Introduction
AXL is a member of the TAM (Tyro3, AXL, Mer) family
of receptor tyrosine kinases (RTKs), and is activated by its ligand
Gas6, which leads to activation of several downstream signaling
pathways, depending on the cell types, mainly
phosphoinositide-3-kinase (PI3K)/protein kinase B (AKT) and
mitogen-activated protein kinase
(MEK)/extracellular-signal-regulated kinase (ERK). AXL has also
been implicated in the pathophysiology of numerous cancers
including breast, gastric, prostate, ovarian and lung (1-4).
It enhances cancer cell survival, angiogenesis, metastasis and
drug-resistance (2,4,5).
c-MYC is a basic helix-loop-helix leucine zipper
transcription factor that dimerizes with MAX to bind DNA and
regulate gene expression (6).
c-Myc plays an essential role in cell cycle progression (7-9),
cellular differentiation (10),
metabolism (11), cancer
progression (12) and drug
resistance (13).
More than 1 million new breast cancer (BC) cases are
diagnosed each year worldwide and account for most cancer-related
deaths in women (14). Among BCs,
some basal-like BCs are estrogen receptor negative, progesterone
receptor negative and HER2 negative, causing triple-negative BC
(TNBC), which accounts for 10-20% of all BC cases. These tumors are
aggressive with poor prognosis due to ineffective target
therapies.
In BC, 30-50% of tumors overexpress c-MYC (15), and it is considered that c-Myc is
an enhancer of tumorigenesis in BC. Notably, c-MYC is elevated in
TNBC compared with other cancer subtypes (ER+ (estrogen
receptor alpha positive), HER2+ (human epidermal growth
factor receptor 2 positive) (16).
AXL has been reported to be overexpressed in BC, and
is a negative prognostic indicator for survival in patients with BC
(17). AXL is also an inducer of
Epithelial-to-mesenchymal transition-induced downstream effector,
that is required for BC metastasis and progression (1,18).
AXL also mediates BC cells resistance to epidermal growth factor
receptor-targeted therapies (19).
Thus, AXL represents a promising target in BC therapies (20).
Based on the observation that both c-Myc and AXL
overexpressed in TNBC, it was hypothesized that as a receptor
tyrosine receptor, AXL maybe regulate c-Myc expression in BC. It
was revealed that AXL could upregulate c-Myc expression in an AKT-
and ERK-dependent manner.
Materials and methods
Cell lines and reagents
293T cells, HeLa cells, MDA-MB-231 cells were
purchased from The Cell Bank of Type Culture Collection of the
Chinese Academy of Sciences. 293T cells, HeLa cells, MDA-MB-231
cells were cultured in DMEM medium (Gibco; Thermo Fisher
Scientific, Inc.), supplemented with 10% fetal bovine serum (FBS)
(Zhejiang Tianhang Biotechnology Co., Ltd.). MCF7 cells were
purchased from Procell Life Science & Technology and cultured
in MEM medium (cat. no. PM150410; Procell Life Science &
Technology) supplemented with 0.01 mg/ml insulin (cat. no.
PB180432; Procell Life Science & Technology) and 10% FBS (cat.
no. 164210-500; Procell Life Science & Technology). All the
cells were maintained in a humidified atmosphere at 37˚C with 5%
CO2. Cell line authentication was achieved by genetic
profiling using the polymorphic short tandem repeat (STR) method at
the Forensic Science Center, Jining Medical University (Shandong,
China). R428 was purchased from MedChemExpress and used as
previously described (21).
LY294002 and U0126 were purchased from Beyotime Institute of
Biotechnology.
Western blotting
Total cellular proteins were extracted from cells
with RIPA buffer containing protease inhibitors and phosphatase
inhibitors (all from Beyotime Institute of Biotechnology), and the
quantity was determined by BCA Protein Assay Kit (Beyotime
Institute of Biotechnology). Then 10 µg proteins were subjected to
10% SDS-polyacrylamide gel electrophoresis and transferred onto
PVDF membranes (Beyotime Institute of Biotechnology). The membrane
was blocked for 1 h at room temperature in 5% non-fat milk solution
and incubated overnight at 4˚C with the following primary
antibodies: AXL (1:1,000; cat. no. 8661), phosphorylated (p)-AKT
(1:1,000; cat. no. 4060), AKT (cat. no. 4691; all from Cell
Signaling Technology, Inc.), ERK1 + ERK2 (1:10,000; cat. no.
ab184699), p-ERK1 + ERK2 (1:10,000; cat. no. ab76299; both from
Abcam) and c-Myc antibody (1:1,000; cat. no. ab32072; Abcam).
Following incubation with primary antibodies, membranes were
incubated with peroxidase-conjugated goat anti-rabbit (cat. no.
ZB-2301) or goat anti-mouse (cat. no. ZB-2305; both from OriGene
Technologies, Inc.) secondary antibodies (1:5,000) at room
temperature for 1 h. Immunoblot bands were visualized using ECL
reagent (Beyotime Institute of Biotechnology). GAPDH (1:1,000; cat.
no. TA-08; OriGene Technologies, Inc.) was included as an
endogenous protein loading control.). All the experiments were
repeated three times. ImageJ software (National Institutes of
Health, version 1.53m) was used for the densitometric analysis.
Nuclear and cytoplasmic protein
extraction
Nuclear and cytoplasmic protein were extracted using
nuclear and cytoplasmic protein extraction kit (cat. no. P0027)
from Beyotime Institute of Biotechnology according to the
manufacturer's instructions. These experiments were repeated three
times.
Plasmids, short hairpin (sh)RNA and
transfection
AXL plasmid (cat. no. 105932) or AXL kinase-dead
plasmid (cat. no. 105934; both from Addgene, Inc.) were gifts from
Rosa Marina Melillo (22).
Transfection of plasmids (AXL or empty vector) was performed using
Lipo8000 (cat. no. C053; Beyotime Institute of Biotechnology)
according to the manufacturer's protocol. Briefly, Lipo8000 and
vector were incubated in DMEM without FBS for 5 min and added the
cell medium for 4 h. ~44-48 h later, cells were harvested for
analysis. Lentivirus for shRNA targeting AXL (shAXL-3)
(5'-GGGTGACAATGTGGGAGATTG-3') and Control viruses (Cont;
5'-TTCTCCGACGTGTCACGT-3') were purchased from Shanghai GenePharma
Co., Ltd., and the titer for both of them was 109 TU/ml.
For viral transfection, viruses (5 µl/ml) and polybrene (2 µg/ml)
were added to the cell medium (MOI=~5 for HeLa and MDA-MB-231
cells), after 3 days cells were treated with puromycin (2 µg/ml)
for ~1 week to select transfected cells. Then, cells were cultured
at puromycin (0.5 µg/ml) for maintenance.
Analysis of protein expression of AXL
and c-Myc from The Cancer Proteome Atlas (TCPA)
TCPA is a resource for cancer functional proteomics
data over a large number of tumor and cell line samples using
reverse-phase protein arrays as a part of The Cancer Genome Atlas
Project (23). To analyze the
association between AXL and c-Myc in BCs, their protein expression
data was downloaded from TCPA (n=901) (https://www.tcpaportal.org/tcpa/download.html). The
samples whose AXL expression marked with NA (n=156) were excluded.
The expression association between AXL and c-Myc was analyzed by
SPSS (version 13.0; SPSS, Inc.) using Pearson's correlation
analysis (n=745). To further analyze the samples without c-Myc
amplification, data of c-Myc amplification of the samples were
extracted from cBioportal (https://www.cbioportal.org/). After excluding these
samples with c-Myc amplification, the expression association
between AXL and c-Myc was analyzed by SPSS using Pearson's
correlation analysis (n=648).
Statistical analysis
Data are presented as mean ± standard deviation
(SD). Statistical differences between experimental and control
groups were analyzed using an unpaired two-tailed Student's t-test
(2 groups) or a one-way ANOVA (>2 groups) followed by a multiple
post hoc comparisons test (Dunnett's test) using GraphPad Prism 8
software (GraphPad Software, Inc.). P<0.05 was considered to
indicate a statistically significant difference.
Results
AXL increases c-Myc expression
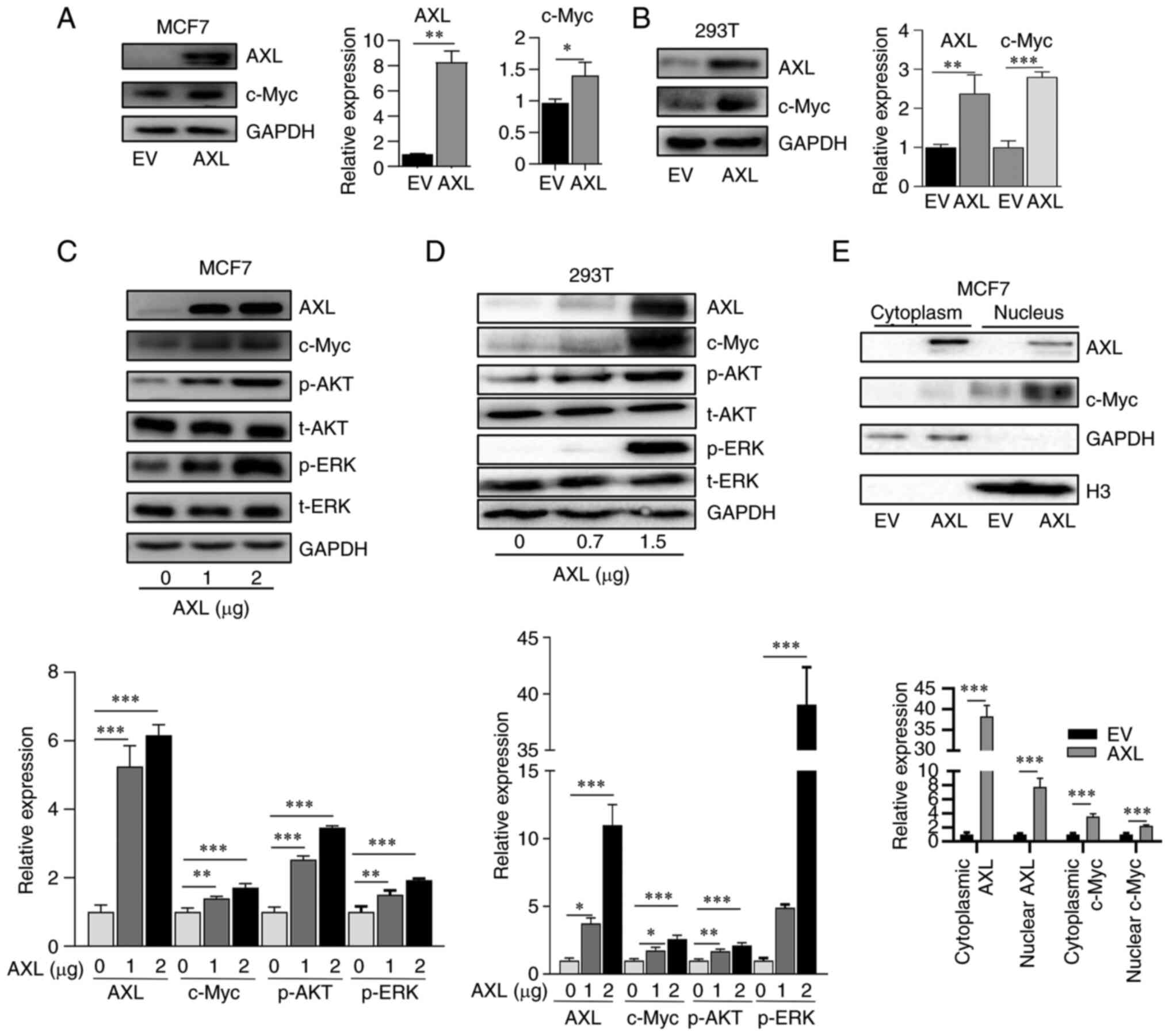
To determine the association of AXL and c-Myc, AXL
was overexpressed in MCF7 and 293T cells which did not express AXL,
and it was revealed that exogenous AXL increased c-Myc expression
(Fig. 1A and B). Furthermore, c-Myc expression was
revealed to be dependent on AXL amount transfected; increasing AXL
transfection led to elevated c-Myc expression (Fig. 1C and D). c-Myc is located in the nucleus,
therefore the expression of cytoplasmic and nuclear c-Myc was
explored. It was identified that nuclear c-Myc was upregulated in
response to AXL overexpression (Fig.
1E). The present results also showed a part of AXL was also
located in the nucleus, which was consistent with previous results
(24).
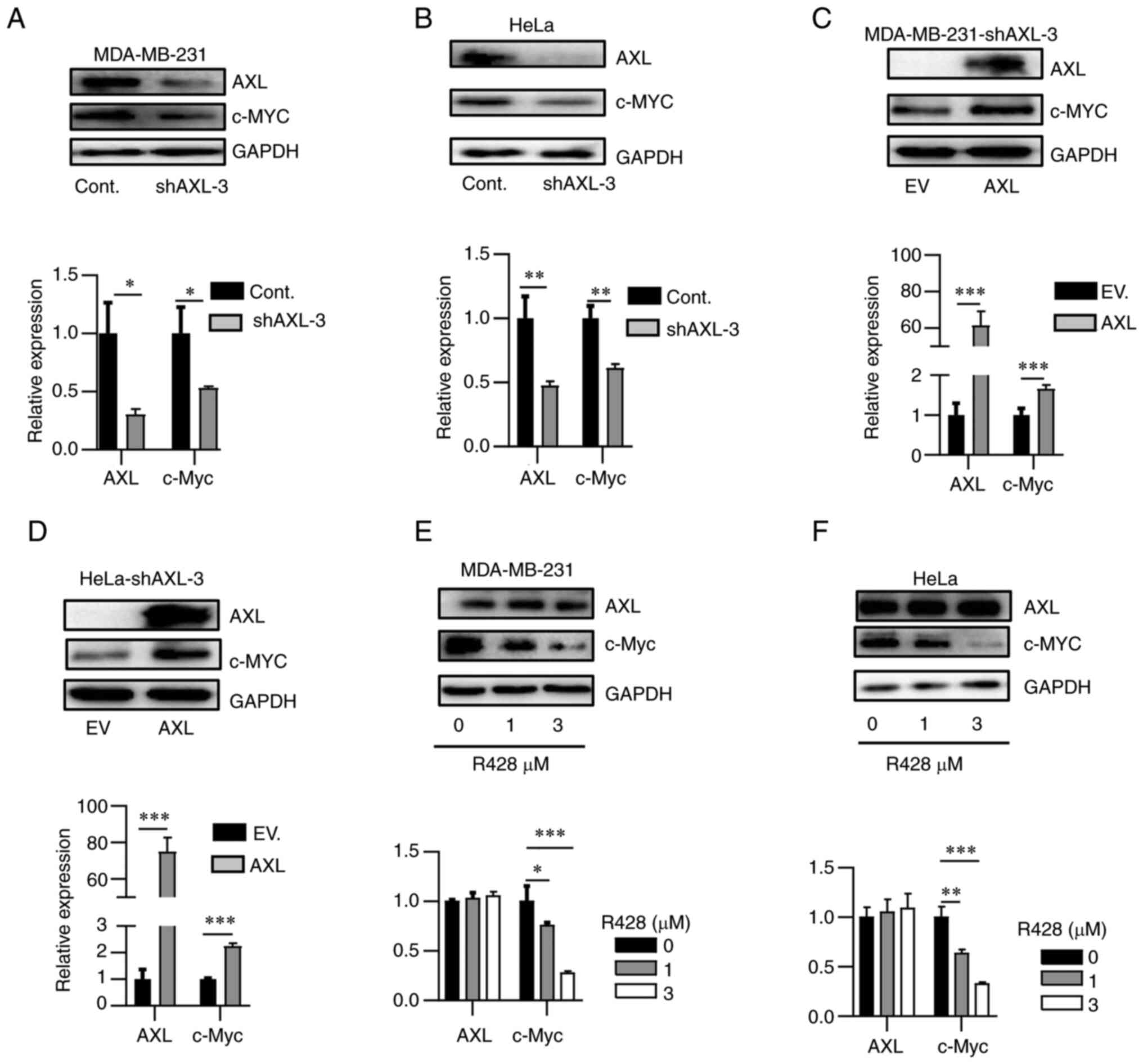
To further determine the association between AXL and
c-Myc, AXL expression was knocked down in two AXL-high expression
cell lines, HeLa and MDA-MB-231. AXL knockdown using shRNA
decreased c-Myc expression in both cell lines (Fig. 2A and B). Re-overexpression of AXL in
shRNA-knockdown HeLa and MDA-MB-231 recovered c-Myc expression
(Fig. 2C and D). R428, a selective AXL inhibitor
(25), suppressed c-Myc expression
in a dose-dependent manner (Fig.
2E and F). Thus, it was
concluded that AXL could increase c-Myc expression.
AXL increases c-Myc expression through
AKT and ERK pathway
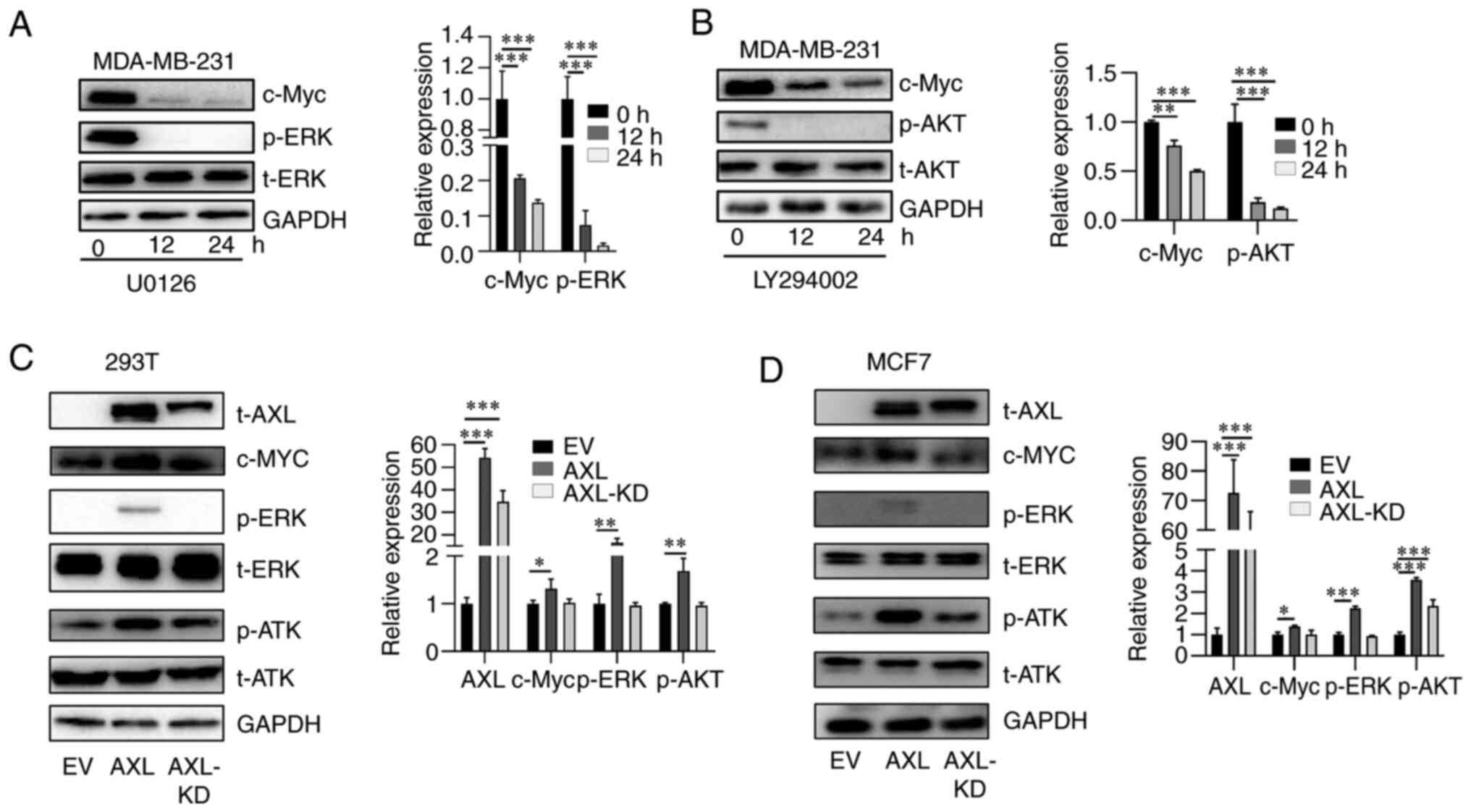
AKT and ERK are the main downstream signaling
pathways of AXL (4,26). It was observed that overexpression
of AXL is accompanied by activation of AKT and ERK pathways, which
are manifested by p-AKT and p-ERK, respectively (Fig. 1C and D). Thus, to understand the mechanisms
underlying AXL-upregulated c-Myc, MDA-MB-231 cells were first
treated with AKT and ERK inhibitors LY294002 and U0126,
respectively, and it was revealed that LY294002 and U0126
suppressed the activation of AKT and ERK, respectively (Fig. 3A and B). Moreover, both inhibitors suppressed
c-Myc expression in a time-dependent manner, suggesting that both
pathways are important in maintaining c-Myc expression (Fig. 3A and B). Subsequently, it was investigated
whether both signaling pathways are involved in AXL-upregulated
c-Myc. Overexpression of AXL led to c-Myc upregulation; however,
overexpression of kinase-dead AXL (K567R) did not increase c-Myc
expression as markedly as AXL. AXL activated AKT and ERK signaling
while AXL-KD activated AKT weakly and did not activate ERK
signaling (Fig. 3C and D). These results suggested both AKT and
ERK signaling are important for c-Myc upregulation by AXL.
AXL is associated with c-Myc
expression in patients with BC
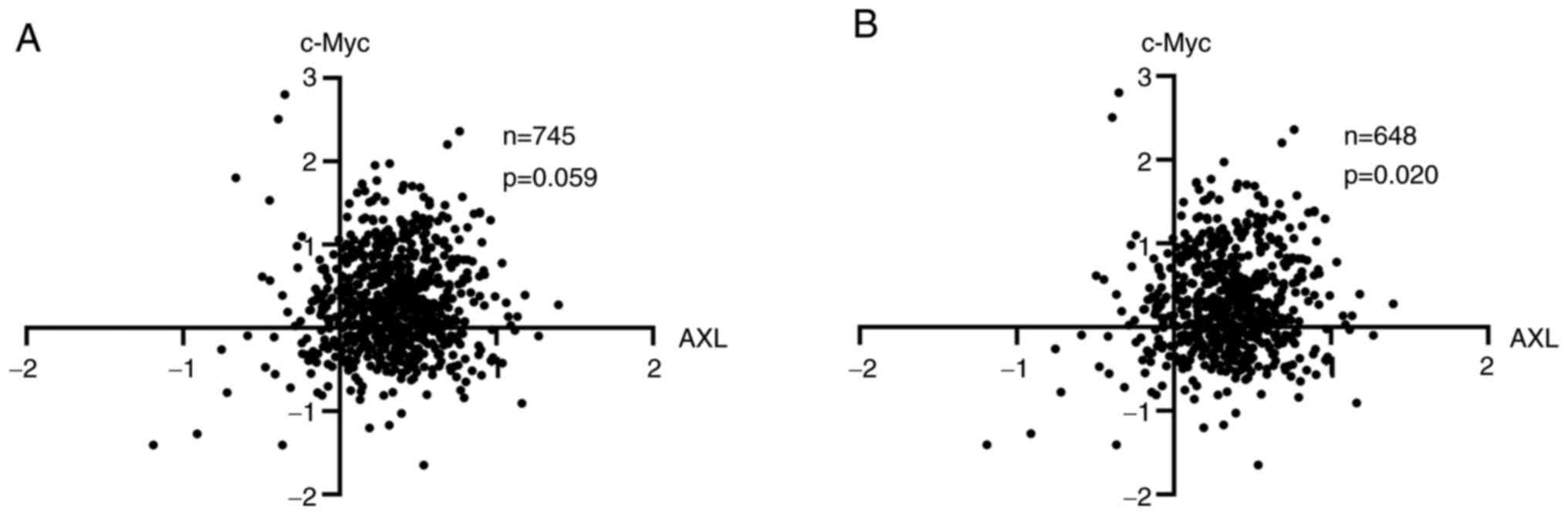
To further study the association between AXL and
c-Myc, protein expression data from patients with BC of TCPA
(23) was examined. It was
revealed that there is a trend association between AXL and c-Myc
(P=0.059) (Fig. 4). In BCs, c-Myc
protein expression was closely correlated with c-Myc amplification
(27), suggesting a subset of
c-Myc overexpression is caused by c-Myc amplification, and it was
hypothesized that c-Myc in this part of tumors was more dependent
on amplification but not signaling pathways. Indeed, when this
subset of samples was excluded, AXL and c-Myc were associated with
each other significantly (P=0.020) (Fig. 4).
Discussion
c-Myc is now known to be one of the most frequently
dysregulated oncogenes. It is overexpressed by different mechanisms
in 60-70% of human solid and hematopoietic tumors (7,11,12).
It was found in a pan-cancer copy-number analysis to be the third
most amplified gene in human cancers (11). Besides copy-number amplification,
numerous dysregulated signaling pathways also contribute to the
overexpression of c-Myc, such as Wnt-APC pathway found in human
colon carcinoma (28) and NOTCH
signaling pathway found in T cell leukemia (29). c-MYC contributes to the genesis of
numerous human cancers. It has been revealed to be linked to
numerous key biological processes of cancer cells including
proliferation, stem cell properties, metabolism, metastasis, cancer
immune and genome instability (11,12,30).
In the present study, a new mechanism of c-Myc
overexpression in BC cells was demonstrated, in which AXL
upregulates c-Myc expression through the AKT and ERK signaling
pathways. The results are in accordance with those of Hong et
al (13), which displayed a
close association between AXL and c-Myc in esophageal
adenocarcinoma.
In the present study, to the best of our knowledge,
exogeneous AXL was first overexpressed in 293T and MCF7 cells, both
of which do not express AXL. As a result, c-Myc expression was
upregulated in a dose-dependent manner. Conversely, knockdown of
AXL expression in MDA-MB-231 and HeLa cells decreased c-Myc
expression. Furthermore, R428, a selective AXL inhibitor, also
suppressed c-Myc expression. Collectively, these results supported
a role of AXL in c-Myc upregulation in BC cells. Furthermore, the
association between AXL and c-Myc was supported by data from tumor
tissues of patients with BC. To understand the mechanism underlying
c-Myc upregulation by AXL, the AKT and ERK pathways were examined.
It was revealed that both of the two pathways are important for
c-Myc upregulation by AXL. Nevertheless, the detailed mechanisms
must be further studied. These experiments were not performed,
which is a limitation to the present study.
In conclusion, in the present study, it was
demonstrated that AXL upregulates c-MYC expression through
activation of the AKT and ERK signaling in BC cells.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by innovation and
entrepreneurship training program for college students of Jining
Medical University (grant no. cx2021138), the Enterprise Technology
Innovation Project of Jinan (grant no. 201940901085) and the
Project of Medicine and Health Development Plan of Shandong (grant
no. 2019WS176).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
GZ, HoZ and QG designed the study. XS, HC, SY, ZT,
ZW, FL, WH and HaZ performed the experiments and analyzed the data.
GZ, QG and HoZ prepared the manuscript. All authors read and
approved the final manuscript. HoZ and QG confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Paccez JD, Vogelsang M, Parker MI and
Zerbini LF: The receptor tyrosine kinase Axl in cancer: Biological
functions and therapeutic implications. Int J Cancer.
134:1024–1033. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Graham DK, DeRyckere D, Davies KD and Earp
HS: The TAM family: Phosphatidylserine sensing receptor tyrosine
kinases gone awry in cancer. Nat Rev Cancer. 14:769–785.
2014.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Qu X, Liu G, Zhong X, Li X and Zhang Q:
Role of AXL expression in non-small cell lung cancer. Oncol Lett.
12:5085–5091. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Zhang G, Wang M, Zhao H and Cui W:
Function of Axl receptor tyrosine kinase in non-small cell lung
cancer. Oncol Lett. 15:2726–2734. 2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zhang Z, Lee JC, Lin L, Olivas V, Au V,
LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, et al:
Activation of the AXL kinase causes resistance to EGFR-targeted
therapy in lung cancer. Nat Genet. 44:852–860. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
6
|
Conacci-Sorrell M, McFerrin L and Eisenman
RN: An overview of MYC and its interactome. Cold Spring Harb
Perspect Med. 4(a014357)2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
García-Gutiérrez L, Delgado MD and León J:
MYC oncogene contributions to release of cell cycle brakes. Genes
(Basel). 10(244)2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bretones G, Delgado MD and León J: Myc and
cell cycle control. Biochim Biophys Acta. 1849:506–516.
2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Singh AM and Dalton S: The cell cycle and
Myc intersect with mechanisms that regulate pluripotency and
reprogramming. Cell Stem Cell. 5:141–149. 2009.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Gómez-Casares MT, García-Alegria E,
López-Jorge CE, Ferrándiz N, Blanco R, Alvarez S, Vaqué JP,
Bretones G, Caraballo JM, Sánchez-Bailón P, et al: MYC antagonizes
the differentiation induced by imatinib in chronic myeloid leukemia
cells through downregulation of p27(KIP1.). Oncogene. 32:2239–2246.
2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Stine ZE, Walton ZE, Altman BJ, Hsieh AL
and Dang CV: MYC, metabolism, and cancer. Cancer Discov.
5:1024–1039. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Dang CV: MYC on the path to cancer. Cell.
149:22–35. 2012.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hong J, Maacha S and Belkhiri A:
Transcriptional upregulation of c-MYC by AXL confers epirubicin
resistance in esophageal adenocarcinoma. Mol Oncol. 12:2191–2208.
2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Padayachee J, Daniels A, Balgobind A,
Ariatti M and Singh M: HER-2/neu and MYC gene silencing in breast
cancer: Therapeutic potential and advancement in nonviral
nanocarrier systems. Nanomedicine (Lond). 15:1437–1452.
2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Cancer Genome Atlas Network. Comprehensive
molecular portraits of human breast tumours. Nature. 490:61–70.
2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Gjerdrum C, Tiron C, Hoiby T, Stefansson
I, Haugen H, Sandal T, Collett K, Li S, McCormack E, Gjertsen BT,
et al: Axl is an essential epithelial-to-mesenchymal
transition-induced regulator of breast cancer metastasis and
patient survival. Proc Natl Acad Sci USA. 107:1124–1129.
2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chang H, An R, Li X, Lang X, Feng J and Lv
M: Anti-Axl monoclonal antibodies attenuate the migration of
MDA-MB-231 breast cancer cells. Oncol Lett. 22(749)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Meyer AS, Miller MA, Gertler FB and
Lauffenburger DA: The receptor AXL Diversifies EGFR signaling and
limits the response to EGFR-Targeted inhibitors in triple-negative
breast cancer cells. Sci Signal. 6(ra66)2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Khera L and Lev S: Accelerating AXL
targeting for TNBC therapy. Int J Biochem Cell Biol.
139(106057)2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Han S, Wang Y, Ge C, Gao M, Wang X, Wang
F, Sun L, Li S, Dong T, Dang Z, et al: Pharmaceutical inhibition of
AXL suppresses tumor growth and invasion of esophageal squamous
cell carcinoma. Exp Ther Med. 20(41)2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Avilla E, Guarino V, Visciano C, Liotti F,
Svelto M, Krishnamoorthy G, Franco R and Melillo RM: Activation of
TYRO3/AXL tyrosine kinase receptors in thyroid cancer. Cancer Res.
71:1792–1804. 2011.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Li J, Lu Y, Akbani R, Ju Z, Roebuck PL,
Liu W, Yang JY, Broom BM, Verhaak RG, Kane DW, et al: TCPA: A
resource for cancer functional proteomics data. Nat Methods.
10:1046–1047. 2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Brand TM, Iida M, Corrigan KL, Braverman
CM, Coan JP, Flanigan BG, Stein AP, Salgia R, Rolff J, Kimple RJ
and Wheeler DL: The receptor tyrosine kinase AXL mediates nuclear
translocation of the epidermal growth factor receptor. Sci Signal.
10(eaag1064)2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Holland SJ, Pan A, Franci C, Hu Y, Chang
B, Li W, Duan M, Torneros A, Yu J, Heckrodt TJ, et al: R428, a
selective small molecule inhibitor of Axl kinase, blocks tumor
spread and prolongs survival in models of metastatic breast cancer.
Cancer Res. 70:1544–1554. 2010.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Linger RM, Keating AK, Earp HS and Graham
DK: TAM receptor tyrosine kinases: Biologic functions, signaling,
and potential therapeutic targeting in human cancer. Adv Cancer
Res. 100:35–83. 2008.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Blancato J, Singh B, Liu A, Liao DJ and
Dickson RB: Correlation of amplification and overexpression of the
c-myc oncogene in high-grade breast cancer: FISH, in situ
hybridisation and immunohistochemical analyses. Br J Cancer.
90:1612–1619. 2004.PubMed/NCBI View Article : Google Scholar
|
|
28
|
He TC, Sparks AB, Rago C, Hermeking H,
Zawel L, da Costa LT, Morin PJ, Vogelstein B and Kinzler KW:
Identification of c-MYC as a target of the APC pathway. Science.
281:1509–1512. 1998.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Palomero T, Lim WK, Odom DT, Sulis ML,
Real PJ, Margolin A, Barnes KC, O'Neil J, Neuberg D, Weng AP, et
al: NOTCH1 directly regulates c-MYC and activates a
feed-forward-loop transcriptional network promoting leukemic cell
growth. Proc Natl Acad Sci USA. 103:18261–18266. 2006.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Casey SC, Baylot V and Felsher DW: The MYC
oncogene is a global regulator of the immune response. Blood.
131:2007–2015. 2018.PubMed/NCBI View Article : Google Scholar
|