Introduction
KRAS is a small 21 kDa GTP-binding protein that
plays a role in transmitting growth signals downstream of EGFR.
KRAS mutations are found in 45-50% of colorectal cancers, of which
~90% are found at codon 12 and codon 13 (1-3).
It is thought that the presence of KRAS mutation maintains
GTP-binding activity and enhances downstream growth signals
(4). Subgroup analysis of
large-scale Randomized Controlled Trials showed that cetuximab and
panitumumab were effective in wild-type patients with no mutations
in exon 2 (codons 12 and 13) of the KRAS gene, whereas anti-EGFR
antibody treatment was not effective in the KRAS mutant (1,2,5).
Currently, administration of anti-EGFR antibody is not indicated
for colorectal cancer patients with any KRAS mutation. However, it
has been reported that treatment with cetuximab prolonged
progression-free survival (PFS) and overall survival (OS) in
patients with KRAS G13D mutation colorectal cancer (6). In addition, there are a few reports
that KRAS G13D mutation colorectal cancer may be sensitive to
anti-EGFR antibodies in vitro (6,7). As
other KRAS mutations, codon 61 and 146 mutations (with frequencies
of ~2%) are known. Imamura et al reported that colorectal
cancer with codon 61 and 146 mutations have similar
clinicopathological features to exon 2 (codons 12, 13) mutations
(3). In the report, anti-EGFR
antibody treatment was ineffective in all colorectal cancers with
codon 61 mutations, whereas it was effective in some codon 146
mutation cases. KRAS mutations are more frequent in the order of
G12D, G12V and G13D, three of which account for approximately 75%
(1-3).
In our report, these three mutations are referred to as major
mutations. Otherwise, the next most frequent G12A, G12C, G12S, Q61H
and A146T were described as minor KRAS mutations. To assess the
sensitivity of molecularly targeted drugs for KRAS mutations, Mix
Culture Assays (8,9) were performed. First, we evaluated the
resistance of EGFR drugs to minor KRAS mutations in colorectal
cancer cells, the sensitivity of MEK and BCL-XL inhibitors, and
their combined effects. Furthermore, we evaluated the effect of a
novel KRAS-G12C selective inhibitor, AMG510, and its combination
effects with MEK and BCL-XL inhibitors in colorectal cancer
cells.
Materials and methods
Cell culture
CACO-2 cells, a human colorectal cancer cell line,
were purchased from RIKEN Cell Bank and maintained in DMEM (Gibco;
Thermo Fisher Scientific, Inc.). Cells were incubated with 10%
fetal bovine serum and penicillin/streptomycin at 37˚C and 5%
CO2.
Antibodies and reagents
The following antibodies were used: Monoclonal mouse
FLAG (cat. no. 014-22383; 1:1,000 for western blotting; FUJIFILM
Wako Pure Chemicals Corporation); monoclonal rabbit ERK; cat. no.
4695; 1:1,000), monoclonal rabbit p-ERK (cat. no. 4376; 1:1,000),
monoclonal mouse MEK1/2 (cat. no. 4694; 1:1,000) and monoclonal
rabbit p-MEK1/2 (cat. no. 9121; 1:1,000) all purchased from Cell
Signaling Technology, Inc.; monoclonal mouse β-actin (cat. no.
sc-47787; 1:2,000) purchased from Santa Cruz Biotechnology, Inc.
The secondary antibodies polyclonal goat anti-mouse (cat. no.
P0447; 1:5,000) IgG and polyclonal goat anti-rabbit (cat. no.
P0448; 1:5,000) IgG conjugated with HRP were obtained from Dako;
Agilent Technologies, Inc. Cetuximab and panitumumab were purchased
from Merck and Takeda Pharmaceutical Company and 7-aminoactinomycin
D (7-AAD) was purchased from BioLegend. Trametinib, ABT263 and
AMG510 were purchased from Cayman Chemical, LC Laboratories and
Selleck Chemicals.
Construction and sequencing of
vectors
Total mRNA of CACO-2 cells was extracted using
NucleoSpin RNAplus (Takara Bio, Inc.) and cDNA was synthesized by
using PrimeScript™ RT reagent Kit and PrimeScript RT Master Mix
(Takara Bio, Inc.). KRAS-4B carrying a C-terminal FLAG was
amplified using PCR and KRAS mutants of G12D, G12V, G13D, G12A,
G12C, G12S, Q61H and A146T were created using In-Fusion®
HD Cloning Kit (Takara Bio, Inc.). DNA sequences of all the
constructs were confirmed using ABI 3130xl Genetic Analyzer using
BigDye® Terminator v3.1 Cycle Sequencing Kit (Thermo
Fisher Scientific, Inc.).
The method of creating these vectors is shown in the
paper by Koyama et al (9).
Retroviral transduction of the KRAS
mutations
KRAS wild and mutated genes, G12D, G12V, G13D, G12A,
G12C, G12S, Q61H, and A146T, were inserted into the multiple
cloning site of pMXs-IRES-GFP vector (Cell Bio-Lab, Inc.). For
retroviral transduction, these vectors were transfected into the
amphotropic packaging cells, Phoenix, using PEI MAX (Polysciences
Inc.). The virus-containing supernatants were harvested 24 and 48 h
after gene transduction, and CACO-2 cells were infected with the
retroviral particles on RetroNectin (Takara Bio, Inc.) coated
plates. We confirmed transduction efficiency of pMXs-IRES-GFP
vector as a GFP-positive ratio measured using a flow cytometer (BD
FACSCanto II) and analyzed with Kaluza 2.1 software (Beckman
Coulter, Inc.), and cells from the 10th passage were used for the
Mix Culture Assay, as shown below.
Protein sample preparation and western
blotting
After introducing the KRAS wild and mutant genes
into CACO-2, western blotting was performed to confirm the gene
transfer. CACO-2 cells were lysed in RIPA lysis buffer (cat. no.
sc-24948; Santa Cruz Biotechnology, Inc.) containing 1 mM PMSF on
ice for 30 min. The lysates were separated by centrifugation at
10,000 x g for 10 min at 4˚C and the resultant supernatant was
collected as the total cell lysate. Protein was quantified using a
Pierce BCA Protein assay kit (Thermo Fisher Scientific, Inc.) and
10 µg protein was separated using SDS-PAGE gel and then
electroblotted onto a PVDF membrane. The membrane was blocked with
Tris-buffered saline containing 5% non-fat dry milk and 1% Tween-20
for 1 h at room temperature and then probed using the primary
antibodies at 4˚C overnight. The membrane was then incubated with
horseradish peroxidase-conjugated secondary antibody for 1 h at
4˚C, which was detected by enhanced chemiluminescence using
Immobilon Western HRP (Amersham ECL Prime Western Blotting
Detection Reagent; Cytiva). We used Bolt™ 4-12% Bis-Tris Plus Gels,
12 wells (Invitrogen), and laded 10 µg of protein into one lane in
the order of ladder, Mock, KRAS wild, G12V, G12C, ladder, Mock,
KRAS wild, G12V, G12C in one gel. After transfer to the PVDF
membrane, the membrane was cleaved in the center and reacted
separately with the primary antibodies MEK and pMEK. Similarly, we
cut the same membrane in half and perform Western blotting of ERK
and pERK, FLAG and β-actin. This experiment has been conducted
three times separately.
Mix culture assay
We have developed and reported a mixed culture assay
for stable and reliable screening of effective therapeutic targets,
transduced with the pMXs-IRES-GFP vector, and analyzed using a flow
cytometer (8,9). For this assay, we retrovirally
transduced wild-type KRAS, major mutant KRAS genes (G12D, G12V,
G13D) and minor mutant KRAS genes (G12A, G12C, G12S, Q61H, and
A146) into CACO-2 cells using the pMX-IRES-GFP vector. A high
gene-transduction efficiency of ≥90%, which was determined using
the GFP-positive rate (%) measured using a flow cytometer, was
obtained. After gene transfer, GFP expression in gene transduced
cells stabilizes at ~7 passages, so cells with 7-10 passages are
used in the experiment. Parental cells (GFP negative) and
gene-transduced cells (GFP positive) were mixed ideally at a 1:1
ratio. It is impossible to keep the GFP positive rate constant at
50%. Approximately plus or minus 10% is considered to be an
acceptable range (8,9). On the first day, the mixed cells were
seeded at a 20% confluency on a 12-well plate and were cultured for
12 days with molecular targeting agents. They were then passaged at
a 5:1 ratio before reaching confluence. On day 12, the cells were
harvested and stained with 7-AAD. The population that was
7-AAD-negative, which represents viable cells, was gated and the
GFP-positive ratio of these populations was determined using a flow
cytometer. We calculated the relative proliferation ratio (RPR)
using the following formula, the day 0 GFP-positive rate (%) (A),
and the day 12 GFP-positive rate (%) (B). The outline of this
experimental system, the calculation method for RPR, and the
experimental example are shown in our previous report (9).
Here, a low RPR indicates that the GFP-positive cell
population was sensitive to the drug, while a high RPR indicates
drug resistance. Using this system, we evaluated the drug
sensitivities of KRAS-transduced cells to several molecular
targeting drugs.
Statistical analysis
The data are expressed as mean ± standard deviation
of 4 experiments for each assay. Statistical significance of the
results was evaluated using Student's t-test and one-way ANOVA,
followed by Bonferroni correction. Differences were considered
significant at a P<0.05.
Results
Sequence analysis confirmed the
correct gene sequence of the KRAS mutations inserted in the
pMXs-IRES vector
G12D, G12V, and G13D KRAS mutations, which are
frequently found in colorectal cancer, are referred to as KRAS
major mutations, and other mutations (G12A, G12C, G12S, Q61H, and
A146T) are referred to as KRAS minor mutations in this study. It
was confirmed that correctly sequenced wild KRAS, major KRAS
mutations (G12D, G12V, and G13D) and minor KRAS mutations (G12A,
G12C, G12S, Q61H, and A146T) had been inserted into pMX-IRES-GFP
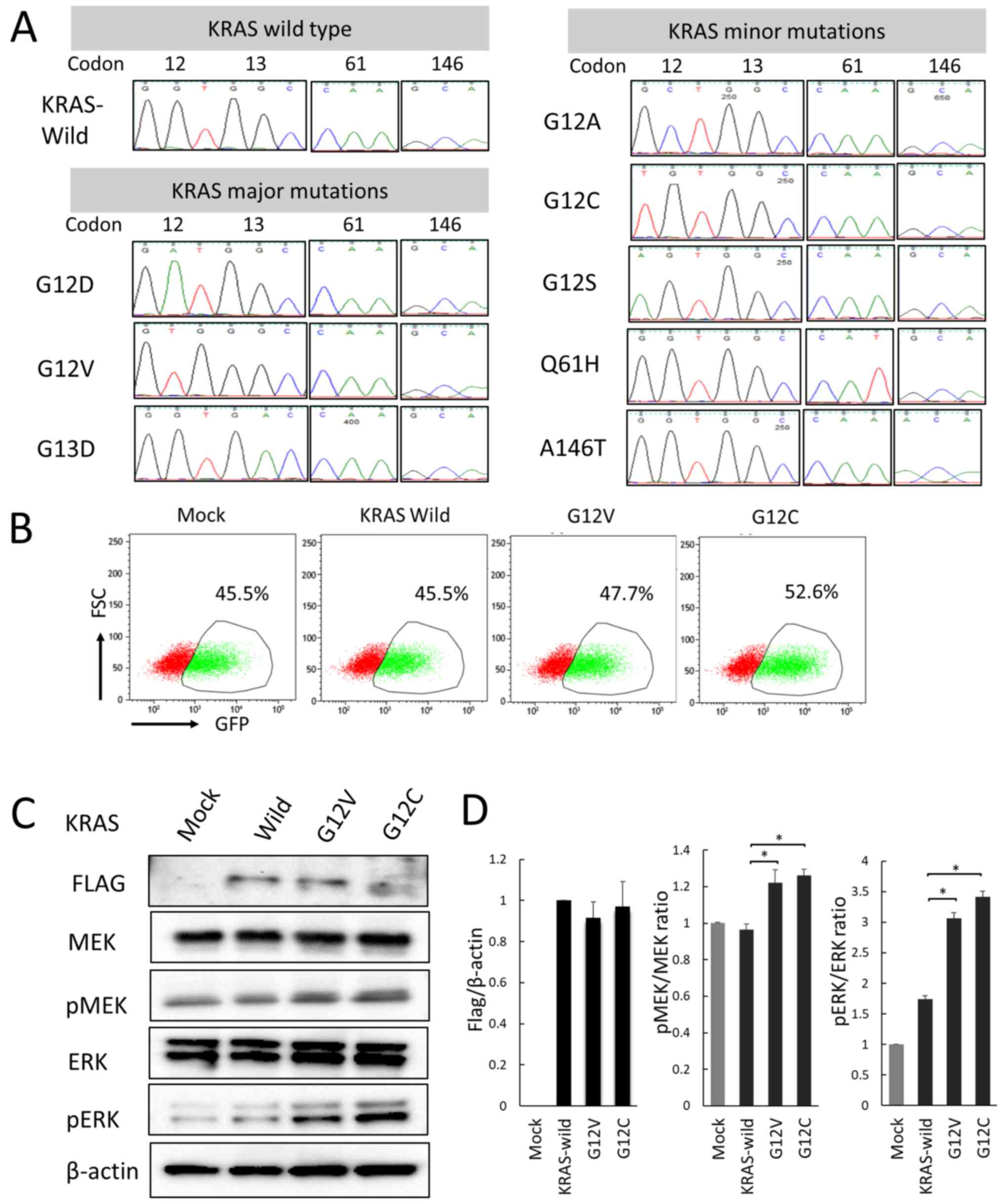
using an ABI 3130xl Genetic Analyzer (Fig. 1A). GFP-positive rates in a mixed
state in Mock and KRAS mutations were confirmed by flow cytometer
(Fig. 1B). Transfection efficiency
of KRAS wild and mutant genes was confirmed by Western blotting
with FLAG-specific antibodies. Furthermore, it was confirmed by
western blotting that MEK and ERK were activated by the
introduction of the mutant KRAS gene (Fig. 1C).
Flow cytometers and western blots
showed successful ectopic KRAS gene transduction
To confirm successful transduction with pMX-IRES
GFP, Mutant KRAS promotes cell proliferation via the upregulation
of ERK phosphorylation., wild-type and mutant KRAS genes carrying a
C-terminal FLAG were inserted into pDON-5Neo DNA vectors, which
were retrovirally transduced into CACO-2 and SW48 human CRC cells
expressing wild-type KRAS. Transduction was confirmed using an
anti-FLAG antibody (Fig. 1C). The
effect of KRAS gene mutation on cell proliferation were then
examined. Mutant KRAS-transduced CACO-2 cells exhibited a
significantly higher rate of proliferation compared with cells
transduced with wild-type KRAS. The RAS/MEK/ERK signaling pathway
is known to act downstream of the KRAS gene and regulate key
cellular activities including differentiation, proliferation and
survival. Therefore, p-MEK and p-ERK expression was examined using
western blotting, and the ratios of p-protein/total protein were
significantly higher in all mutant KRAS cells compared with in
wild-type cells (Fig. 1C and
D). Subsequently, other pathways
associated with the RAS/MEK/ERK pathway were examined using the Mix
Culture assay system.
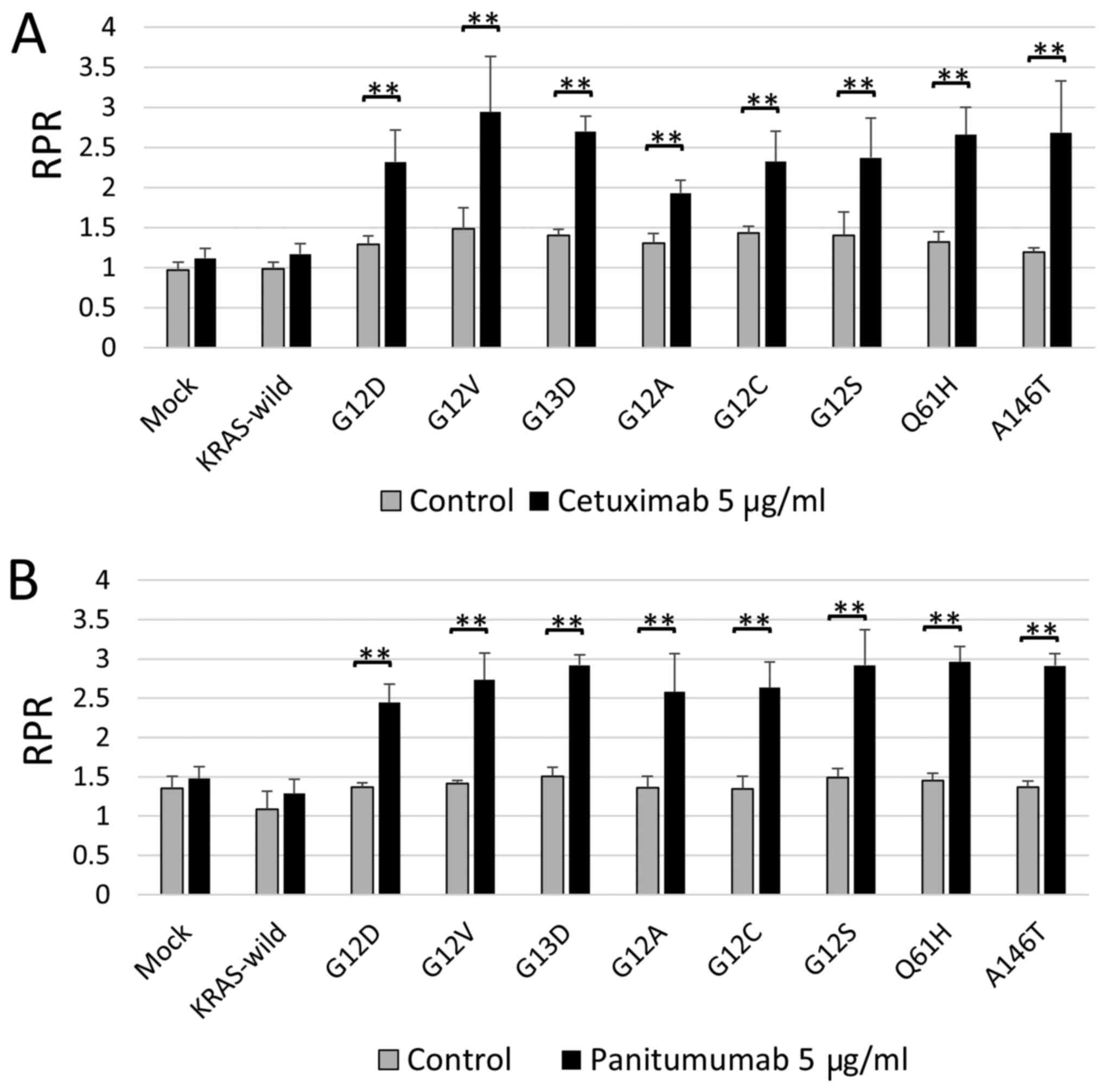
All major and minor KRAS mutations in
CACO-2 cells contributed to resistance to anti-EGFR agents,
cetuximab, and panitumumab
Do G13D and minor KRAS mutations really contribute
to resistance to anti-EGFR agents? The Mix Culture Assay was used
to answer this clinical question. As a result, RPR did not change
significantly in KRAS wild type, but in all KRAS mutations, RPR
increased significantly in the anti-EGFR-administered group,
compared to the non-treated group. That is, it is suggested that
all KRAS mutations, including G13D and minor mutations, contribute
to anti-EGFR antibody resistance (Fig.
2).
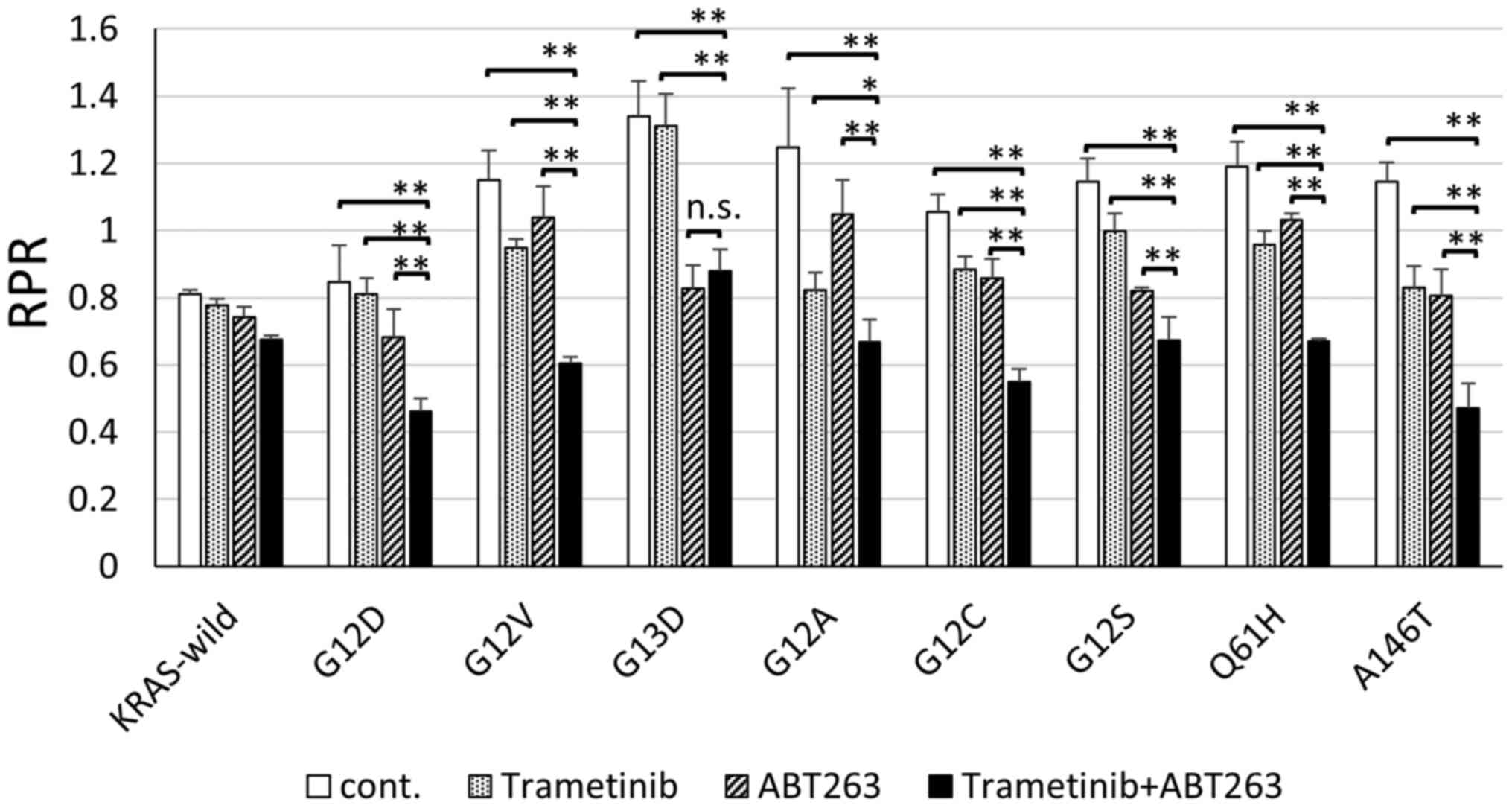
The Mix Culture Assay showed that the
combination of MEK and BCL-XL inhibition was effective as a
treatment target for all KRAS mutations
We reported that the combination of MEK and BCL-XL
inhibition was effective as a targeting treatment for KRAS major
mutations of colorectal cancer cells (9). We also analyzed whether simultaneous
inhibition of MEK and BCL-XL had potent tumor suppressor effects on
KRAS minor mutant colorectal cancer cells. In all minor KRAS
mutations, the combination of MEK and BCL-XL inhibitors
significantly reduced RPR, compared to drug-free controls. It was
suggested that MEK and BCL-XL inhibition could be effective
treatments, even for minor KRAS mutations (Fig. 3).
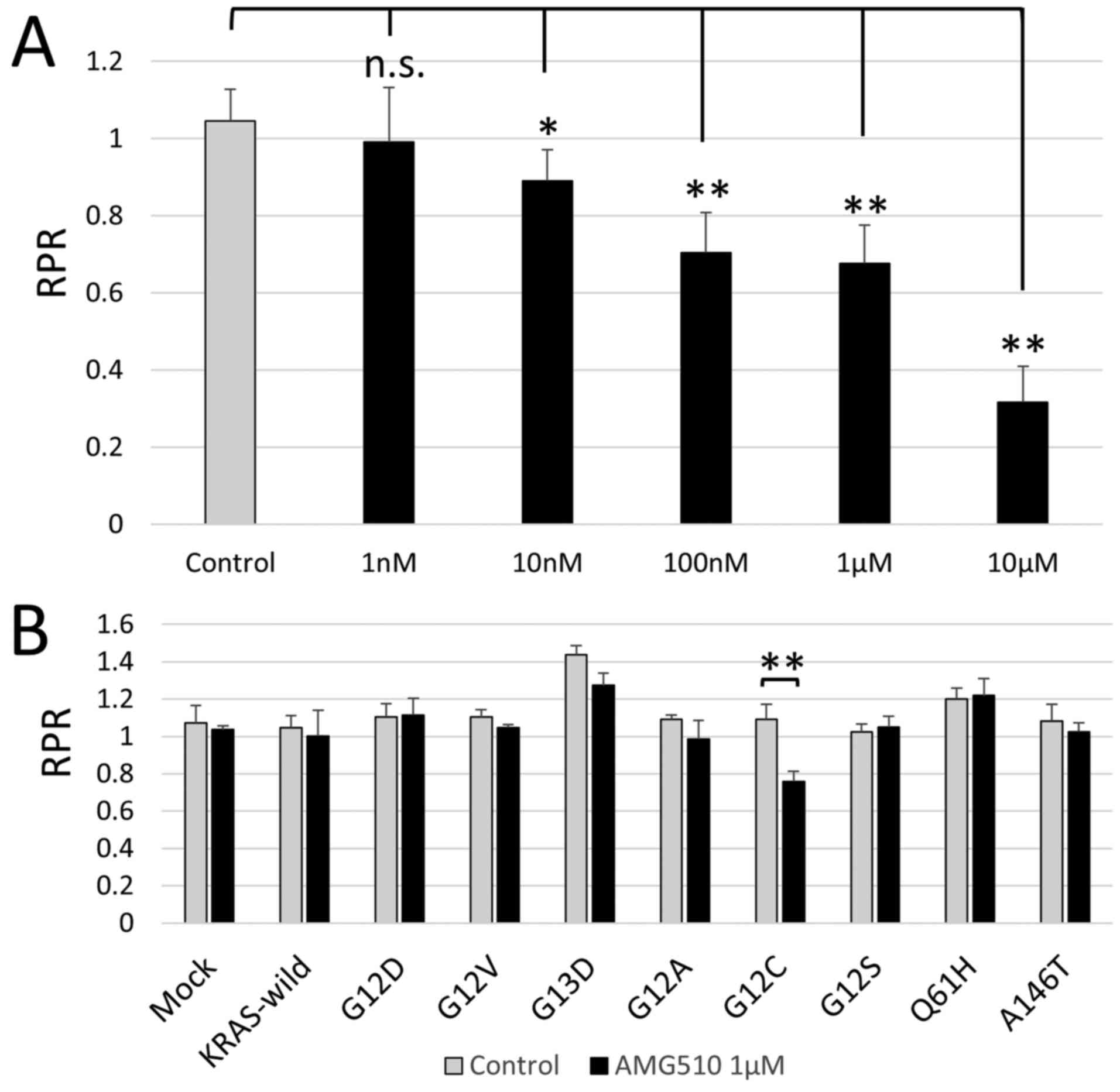
AMG510 selectively suppresses the
growth of colorectal cancer cells with G12C KRAS mutation in
vitro
The results of the phase I clinical trial for the
solid tumor of AMG510, a novel KRAS G12C inhibitor, was reported at
the 2019 American Oncology Society. AMG510 showed an antitumor
effect on KRAS G12C mutation in non-small cell lung cancer (NSCLC),
but unfortunately it did not have sufficient clinical effects on
colorectal cancer (Trial ID: NCT03600883). The frequency of G12C in
colorectal cancer is ~3-4% of KRAS mutations (3,10), the
fourth highest, and finding an effective treatment for it is very
important, from a clinical standpoint. First, using Mix Culture
Assay, it was examined whether AMG510 has an antitumor effect on
the CACO-2 cells into which the KRAS G12C gene was introduced. The
results showed that there was a clear concentration dependent RPR
decrease and that AMG 510 selectively inhibited KRAS G12C in colon
cancer cells in vitro (Fig.
4A). Next, in order to evaluate the selectivity of AMG510 for
KRAS G12C, Mix Culture Assays of KRAS-wild, G12D, G12V, G13D, G12A,
G12C, G12S, Q61H and A146T were performed. The results showed that
RPR was only significantly reduced in G12C (n=4, P<0.01). It was
confirmed that the effect of AMG510 is selective for G12C and does
not cross-react with other KRAS mutations. This result also
demonstrates that the Mix Culture Assay is a powerful experimental
tool for screening effective drugs that target gene mutations
(Fig. 4B).
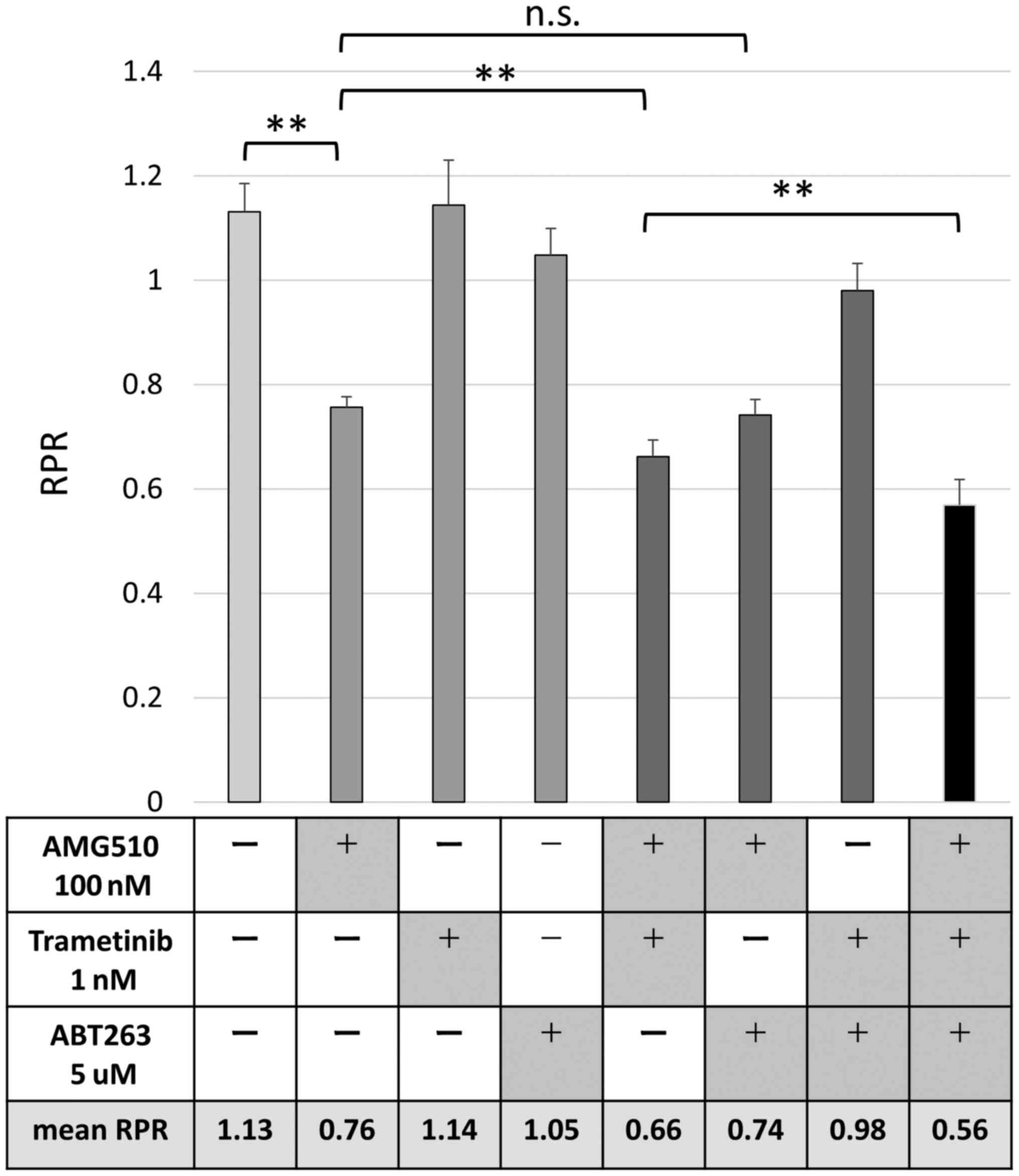
The combination of AMG510, MEK
inhibitor, and BCL-XL inhibitor has potent anti-tumor effects on
colorectal cancer cells with G12C KRAS mutation
The results of the Phase Ⅰ trial were presented at
American Society of Clinical Oncology (ASCO) in 2019, and AMG510
proved to be effective for NSCLC, but it is not expected to be as
effective in colon cancer. Since the effects of AMG510 are observed
in CACO-2 cells in vitro, we hypothesized that clinical
effects could be expected if there is a drug that enhances the
effects of AMG510. In our previous study, we demonstrated that
inhibition of MEK and BCL-XL was an effective targeting therapy for
colorectal cancer cells with KRAS mutation (9). We evaluated the combined effect of
AMG510, MEK inhibitor, and BCL-XL inhibitor in colorectal cancer
cells using a Mix Culture Assay. Finally, G12C-transfected CACO-2
cells were used to evaluate the changes in RPR due to the
combination of AMG510, MEK inhibitor, and BCL-XL inhibitor. The
combination of MEK inhibitor, trametinib, significantly enhanced
the RPR-lowering effect of AMG510, but BCL-XL inhibitor, ABT263,
did not show such combination effect. Surprisingly, the combined
use of the three drugs showed a further RPR-lowering effect,
suggesting that simultaneous inhibition of KRAS, MEK, and BCL-XL
may contribute to a further antitumor effect (Fig. 5).
Discussion
Our study focused on minor KRAS mutations and
evaluated drug sensitivity using a unique method called Mix Culture
Assay (9). There are three
important points in our research. The first is the selection of
KRAS mutations G12D, G12V, G13D, G12A, G12C, G12S, Q61H, and A146T.
The sum of these eight mutations accounts for over 90% of KRAS
mutations and can be considered the most clinically encountered
KRAS mutations. The second point is the selection of a cell line
without mismatch repair (MMR) deficiency. Many colorectal cancer
studies use MMR-deficient colorectal cancer cell lines. Since
MMR-deficient colorectal cancer has the characteristic of being
clinically resistant to anticancer drugs (11), we believe that it may have a
completely different phenotype from the usual colorectal cancer. In
fact, the studies above mentioned (6,7) use
MMR-deficient colon cancer cell lines, such as SW48, HT-116, and
Lovo, which suggests that colorectal cancer cells with KRAS G13D
mutation were sensitive to anti-EGFR antibody. In our study, CACO-2
cells were selected because the use of a cell line with the common
genetic alterations of colorectal cancer was thought to have
clinically significant consequences. The genetic status of CACO-2
cells was considered a usual colorectal cancer with
MMR-proficiency, TP53 mutation, and APC mutation. Although we
needed to study using multiple cell lines, only CACO-2 was
available for human colorectal cell lines that meet the two
conditions of RAS wild-type and MSS.A third important point is the
use of a unique experimental procedure, called Mix Culture Assay,
to evaluate the therapeutic targets for KRAS mutations. It may seem
complicated at a first glance, but it is a simple research method
to evaluate whether the GFP-positive rate increases or decreases by
co-culturing KRAS-mutant-gene transfected cells (GFP-positive) and
parental cells (GFP-negative) for a certain period under drug
administration. RPR is a mathematical formula created to quantify
the experimental results. In this assay, the number of cells is
measured using a flow cytometer, and stable results are obtained.
Details of this method can be found in the reported study by Koyama
(9). The usual proliferation assay
is carried out in different cell dishes or different mice under the
same conditions, and the evaluation method is performed by counting
the total cell number or measuring tumor size. In the Mix Culture
Assay, KRAS mutant cells are cultured with internal control cells
in the same cell culture dish with the same drug concentration, and
the results are calculated as relative ratios using a flow
cytometer. We believe that the Mix Culture Assay is an extremely
useful tool to screen effective drugs targeting a certain gene
mutation. Furthermore, we believe that the Mix Culture Assay is a
very useful experimental tool for verifying the combined effect of
multiple antitumor agents. On the other hand, the limitation of
this assay is only in vitro validation, which only reveals
the relative difference in drug susceptibility between transgenic
GFP-positive cells and control GFP-negative cells. In other words,
even if the RPR is low and the agent is expected to be highly
effective, it needs to be further verified in vitro and
further confirmed in vivo. On the contrary, even if the RPR
is high and resistance can be expected, it is necessary to further
verify whether the drug is really ineffective in vitro and
in vivo (8,9).
The majority of KRAS mutations in colorectal cancer
includes G12D, G12V and G13D, which are reported to account for
approximately 75% of the total mutations (1-3).
Based on clinical and basic research data, there is no doubt that
G12D and G12V mutations contribute to anti-EGFR antibody
resistance. On the other hand, it has been reported that anti-EGFR
antibody may be effective for colorectal cancers with KRAS G13D
mutation (6,7). For the 61 and 146 codon mutations,
clinical data suggested that 61 codon-mutated colorectal cancers
may be resistant to anti-EGFR; on the other hand, 146 codon-mutated
colorectal cancers may be susceptible to anti-EGFR (3). However, this report is a very limited
analysis of the few patient data, and this susceptibility cannot be
further clarified. Clinically, all colorectal cancers with KRAS
mutations are considered resistant to anti-EGFR antibodies, and
cetuximab and panitumumab have not been used for them. Are all KRAS
mutations contributing to anti-EGFR antibody resistance? Our study
gave a clear answer to that question. All KRAS mutations at codons
12, 13, 61, and 146 have clearly been shown to contribute to
resistance to the anti-EGFR drugs, cetuximab and panitumumab.
(Fig. 2).
Previously, we reported that the combined inhibition
of MEK and BCL-XL was effective against major mutations in KRAS:
G12D and G12V (9). This study
showed that MEK inhibitors and BCL-XL had similar effects on all
other KRAS minor mutations.
Furthermore, we also evaluated the effect of a novel
G12C-specific inhibitor, AMG510, by using the Mix Culture Assay.
G12C is the most common KRAS mutation in NSCLC. Why are the
treatment effects of colorectal cancer and lung cancer different? A
similar phenomenon is observed in BRAF-mutant colorectal cancer.
BRAF inhibitors are effective in treating BRAF-mutant lung cancer
and melanoma, but not in treating colorectal cancer. This happens
due to the previously reported reason that inhibition of BRAF
activates CRAF and enhances downstream proliferation and
anti-apoptotic signals (12,13).
Since KRAS mutation activates downstream MEK, it has been thought
that it could be an effective therapeutic target for KRAS mutant
cancer; however, at present, MEK inhibition alone has not been
established as an effective therapeutic method. MEK inhibition has
been reported to activate other molecules, such as PI3K (14,15),
ERBB2(16), FGFR (17), YAP (18), BCL-XL (9,19), and
CDK4/6 (20-22)
to amplify proliferative and anti-apoptotic signals. This
phenomenon is called feedback/paradoxical activation and it is
expected that this signal may also be induced by AMG510(23). Blocking this signal under
administration of AMG510 enhances the effect of AMG510 and could be
an effective clinical treatment. We developed this Mix Culture
Assay to stably evaluate the combined effect of multiple drugs on
KRAS mutations. Koyama et al screened for effective drugs in
combination with MEK inhibitors in KRAS mutant colorectal cancer
cells (9). As a result, it was
reported that BCL-XL inhibitors potentiated the effects of MEK
inhibitors, while BRAF inhibitor, PI3K inhibitor, and CDK4/6
inhibitor had no effects on MEK actuation. In our study, it was
confirmed that the combination of MEK and BCL-XL inhibitors was
effective for minor KRAS mutations. Furthermore, it was clarified
that the anti-tumor effect of AMG510 on KRAS G12C mutant colorectal
cancer was enhanced by MEK inhibitor, but not by BCL-XL.
Surprisingly, it was shown that the three-drug combination has a
dramatic antitumor effect on G12C KRAS colorectal cancer.
Mix Culture Assays demonstrated that all KRAS
mutations, including minor KRAS mutations, contributed to anti-EGFR
antibody drug resistance. It was also demonstrated that the
combination of MEK and BCL-XL inhibitors has antitumor effects on
all minor KRAS mutations. Furthermore, it was revealed in
vitro that MEK inhibitor enhanced the selective antitumor
effect of AMG510, a novel KRAS G12C selective inhibitor, in
colorectal cancer cells. Furthermore, the combination of AMG510,
MEK inhibitor, and BCL-XL inhibitor showed a strong antitumor
effect on G12C colon cancer in vitro.
Acknowledgements
Not applicable.
Funding
This work was supported by the Japan Society for the Promotion
of Science KAKENHI (grant no. JP18K15238).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MKi, DK, MT and YS designed the current study. MKi,
MKo, SN, SM, NH and TE performed the experiments, analyzed the data
and wrote the manuscript. YM, FM, ST, MKu, HT and YY designed the
experiments, and interpreted the data. All authors read and
approved the final manuscript. YM and MKo confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Karapetis CS, Khambata-Ford S, Jonker DJ,
O'Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD,
Robitaille S, et al: K-ras Mutations and benefit from cetuximab in
advanced colorectal cancer. N Engl J Med. 359:1757–1765.
2008.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Amado RG, Wolf M, Peeters M, Van Cutsem E,
Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et
al: Wild-type KRAS is required for panitumumab efficacy in patients
with metastatic colorectal cancer. J Clin Oncol. 26:1626–1634.
2008.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Imamura Y, Lochhead P, Yamauchi M, Kuchiba
A, Qian ZR, Liao X, Nishihara R, Jung S, Wu K, Nosho K, et al:
Analyses of clinicopathological, molecular, and prognostic
associations of KRAS codon 61 and codon 146 mutations in colorectal
cancer: Cohort study and literature review. Mol Cancer.
13(135)2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Lièvre A, Bachet JB, Le Corre D, Boige V,
Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, et al:
KRAS mutation status is predictive of response to cetuximab therapy
in colorectal cancer. Cancer Res. 66:3992–3995. 2006.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Dahabreh IJ, Terasawa T, Castaldi PJ and
Trikalinos TA: Systematic review: Anti-epidermal growth factor
receptor treatment effect modification by KRAS mutations in
advanced colorectal cancer. Ann Intern Med. 154:37–49.
2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
De Roock W, Jonker DJ, Di Nicolantonio F,
Sartore-Bianchi A, Tu D, Siena S, Lamba S, Arena S, Frattini M,
Piessevaux H, et al: Association of KRAS p.G13D mutation with
outcome in patients with chemotherapy-refractory metastatic
colorectal cancer treated with cetuximab. JAMA. 304:1812–1820.
2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kumar SS, Price TJ, Mohyieldin O, Borg M,
Townsend A and Hardingham JE: KRAS G13D mutation and sensitivity to
cetuximab or panitumumab in a colorectal cancer cell line model.
Gastrointest Cancer Res. 7:23–26. 2014.PubMed/NCBI
|
|
8
|
Kitazawa M, Hida S, Fujii C, Taniguchi S,
Ito K, Matsumura T, Okada N, Sakaizawa T, Kobayashi A, Takeoka M
and Miyagawa SI: ASC induces apoptosis via activation of caspase-9
by enhancing Gap junction-mediated intercellular communication.
PLoS One. 12(e0169340)2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Koyama M, Kitazawa M, Nakamura S,
Matsumura T, Miyazaki S, Miyagawa Y, Muranaka F, Tokumaru S,
Okumura M, Yamamoto Y, et al: Low-dose trametinib and Bcl-xl
antagonist have a specific antitumor effect in KRAS-mutated
colorectal cancer cells. Int J Oncol. 57:1179–1191. 2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
AACR Project GENIE Consortium. AACR
Project GENIE: Powering precision medicine through an International
Consortium. Cancer Discov. 7:818–831. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Des Guetz G, Schischmanoff O, Nicolas P,
Perret GY, Morere JF and Uzzan B: Does microsatellite instability
predict the efficacy of adjuvant chemotherapy in colorectal cancer?
A systematic review with meta-analysis. Eur J Cancer. 45:1890–1896.
2009.PubMed/NCBI
|
|
12
|
Poulikakos PI, Zhang C, Bollag G, Shokat
KM and Rosen N: RAF inhibitors transactivate RAF dimers and ERK
signalling in cells with wild-type BRAF. Nature. 464:427–430.
2010.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Prahallad A, Sun C, Huang S, Di
Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A
and Bernards R: Unresponsiveness of colon cancer to BRAF(V600E)
inhibition through feedback activation of EGFR. Nature.
483:100–103. 2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Engelman JA, Chen L, Tan X, Crosby K,
Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y,
et al: Effective use of PI3K and MEK inhibitors to treat mutant
Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med.
14:1351–1356. 2008.PubMed/NCBI View
Article : Google Scholar
|
|
15
|
Ebi H, Corcoran RB, Singh A, Chen Z, Song
Y, Lifshits E, Ryan DP, Meyerhardt JA, Benes C, Settleman J, et al:
Receptor tyrosine kinases exert dominant control over PI3K
signaling in human KRAS mutant colorectal cancers. J Clin Invest.
121:4311–4321. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
16
|
Turke AB, Song Y, Costa C, Cook R, Arteaga
CL, Asara JM and Engelman JA: MEK inhibition leads to PI3K/AKT
activation by relieving a negative feedback on ERBB receptors.
Cancer Res. 72:3228–3237. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Manchado E, Weissmueller S, Morris JP IV,
Chen CC, Wullenkord R, Lujambio A, de Stanchina E, Poirier JT,
Gainor JF, Corcoran RB, et al: A Combinatorial strategy for
treating KRAS-mutant lung cancer. Nature. 534:647–651.
2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Lin L, Sabnis AJ, Chan E, Olivas V, Cade
L, Pazarentzos E, Asthana S, Neel D, Yan JJ, Lu X, et al: The Hippo
effector YAP promotes resistance to RAF- and MEK-targeted cancer
therapies. Nat Genet. 47:250–256. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
19
|
Corcoran RB, Cheng KA, Hata AN, Faber AC,
Ebi H, Coffee EM, Greninger P, Brown RD, Godfrey JT, Cohoon TJ, et
al: Synthetic lethal interaction of combined BCL-XL and MEK
inhibition promotes tumor regressions in KRAS mutant cancer models.
Cancer Cell. 23:121–128. 2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ziemke EK, Dosch JS, Maust JD, Shettigar
A, Sen A, Welling TH, Hardiman KM and Sebolt-Leopold JS:
Sensitivity of KRAS-mutant colorectal cancers to combination
therapy that cotargets MEK and CDK4/6. Clin Cancer Res. 22:405–414.
2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Tao Z, Le Blanc JM, Wang C, Zhan T, Zhuang
H, Wang P, Yuan Z and Lu B: Coadministration of Trametinib and
Palbociclib radiosensitizes KRAS-mutant non-small cell lung cancers
in vitro and in vivo. Clin Cancer Res. 22:122–133. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Lee MS, Helms TL, Feng N, Gay J, Chang QE,
Tian F, Wu JY, Toniatti C, Heffernan TP, Powis G, et al: Efficacy
of the combination of MEK and CDK4/6 inhibitors in vitro and in
vivo in KRAS mutant colorectal cancer models. Oncotarget.
7:39595–39608. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ryan MB, Fece de la Cruz F, Phat S, Myers
DT, Wong E, Shahzade HA, Hong CB and Corcoran RB: Vertical pathway
inhibition overcomes adaptive feedback resistance to KRAS G12C
inhibition. Clin Cancer Res. 26:1633–1643. 2020.PubMed/NCBI View Article : Google Scholar
|