 1939). Lancet.
355(1106)2000.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Temin HM: Origin of retroviruses from
cellular moveable genetic elements. Cell. 21:599–600.
1980.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Jove R and Hanafusa H: Cell transformation
by the viral src oncogene. Annu Rev Cell Biol. 3:31–56.
1987.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Frame MC: Newest findings on the oldest
oncogene; how activated src does it. J Cell Sci. 117:989–998.
2004.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Oneyama C and Okada M: MicroRNAs as the
fine-tuners of Src oncogenic signalling. J Biochem. 157:431–438.
2015.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Ha M and Kim VN: Regulation of microRNA
biogenesis. Nat Rev Mol Cell Biol. 15:509–524. 2014.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Zhang B, Pan X, Cobb GP and Anderson TA:
microRNAs as oncogenes and tumor suppressors. Dev Biol. 302:1–12.
2007.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Mamane Y, Petroulakis E, LeBacquer O and
Sonenberg N: mTOR, translation initiation and cancer. Oncogene.
25:6416–6422. 2006.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Wullschleger S, Loewith R and Hall MN: TOR
signaling in growth and metabolism. Cell. 124:471–484.
2006.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Engelman JA: Targeting PI3K signalling in
cancer: Opportunities, challenges and limitations. Nat Rev Cancer.
9:550–562. 2009.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Oneyama C, Ikeda J, Okuzaki D, Suzuki K,
Kanou T, Shintani Y, Morii E, Okumura M, Aozasa K and Okada M:
MicroRNA-mediated downregulation of mTOR/FGFR3 controls tumor
growth induced by Src-related oncogenic pathways. Oncogene.
30:3489–3501. 2011.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Doghman M, El Wakil A, Cardinaud B, Thomas
E, Wang J, Zhao W, Peralta-Del Valle MH, Figueiredo BC, Zambetti GP
and Lalli E: Regulation of insulin-like growth factor - mammalian
target of rapamycin signaling by microRNA in childhood
adrenocortical tumors. Cancer Res. 70:4666–4675. 2010.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Fornari F, Milazzo M, Chieco P, Negrini M,
Calin GA, Grazi GL, Pollutri D, Croce CM, Bolondi L and Gramantieri
L: miR-199a-3p regulates mTOR and c-Met to influence the
doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res.
70:5184–5193. 2010.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Hannigan G, Troussard AA and Dedhar S:
Integrin-linked kinase: A cancer therapeutic target unique among
its ILK. Nat Rev Cancer. 5:51–63. 2005.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Hannigan GE, Leung-Hagesteijn C,
Fitz-Gibbon L, Coppolino MG, Radeva G, Filmus J, Bell JC and Dedhar
S: Regulation of cell adhesion and anchorage-dependent growth by a
new beta 1-integrin-linked protein kinase. Nature. 379:91–96.
1996.PubMed/NCBI View Article : Google Scholar
|
|
62
|
McDonald PC, Fielding AB and Dedhar S:
Integrin-linked kinase - essential roles in physiology and cancer
biology. J Cell Sci. 121:3121–3132. 2008.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Oneyama C, Morii E, Okuzaki D, Takahashi
Y, Ikeda J, Wakabayashi N, Akamatsu H, Tsujimoto M, Nishida T,
Aozasa K and Okada M: MicroRNA-mediated upregulation of
integrin-linked kinase promotes Src-induced tumor progression.
Oncogene. 31:1623–1635. 2012.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Donadelli M, Dando I, Fiorini C and
Palmieri M: Regulation of miR-23b expression and its dual role on
ROS production and tumour development. Cancer Lett. 349:107–113.
2014.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Azuma K, Tanaka M, Uekita T, Inoue S,
Yokota J, Ouchi Y and Sakai R: Tyrosine phosphorylation of paxillin
affects the metastatic potential of human osteosarcoma. Oncogene.
24:4754–4764. 2005.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Navon R, Wang H, Steinfeld I, Tsalenko A,
Ben-Dor A and Yakhini Z: Novel rank-based statistical methods
reveal microRNAs with differential expression in multiple cancer
types. PLoS One. 4(e8003)2009.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Shi J, Wang S, Zhao E, Shi L, Xu X and
Fang M: Paxillin expression levels are correlated with clinical
stage and metastasis in salivary adenoid cystic carcinoma. J Oral
Pathol Med. 39:548–551. 2010.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Taylor BS, Schultz N, Hieronymus H,
Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva
B, et al: Integrative genomic profiling of human prostate cancer.
Cancer Cell. 18:11–22. 2010.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Majid S, Dar AA, Saini S, Arora S,
Shahryari V, Zaman MS, Chang I, Yamamura S, Tanaka Y, Deng G, et
al: miR-23b represses proto-oncogene Src kinase and functions as
methylation-silenced tumor suppressor with diagnostic and
prognostic significance in prostate cancer. Cancer Res.
72:6435–6446. 2012.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Kokuda R, Watanabe R, Okuzaki D, Akamatsu
H and Oneyama C: MicroRNA.137.mediated Src oncogenic signaling
promotes cancer progression. Genes Cells: Jul 2, 2018 (Epub ahead
of print).
|
|
71
|
Okuzaki D, Yamauchi T, Mitani F, Miyata M,
Ninomiya Y, Watanabe R, Akamatsu H and Oneyama C: c-Src promotes
tumor progression through downregulation of microRNA-129-1-3p.
Cancer Sci. 111:418–428. 2020.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Kennedy S, Clynes M, Doolan P, Mehta JP,
Rani S, Crown J and O'Driscoll L: SNIP/p140Cap mRNA expression is
an unfavourable prognostic factor in breast cancer and is not
expressed in normal breast tissue. Br J Cancer. 98:1641–1645.
2008.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Yu Z, Ye S, Hu G, Lv M, Tu Z, Zhou K and
Li Q: The RAF-MEK-ERK pathway: Targeting ERK to overcome obstacles
to effective cancer therapy. Future Med Chem. 7:269–289.
2015.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Zhou P, Xiong T, Yao L and Yuan J:
MicroRNA-665 promotes the proliferation of ovarian cancer cells by
targeting SRCIN1. Exp Ther Med. 19:1112–1120. 2020.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Cao M, Hou D, Liang H, Gong F, Wang Y, Yan
X, Jiang X, Wang C, Zhang J, Zen K, et al: miR-150 promotes the
proliferation and migration of lung cancer cells by targeting SRC
kinase signalling inhibitor 1. Eur J Cancer. 50:1013–1024.
2014.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Zhao X, Xu Y, Sun X, Ma Y, Zhang Y and
Wang Y, Guan H, Jia Z, Li Y and Wang Y: miR-17-5p promotes
proliferation and epithelial-mesenchymal transition in human
osteosarcoma cells by targeting SRC kinase signaling inhibitor 1. J
Cell Biochem. 120:5495–5504. 2019.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Yang F, Luo LJ, Zhang L, Wang DD, Yang SJ,
Ding L, Li J, Chen D, Ma R, Wu JZ and Tang JH: miR-346 promotes the
biological function of breast cancer cells by targeting SRCIN1 and
reduces chemosensitivity to docetaxel. Gene. 600:21–28.
2017.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Xu X, Wang W, Su N, Zhu X, Yao J, Gao W,
Hu Z and Sun Y: miR-374a promotes cell proliferation, migration and
invasion by targeting SRCIN1 in gastric cancer. FEBS Lett.
589:407–413. 2015.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Ma L, Shao Z and Zhao Y: MicroRNA-374a
promotes pancreatic cancer cell proliferation and epithelial to
mesenchymal transition by targeting SRCIN1. Pathol Res Pract.
215(152382)2019.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Yuan XL, Wen FQ, Chen XW, Jiang XP and Liu
SX: miR-373 promotes neuroblastoma cell proliferation, migration,
and invasion by targeting SRCIN1. Onco Targets Ther. 12:4927–4936.
2019.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev Cancer. 6:259–269.
2006.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Lin PY, Yu SL and Yang PC: MicroRNA in
lung cancer. Br J Cancer. 103:1144–1148. 2010.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Pal MK, Jaiswar SP, Dwivedi VN, Tripathi
AK, Dwivedi A and Sankhwar P: MicroRNA: A new and promising
potential biomarker for diagnosis and prognosis of ovarian cancer.
Cancer Biol Med. 12:328–341. 2015.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Nada S, Okada M, MacAuley A, Cooper JA and
Nakagawa H: Cloning of a complementary DNA for a protein-tyrosine
kinase that specifically phosphorylates a negative regulatory site
of p60c-src. Nature. 351:69–72. 1991.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Nada S, Yagi T, Takeda H, Tokunaga T,
Nakagawa H, Ikawa Y, Okada M and Aizawa S: Constitutive activation
of Src family kinases in mouse embryos that lack Csk. Cell.
73:1125–1135. 1993.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Mitra SK and Schlaepfer DD:
Integrin-regulated FAK-Src signaling in normal and cancer cells.
Curr Opin Cell Biol. 18:516–523. 2006.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Fincham VJ, Unlu M, Brunton VG, Pitts JD,
Wyke JA and Frame MC: Translocation of Src kinase to the cell
periphery is mediated by the actin cytoskeleton under the control
of the Rho family of small G proteins. J Cell Biol. 135:1551–1564.
1996.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Harris KF, Shoji I, Cooper EM, Kumar S,
Oda H and Howley PM: Ubiquitin-mediated degradation of active Src
tyrosine kinase. Proc Natl Acad Sci USA. 96:13738–13743.
1999.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Reinecke J and Caplan S: Endocytosis and
the Src family of non-receptor tyrosine kinases. Biomol Concepts.
5:143–155. 2014.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Tu C, Ortega-Cava CF, Winograd P, Stanton
MJ, Reddi AL, Dodge I, Arya R, Dimri M, Clubb RJ, Naramura M, et
al: Endosomal-sorting complexes required for transport (ESCRT)
pathway-dependent endosomal traffic regulates the localization of
active Src at focal adhesions. Proc Natl Acad Sci USA.
107:16107–16112. 2010.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Tanaka K, Ito Y, Kajiwara K, Nada S and
Okada M: Ubiquitination of Src promotes its secretion via small
extracellular vesicles. Biochem Biophys Res Commun: Feb 18, 2020
(Epub ahead of print).
|
|
92
|
Ji H, Greening DW, Barnes TW, Lim JW,
Tauro BJ, Rai A, Xu R, Adda C, Mathivanan S, Zhao W, et al:
Proteome profiling of exosomes derived from human primary and
metastatic colorectal cancer cells reveal differential expression
of key metastatic factors and signal transduction components.
Proteomics. 13:1672–1686. 2013.PubMed/NCBI View Article : Google Scholar
|
|
93
|
DeRita RM, Zerlanko B, Singh A, Lu H,
Iozzo RV, Benovic JL and Languino LR: c-Src, insulin-like growth
factor I receptor, G-protein-coupled receptor kinases and focal
adhesion kinase are enriched into prostate cancer cell exosomes. J
Cell Biochem. 118:66–73. 2017.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Imjeti NS, Menck K, Egea-Jimenez AL,
Lecointre C, Lembo F, Bouguenina H, Badache A, Ghossoub R, David G,
Roche S, et al: Syntenin mediates SRC function in exosomal
cell-to-cell communication. Proc Natl Acad Sci USA.
114:12495–12500. 2017.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Hikita T, Kuwahara A, Watanabe R, Miyata M
and Oneyama C: Src in endosomal membranes promotes exosome
secretion and tumor progression. Sci Rep. 9(3265)2019.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Roskoski R Jr: Src protein-tyrosine kinase
structure, mechanism, and small molecule inhibitors. Pharmacol Res.
94:9–25. 2015.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Daud AI, Krishnamurthi SS, Saleh MN,
Gitlitz BJ, Borad MJ, Gold PJ, Chiorean EG, Springett GM, Abbas R,
Agarwal S, et al: Phase I study of bosutinib, a src/abl tyrosine
kinase inhibitor, administered to patients with advanced solid
tumors. Clin Cancer Res. 18:1092–1100. 2012.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Moy B, Neven P, Lebrun F, Bellet M, Xu B,
Sarosiek T, Chow L, Goss P, Zacharchuk C, Leip E, et al: Bosutinib
in combination with the aromatase inhibitor letrozole: A phase II
trial in postmenopausal women evaluating first-line endocrine
therapy in locally advanced or metastatic hormone
receptor-positive/HER2-negative breast cancer. Oncologist.
19:348–349. 2014.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Taylor JW, Dietrich J, Gerstner ER, Norden
AD, Rinne ML, Cahill DP, Stemmer-Rachamimov A, Wen PY, Betensky RA,
Giorgio DH, et al: Phase 2 study of bosutinib, a Src inhibitor, in
adults with recurrent glioblastoma. J Neurooncol. 121:557–563.
2015.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Araujo J and Logothetis C: Dasatinib: A
potent SRC inhibitor in clinical development for the treatment of
solid tumors. Cancer Treat Rev. 36:492–500. 2010.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Cortes JE, Kim DW, Pinilla-Ibarz J, Le
Coutre P, Paquette R, Chuah C, Nicolini FE, Apperley JF, Khoury HJ,
Talpaz M, et al: A phase 2 trial of ponatinib in Philadelphia
chromosome-positive leukemias. N Engl J Med. 369:1783–1796.
2013.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Ahn JS, Lee KH, Sun JM, Park K, Kang ES,
Cho EK, Lee DH, Kim SW, Lee GW, Kang JH, et al: A randomized, phase
II study of vandetanib maintenance for advanced or metastatic
non-small-cell lung cancer following first-line platinum-doublet
chemotherapy. Lung Cancer. 82:455–460. 2013.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Gridelli C, Novello S, Zilembo N, Luciani
A, Favaretto AG, De Marinis F, Genestreti G, Crino L, Grossi F,
Caffo O, et al: Phase II randomized study of vandetanib plus
gemcitabine or gemcitabine plus placebo as first-line treatment of
advanced non-small-cell lung cancer in elderly patients. J Thorac
Oncol. 9:733–737. 2014.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Sim MW and Cohen MS: The discovery and
development of vandetanib for the treatment of thyroid cancer.
Expert Opin Drug Discov. 9:105–114. 2014.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Gucalp A, Sparano JA, Caravelli J,
Santamauro J, Patil S, Abbruzzi A, Pellegrino C, Bromberg J, Dang
C, Theodoulou M, et al: Phase II trial of saracatinib (AZD0530), an
oral SRC-inhibitor for the treatment of patients with hormone
receptor-negative metastatic breast cancer. Clin Breast Cancer.
11:306–311. 2011.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Laurie SA, Goss GD, Shepherd FA, Reaume
MN, Nicholas G, Philip L, Wang L, Schwock J, Hirsh V, Oza A, et al:
A phase II trial of saracatinib, an inhibitor of src kinases, in
previously-treated advanced non-small-cell lung cancer: The
Princess Margaret Hospital phase II consortium. Clin Lung Cancer.
15:52–57. 2014.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Molina JR, Foster NR, Reungwetwattana T,
Nelson GD, Grainger AV, Steen PD, Stella PJ, Marks R, Wright J and
Adjei AA: A phase II trial of the Src-kinase inhibitor saracatinib
after four cycles of chemotherapy for patients with extensive stage
small cell lung cancer: NCCTG trial N-0621. Lung Cancer.
85:245–250. 2014.PubMed/NCBI View Article : Google Scholar
|
|
108
|
U.S. National Library of Medicine:
https://clinicaltrials.gov/.
https://clinicaltrials.gov.
Accessed July 1, 2020.
|
|
109
|
U.S. National Library of Medicine: Phase I
Trial of Afatinib (BIBW 2992) and dasatinib in non-small cell lung
cancer (NSCLC). https://clinicaltrials.gov/
Identifier: NCT01999985. https://ClinicalTrials.gov/show/NCT01999985].
Accessed June 14, 2019.
|
|
110
|
U.S. National Library of Medicine:
Dasatinib in combination with trastuzumab and paclitaxel in first
line treatment of Her2-Positive MBC patients. https://clinicaltrials.gov/
Identifier: NCT01306942. https://ClinicalTrials.gov/show/NCT01306942].
Accessed September 9, 2019.
|
|
111
|
U.S. National Library of Medicine: Trial
of dasatinib plus ixabepilone in 2nd or 3rd line metastatic breast
cancer. https://clinicaltrials.gov/
Identifier: NCT00924352. https://ClinicalTrials.gov/show/NCT00924352].
Accessed September 29, 2014.
|
|
112
|
U.S. National Library of Medicine: Study
of dasatinib and docetaxel in metastatic hormone refractory
prostate cancer. https://clinicaltrials.gov/
Identifier: NCT00439270. https://ClinicalTrials.gov/show/NCT00439270].
Accessed November 27, 2013.
|
|
113
|
U.S. National Library of Medicine:
Dasatinib and Erlotinib in Non-Small Cell Lung Cancer (NSCLC).
https://clinicaltrials.gov/
Identifier: NCT00826449. https://ClinicalTrials.gov/show/NCT00826449].
Accessed October 21, 2015.
|
|
114
|
U.S. National Library of Medicine: Study
evaluating bosutinib-exemestane combination vs exemestane alone in
post menopausal women with breast cancer. https://clinicaltrials.gov/
Identifier: NCT00793546. https://ClinicalTrials.gov/show/NCT00793546].
Accessed November 5, 2012.
|
|
115
|
U.S. National Library of Medicine: Study
evaluating bosutinib-letrozole combination versus letrozole alone
in post menopausal women with breast cancer. https://clinicaltrials.gov/
Identifier: NCT00880009. https://ClinicalTrials.gov/show/NCT00880009].
Accessed November 5, 2012.
|
|
116
|
U.S. National Library of Medicine: Study
of bosutinib with capecitabine in solid tumors and locally advanced
or metastatic breast cancer. https://clinicaltrials.gov/
Identifier: NCT00959946. https://ClinicalTrials.gov/show/NCT00959946].
Accessed February 22, 2013.
|
|
117
|
U.S. National Library of Medicine: A
Multicenter Phase 3, Open-label study of bosutinib versus imatinib
in adult patients with newly diagnosed chronic phase chronic
myelogenous leukemia. https://clinicaltrials.gov/
Identifier: NCT02130557. https://ClinicalTrials.gov/show/NCT02130557].
Accessed November 14, 2018.
|
|
118
|
U.S. National Library of Medicine: AZD0530
Phase II study in patients with advanced ovarian cancer. https://clinicaltrials.gov/
Identifier: NCT00610714. https://ClinicalTrials.gov/show/NCT00610714].
Accessed September 20, 2011.
|
|
119
|
U.S. National Library of Medicine:
Saracatinib and paclitaxel in platinum-resistant ovarian cancer.
https://clinicaltrials.gov/
Identifier: NCT01196741. https://ClinicalTrials.gov/show/NCT01196741].
Accessed May 5, 2015.
|