Introduction
Langerhans Cell Histiocytosis (LCH) is a rare
disorder defined as a subgroup of myeloid malignancies consistent
with disseminated infiltration and clonal proliferation of
Langerhans cells, a specific type of immature CD1a-positive cells.
These cells are named after Paul Langerhans, a 19th century young
doctor who first identified them as epidermal cells of
extracutaneous nerves using gold colloid staining technique.
Actually, we now know that epidermal Langerhans cells are dendritic
cells, a heterogeneous group of hematopoietic cells enriched in
interface tissues throughout the body, mainly the skin, lungs,
liver, bone marrow and lymphoid organs. These cells help regulate
the immune system, presenting antigens to and activating
antigen-specific T cells (1). LCH
can appear at any age, but it is found usually during childhood,
mainly between ages 2 and 3, with an annual incidence of 4.6 cases
per 1 million children under 15 years of age. The estimated
incidence among adults is 1 to 2 cases per million, though LCH is
probably underdiagnosed in this population. Race and ethnic
background appear to influence the risk of developing LCH, with a
suspected higher risk among Caucasians, particularly in Northern
Europe (2). LCH can present before,
after, or along with other histologic cancers, frequently with
shared mutations suggesting clonality, though it is not clear
whether a history of LCH confers an increased risk of cancer
(3).
The natural history of LCH consists of an insidious
onset and intermittent remissive pattern, clinical manifestations
of LCH vary from a self-limiting single bone disease to rapidly
fatal multi-systemic one. The prognosis of LCH is closely related
to age of onset (usually better outcome in adult patients),
internal organs involvement and degree of functional impairment
(3).
Case report
A 31-year-old woman was referred to Internal
Medicine consultation because of a 2-month history of polydipsia
(daily water intake around 8 liters per day) and polyuria, lately
associated with exertional dyspnoea and episodic non-productive
cough. She denied weight loss, anorexia and recent use of
medication. She did not present emotional lability or other
psychological distress. The patient was an active smoker, with
familial history of multiple myeloma and Hodgkin disease. During
physical examination, she had some fine crackles at pulmonary
auscultation, with no other significant findings. Respiratory
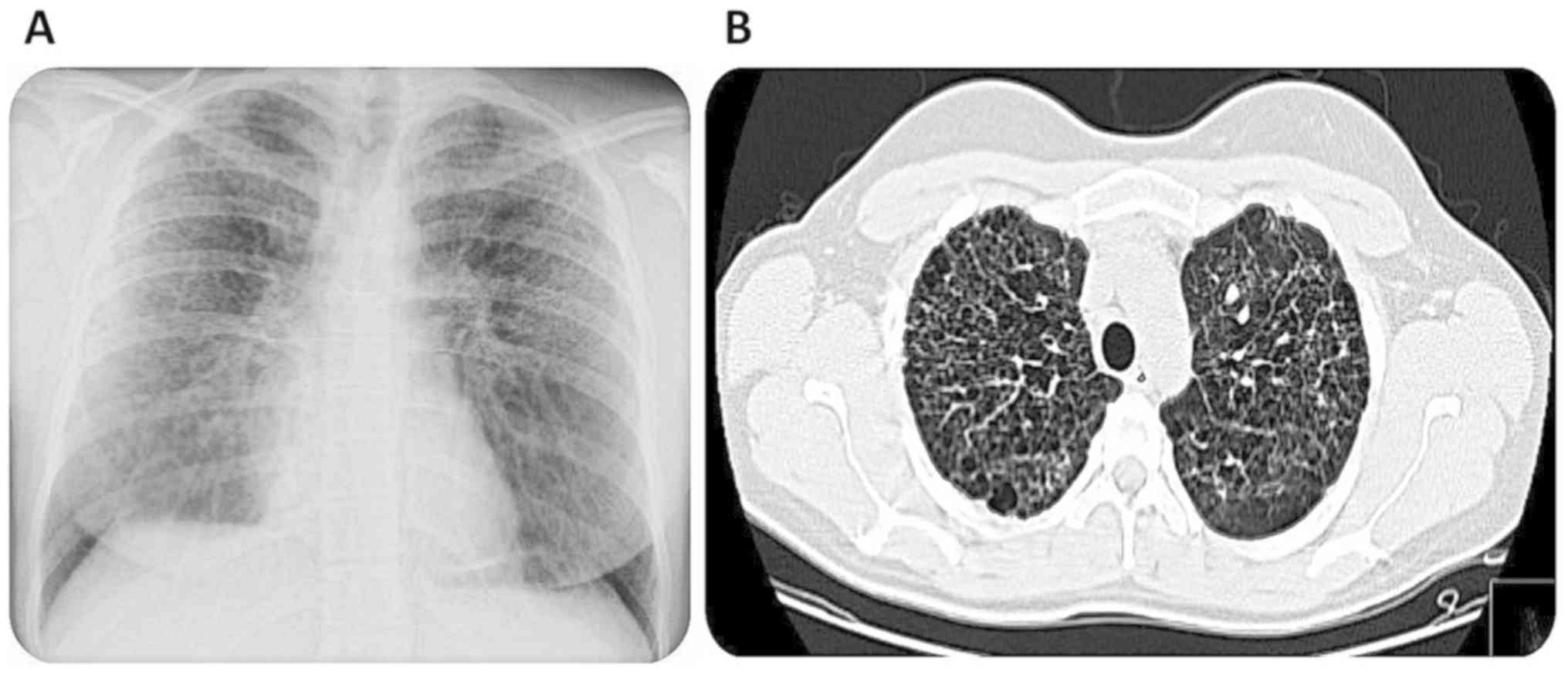
failure was excluded. During ambulatory investigation, the thoracic
x-ray detected a diffuse a reticulonodular pattern (Fig. 1A) and further pulmonary assessment
with thoracic CT scan was performed which showed bilateral
bronchiectasis, interstitial fibrosis and bullous emphysema
(Fig. 1B). Given these findings, a
bronchofibroscopy was performed, which was eventually cancelled
given her intolerance to fasting and her need to maintain water
intake. Instead, it was decided for hospitalization in order to
perform a surgical biopsy of the middle lobe and also to conclude
the diagnostic workup related to her polydipsic and polyuric state.
During hospitalization, the patient presented normal renal function
(serum BUN of 54 mg/dl and serum creatinine of 0.94 mg/dl), normal
levels of adrenocorticotropic hormone (ACTH), and
thyroid-stimulating hormone (TSH) as well as serum eletrolytes
(sodium 137 mmol/l, potassium 3.8 mmol/l and calcium 8.8 mg/dl).
Her biochemical profile also included SACE levels (serum
angiotensin converting enzyme) of 47 U/l (normal value <50 U/l
and serum erythrocyte sedimentation rate of 20 mm/h (normal value
<12 mm/h). She was submitted to a water deprivation test which
lasted for 3 h and presented a serum sodium concentration (sNa) of
145 mmol/l with serum osmolality (SO) of 309 mOsm/kg and urine
osmolality (UO) of 242 mOsm/kg). There was a significant clinical
response after administration of 10 µg intra-nasal desmopressin
(sNa of 138 mmol/l with SO of 272 mOsm/kg and UO of 831 mOsm/kg),
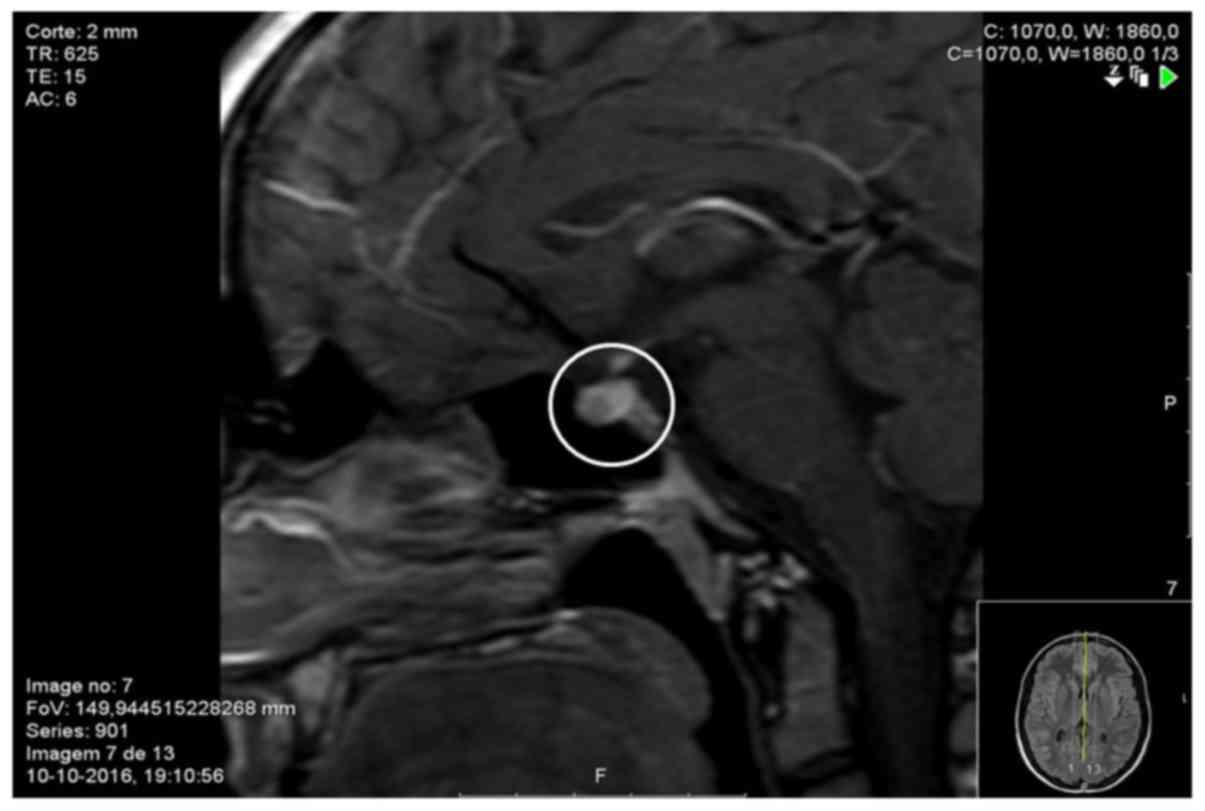
compatible with central diabetes insipidus (CDI). A cranial MRI was
also performed, which showed absence of posterior pituitary
T1-weighted hypersignal and pituitary stalk thickening of 4 mm
(normal value <3.5 mm) (Fig.
2).
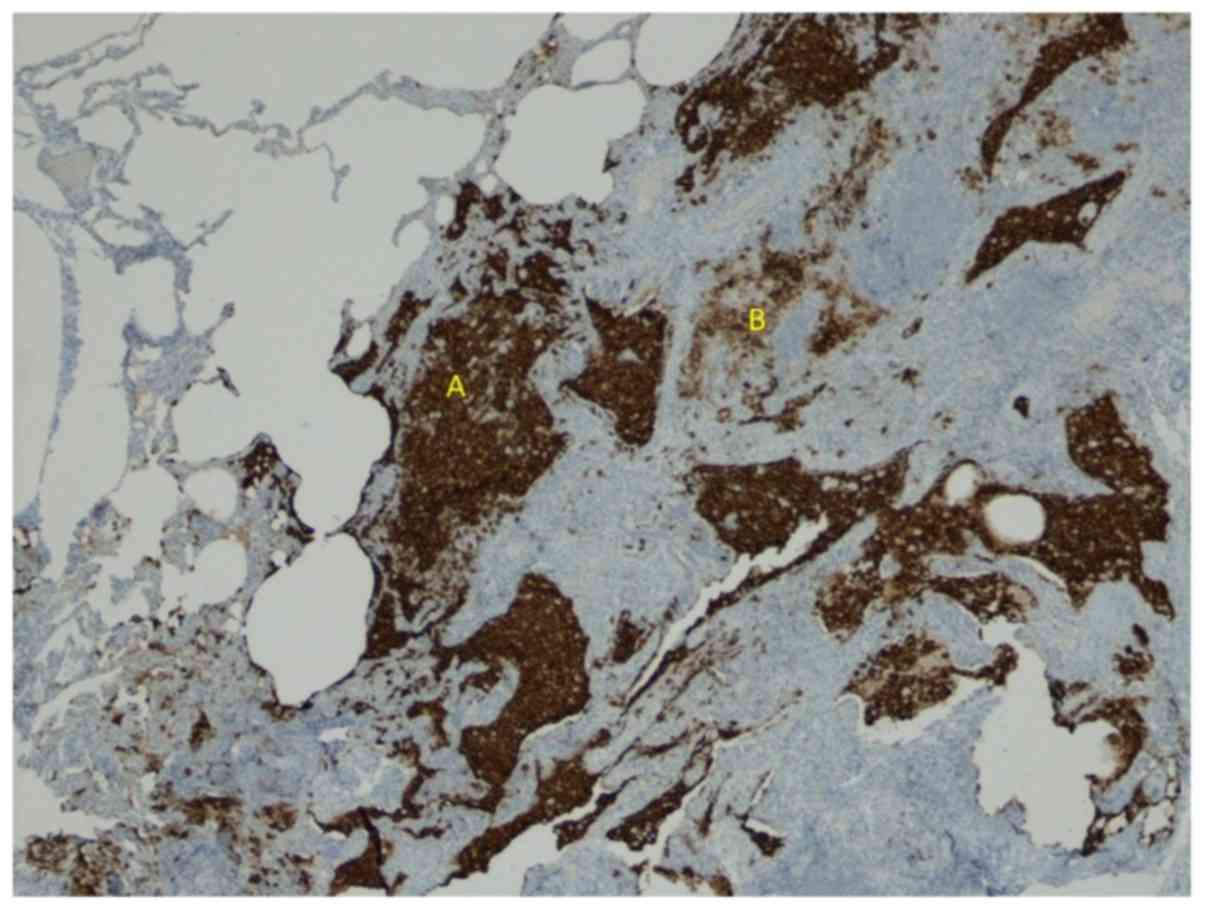
Meanwhile, the histologic exam of lung biopsy
revealed a centrilobular emphysema with intra-alveolar macrophage
desquamation, lymphoplasmocytic infiltration and juxtapleural
confluence of Langerhans cells-CD1a and protein S100 positivity
(Fig. 3).
It was assumed that the water balance disorder and
the respiratory symptoms could be related to LCH affecting both
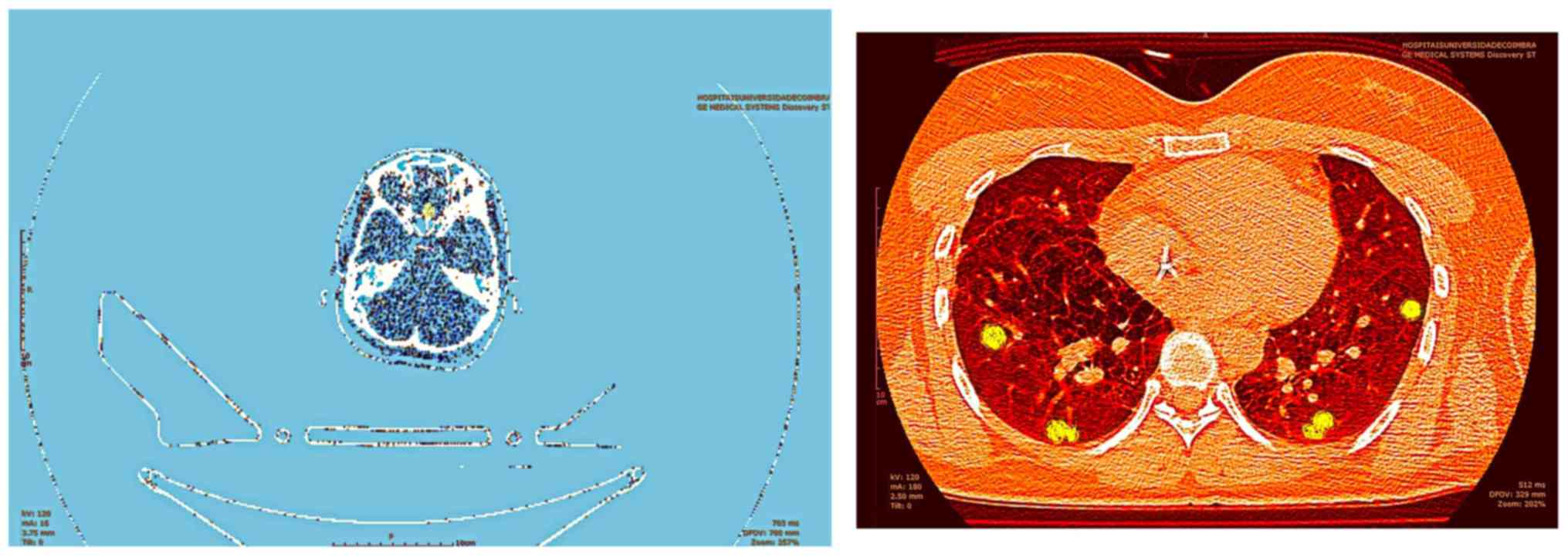
brain and lungs. The patient was discharged maintaining treatment
with desmopressin and underwent ambulatory staging PET-CT which
confirmed pituitary and pulmonary uptake of 18FDG
(Fig. 4). After multidisciplinary
evaluation, the patient began combination therapy with prednisone
and cytarabine. The patient quit smoking and showed complete
resolution of polyuria and polydipsia within the first month of
treatment (oral desmopressin plus cytarabine/prednisone); she also
noticed significant improvements over her respiratory symptoms 6
months later.
Discussion
The Histiocyte Society proposed a classification of
different forms of histiocytosis, according to its cellular
pattern, distinguishing 3 categories: (1) dendritic cells disorders (including
LCH); (2) macrophage-related
disorders; and (3) malignant
histiocytosis (4). At present, LCH
is classified as single-system (SS) and multisystem (MS)
histiocytosis, the latter being divided in two groups depending on
whether risk organs (RO) are involved (liver, lung, spleen and bone
marrow) (5). The most important
clinical LCH syndromes are eosinophilic granuloma (SS),
Hand-Schüller-Christian disease (MS, RO-negative) and Letterer-Siwe
disease (MS, RO-positive) (6). Some
authors assume that histiocytic infiltration appears to be
dominated by regulatory T-cell disfunction (which fail to
neutralize the histiocytes), rather than an hyperproliferative
process alone (7). There is still
an active debate regarding LCH pathogenesis because it appears that
this disease apparently presents features of both chronic
inflammatory disease (presence of circulating pro-inflammatory
cytokines such as TNF, IFN, IL-2, IL-12 and IL-17) (7) and neoplastic disease (presence of
proto-oncogenic BRAF V600 and MEK-1 mutations) (8,9).
The biopsy of one of the affected organs and its
immunohistochemical (IHC) findings are crucial to confirm the
diagnosis of LCH. The main differences between LCH and other forms
of histiocytosis are based on specific profile markers such as CD1a
and CD207 positivity as well as presence of Birbeck granules
(pathognomonic cytoplasmic inclusions viewed by electron microscopy
up to 40% of cases). The major differential diagnosis of adult LCH
is Erdheim-Chester disease (ECD), a CD163 positive and CD1a
negative polyostotic sclerosing form of histiocytosis, which often
affects patients older than 40 years of age and could also develop
CID (9).
In this article we present a rare case of LCH MS
disease presenting with CDI before the beginning of the respiratory
symptoms. Eventually, pulmonary assessment finally led to the
diagnosis.
Despite being found in up to 30% of adult patients,
it has been estimated that only 7% of pathologically-proven
pulmonary LCH develop CDI and, in general, the latter is clinically
evident afterwards (10,11). There are also some reports of
pituitary hormone deficiency in most severe cases (12,13).
The cranial MRI shows absence of physiologic high-T1 signal of
neuro-hypophysis and thickening of pituitary stalk (>3,5 mm)
(12,13). The diagnosis of CDI is made by water
deprivation test or, if not tolerated, using hypersaline infusion
(0,05 ml/kg/min for 2 h). Usually, CDI does not respond to any
LCH-directed treatment and requires long-term replacement therapy
with desmopressin (13). The
respiratory symptoms described by the patient are usually seen in
most cases of adult pulmonary LCH, mainly exertional dyspnoea,
non-productive cough and pleuritic pain. High resolution thoracic
CT scan typically shows interstitial, reticulonodular lesions and
honeycombing pattern, which could contribute to mixed restrictive
and obstructive patterns. In severe cases, pulmonary hypertension
may develop (14,15).
Our patient also reported a history of active
smoking, which is described as a potential risk factor of LCH.
However, the role of cigarette smoke exposure in LCH pathogenesis
and the impact of variable consumption in disease progression is
yet to be fully understood. Smokers with LCH are also at high risk
of developing recurrent pneumothorax (16). Some authors believe that smoking
cessation appears to have significant prognostic impact in
pulmonary LCH patients (16).
Besides pulmonary and pituitary involvement, LCH is characterized
by other important manifestations. Cutaneous and bone involvement
are the most frequent signs of LCH, found in >50% of patients.
This form of LCH could have the potential of spontaneous clinical
remission, particularly in children (17). Bone damage seen in LCH is mainly
related to osteolytic lesions, being the jaw most affected in
adults (18). The osteolytic
lesions seen in LCH are due to osteoclast-like activity of
multinuclear giant cells (18,19)
and different symptoms may develop depending on anatomic location.
In fact, patients could develop conduction deafness (mastoid
involvement), exophthalmia (retro-orbicular involvement) and
paraplegia (vertebral involvement) (20,21).
The PET-CT and axial MRI are most useful for further defining
skeletal lesions and could also be used to evaluate treatment
response (21).
There is no consensus regarding LCH management in
adult patients. In general, the choice of therapeutic regimen is
based on disease severity (22).
Cutaneous form of LCH patients could even benefit from ultraviolet
phototherapy (23,24). The Histiocyte Society and Japan LCH
Study Group have been conducting several prospective, randomized
control trials that studied the effect of several chemotherapy
regimens for LCH (25,26). The LCH-III study was designed for
establishment of a MS LCH treatment strategy, which consisted of
oral prednisone daily and intravenous vimblastine weekly for 6
weeks and repeat the same treatment for another 6 weeks if disease
remains active (27).
Patients in remission after 6-week of induction
therapy should begin maintenance therapy with a 12-month triple
regimen, composed by daily oral 6-mercaptopurine and oral
prednisone associated with weekly intravenous vimblastine. Patients
with multifocal bone disease and/or central nervous system lesions
should be treated with oral prednisone daily and intravenous
vimblastine weekly for 6 months. The addition of methotrexate in
LCH treatment is not recommended in current practice (27). Despite these recommendations, our
patient began combination therapy of cytarabine with oral
prednisone, according to our clinical experience and treatment
protocol developed in our department. This regimen has been studied
in the last few years with promising results. Simko et al
revised data of patients treated with cytarabine for both naïve and
recurrent LCH at Texas Cancer Center from 2005-2013. They concluded
that 88% of LCH achieved remission by the end of first year of
treatment and 59% of patients with recurrent LCH showed significant
improvement in the first three months of therapy (28).
In a retrospective study, there was a significant
survival impact of cytarabine after the first year of remission,
with less toxicity compared with classic regimen
vinblastine/prednisone (29).
Thus, we present a case report that also reaffirms
the therapeutic potential of this alternative treatment as a first
contender to dethrone vinblastine/prednisone. There have been also
some positive results of pulmonary LCH patients being treated with
cladribine, in monotherapy or in association with systemic
glucocorticoids (30). The ongoing
LCH-IV study, a prospective international treatment protocol
sponsored by Dana-Farber Cancer Institute will investigate the
efficiency of second line treatment with cytosine arabinoside and
cladribine (2-chlorodeoxyadenosine) in patients who did not respond
to standard first-line prednisone and vinblastine. In 2017, FDA
approved vemurafenib for ECD with BRAF V600 mutations. There
are some reports of LCH patients who presented this mutation that
showed clinical improvement maintained after 4 months of treatment
with vemurafenib, even though persistent disease activity was still
observed. More clinical trials are needed to validate this
treatment strategy in LCH patients presenting BRAF V600 mutations
(31,32). Bone marrow transplantation or
reduced-intensity condition stem cell transplantation has shown
promise as effective salvage therapy in LCH patients with a very
poor prognosis (rapid disease progression, refractory to
conventional treatment, or with disseminated risk-organ
involvement) (33,34).
In conclusion, the advances over basic knowledge on
LCH poses a huge challenge in clinical practice particularly over
patient care.
Given its low prevalence, there is still the need of
further clinical trials regarding innovative and targeted therapies
that could be used as an alternative to standard care.
There is quite expectations regarding LCH-IV
results, which could consolidate LCH treatment recommendations for
refractory disease.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
Data sharing is not applicable to this article, as
no datasets were generated or analyzed during the current
study.
Authors' contributions
JL concieved and designed the manuscript. JL,CF and
DM acquired the data. CF and DM drafted the manuscript and revised
it critically for important intellectual content. All authors read
and approved the final manuscript. All authors agreed to be
accountable for all aspects of the work in ensuring that questions
related to the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
The patient provided written informed consent for
the publication of any associated data and accompanying images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Stockschlaeder M and Ssucker C: Adult
langerhans cell histiocytosis. Eur J Haematol. 76:363–368.
2006.PubMed/NCBI View Article : Google Scholar
|
|
2
|
WHO Classification of Tumours of
Haematopoietic and Lymphoid Tissue. IARC, Lyon, pp470-472,
2017.
|
|
3
|
Lian C, Lu Y and Shen S: Langerhans cell
histiocytosis in adults: A case report and review of literature.
Oncotarget. 7:18678–18683. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Histiocytosis syndromes in children.
Writing group of the histiocyte society. Lancet. 1:208–209.
1987.PubMed/NCBI
|
|
5
|
Favara BE, Feller AC, Pauli M, Jaffe ES,
Weiss LM, Arico M, Bucsky P, Egeler RM, Elinder G, Gadner H, et al:
Contemporary classification of histiocytic disorders. The WHO
committee on histiocytic/reticulum cell proliferations.
Reclassification working group of the histiocyte society. Med
Pediatr Oncol. 29:157–166. 1997.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Titgemeyer C, Grois N, Minkov M,
Flucher-Wolfram B, Gatterer-Menz I and Gadner H: Pattern and course
of single-system disease in langerhans cell histiocytosis data from
the DAL-HX 83- and 90-study. Med Pediatr Oncol. 37:108–114.
2001.PubMed/NCBI View
Article : Google Scholar
|
|
7
|
Hutter C and Minkov M: Insights into the
pathogenesis of Langerhans cell histiocytosis: The development of
targeted therapies. Immunotargets Ther. 5:81–91. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Tatsuno M, Shioda Y, Iwafuchi H, Yamazaki
S, Iijima K, Takahashi C, Ono H, Uchida K, Okamura O, Matubayashi
M, et al: BRAF V600 mutations in langerhans cell histiocytosis with
a simple and unique assay. Diagn Pathol. 11(39)2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Harmon CM and Brown N: Langerhans cell
histiocytosis: A clinicopathologic review and molecular
pathogenetic update. Arch Pathol Lab Med. 139:1211–1214.
2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kaltsas GA, Powles TB, Evanson J, Plowman
PN, Drinkwater JE, Jenkins PJ, Monson JP, Besser GM and Grossman
AB: Hypothalamo-pituitary abnormalities in adult patients with
langerhans cell histiocytosis: Clinical, endocrinological and
radiological features and response to treatment. J Clin Endocrinol
Metab. 85:1370–1376. 2000.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Choi YS, Lim JS, Kwon W, Jung SH, Park IH,
Lee MK, Lee WY, Yong SJ, Lee SJ, Jung YR, et al: Pulmonary
langerhans cell histiocytosis in an adult male presenting with
central diabetes insipidus and diabetes mellitus: A case report.
Tuberc Respir Dis (Seoul). 78:463–468. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Choi JE, Lee HR, Ohn JH, Moon MK, Park J,
Lee SJ, Choi MG, Yoo HJ, Kim JH and Hong EG: Adult multisystem
langerhans cell histiocytosis presenting with central diabetes
insipidus successfully treated with chemotherapy. Endocrinol Metab
(Seoul). 29:394–399. 2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hong ES, Ohn JH, Kim JH, Hwang-Bo Y, Kim
JJ, Kwon JH, Lee JW, Choi SY, Lee EK, Cho SW, et al: Clinical
characteristics of langerhans cell histiocytosis with
hypothalamo-pituitary involvement. Endocrinol Metab. 26:38–43.
2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wei P, Lu HW, Jiang S, Fan LC, Li HP and
Xu JF: Pulmonary langerhans cell histiocytosis: Case series and
literature review. Medicine (Baltimore). 93:1–7. 2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Tazi A, de Margerie C, Naccache JM, Fry S,
Dominique S, Jouneau S, Lorillon G, Bugnet E, Chiron R, Wallaert B,
et al: The natural history of adult pulmonary langerhans cell
histiocytosis: A prospective multicentre study. Orphanet J Rare
Dis. 10:1–10. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ninaber M, Dik H and Peters E: Complete
pathological resolution of pulmonary Langerhans cell histiocytosis.
Respirol Case Rep. 2:76–78. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
Simko SJ, Garmezy B, Abhyankar H, Lupo PJ,
Chakraborty R, Lim KPH, Shih A, Hicks MJ, Wright TS, Levy ML, et
al: Differentiating skin-limited and multisystem langerhans cell
histiocytosis. J Pediatr. 165:990–996. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Stull MA, Kransdorf MJ and Devaney KO:
Langerhans cell histiocytosis of bone. Radiographics. 12:801–823.
1992.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Kim SH and Choi MY: Langerhans cell
histiocytosis of the rib in an adult: A case report. Case Rep
Oncol. 9:83–88. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Song YS, Lee IS, Yi JH, Cho KH, Kim DK and
Song JW: Radiologic findings of adult pelvis and appendicular
skeletal langerhans cell histiocytosis in nine patients. Skeletal
Radiol. 40:1421–1426. 2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Christopher Z, Binitie O,
Henderson-Jackson E, Perno J and Makanji RJ: Langherhans cell
histiocytosis of bone in an adult: A case report. Radiol Case Rep.
13:310–314. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Aricò M, Girschikofsky M, Généreau T,
Klersy C, McClain K, Grois N, Emile JF, Lukina E, Juli ED and
Danesino C: Langerhans cell histiocytosis in adults: Report from
the international registry of the histiocyte society. Eur J Cancer.
39:2341–2348. 2003.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Do J, Lee J and Kim Y: Successful
treatment of cutaneous langerhans' cell histiocytosis with targeted
narrowband ultraviolet B phototherapy in an infant. Clin Exp
Dermatol. 34(e280-e281)2009.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Vogel C, Aughenbaugh W and Sharata H:
Excimer laser as adjuvant therapy for adult cutaneous langerhans
cell histiocytosis. Arch Dermatol. 144:1287–1290. 2008.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Satter E and High W: Langerhans cell
histiocytosis: A review of current recommendations of the
histiocyte society. Pediatr Dermatol. 25:291–295. 2008.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Gadner H: Treatment of adult-onset
langerhans cell histiocytosis-is it different from the pediatric
approach? Ann Oncol. 21:1141–1142. 2010.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Gadner H, Minkov M, Grois N, Pötschger U,
Thiem E, Aricò M, Astigarraga I, Braier J, Donadieu J, Henter JI,
et al: Therapy prolongation improves outcome in multisystem
langerhans cell histiocytosis. Blood. 121:5006–5004.
2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Simko S, McClain K and Allen C: Up-front
therapy for LCH: Is it time to test an alternative to
vinblastine/prednisone? Br J Haematol. 169:299–301. 2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Cantu MA, Lupo PJ, Bilgi M, Hicks MJ,
Allen CE and McClain KL: Optimal therapy for adults with langerhans
cell histiocytosis bone lesions. PLoS One. 7(e43257)2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
NassEr M, Traclet J and Cottin V: Effect
of cladribine therapy on lung cysts in pulmonary langerhans cell
histiocytosis. ERJ Open Res. 4:00089–2017. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Haroche J, Cohen-Aubart F, Emile JF,
Arnaud L, Maksud P, Charlotte F, Cluzel P, Drier A, Hervier B,
Benameur N, et al: Dramatic efficacy of vemurafenib in both
multisystemc and refractory erdheim-chester disease and langerhans
cell histiocytosis harbouring the BRAF V600E mutation. Blood.
121:1495–1500. 2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Abla O and Weitzman S: Treatment of
langerhans cell histiocytosis: Role of BRAF/MAPK inhibition.
Hematology Am Soc Hematol Educ Program. 2015:565–570.
2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Steiner M, Matthes-Martin S, Attarbaschi
A, Minkov M, Grois N, Unger E, Holter W, Vormoor J, Wawer A,
Ouachee M, et al: Improved outcome of treatment-resistant high-risk
langerhans cell histioytosis after allogenic stem cell
transplantation with reduced-intensity conditioning. Bone Marrow
Transplant. 36:215–225. 2005.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Kesik V, Citak C, Kismet E, Koseoglu V and
Akyuz C: Hematopoietic stem cell transplantation in langerhans cell
histiocytosis: Case report and review of the literature. Pediatr
Transplant. 13:371–374. 2009.PubMed/NCBI View Article : Google Scholar
|