Introduction
Tubular aggregate myopathy (TAM) is a progressive
muscle disease that mainly affects the muscle of the proximal lower
limbs and results in myasthenia, myalgia and exercise intolerance
(1,2). The term Stormorken syndrome (STRMK) is
used when TAM is combined with thrombocytopenia, bleeding tendency,
mild anemia, hypocalcemia, asplenia, miosis, dyslexia, ichthyosis
and a short stature. TAM overlaps with STRMK and differs in
severity and age of onset (3). Some
patients with STRMK may exhibit only TAM-related clinical
manifestations, and the phenotypical spectrum ranges from
asymptomatic hyperCKemia to myalgia, progressive muscle weakness
and the contracture of muscle (1).
Studies have reported that TAM/STRMK is a rare
autosomal-dominant inheritance disease with mutations in stromal
interaction molecule 1 (STIM1) or calcium release-activated
calcium channel protein 1 (ORAI1) (4,5).
STIM1 or ORAI1 both encode key factors of
store-operated Ca2+ entry (SOCE), and thereby direct a plethora of
Ca2+-dependent cellular pathways and functions, including muscle
contraction (6,7). Functional analyses have demonstrated
that TAM/STRMK arise from gain-of-function mutations in
STIM1 or ORAI1 (8,9).
The TAM/STRMK case described in the present study is
the second case in China identified to date, at least to the best
of our knowledge. The present study describes the case of a
pediatric patient with TAM/STRMK at the Children's Hospital of
Soochow University, Suzhou, China and also provides a review of the
literature on TAM/STRMK. The aim of the present study was to
explore the clinical features, diagnosis and treatment of TAM/STRMK
in order to aid in its early identification.
Case report
Clinical assessment
The present study abided by the principles of the
Declaration of Helsinki. All clinical, laboratory and molecular
biological examinations and treatment programs were performed after
obtaining informed consent from the parents of the child and were
approved by the Ethics Committee of the Children's Hospital of
Soochow University.
Clinical general information
A 2-year-old Chinese girl was born to a gravida 2
para 2 (G2P2) woman via caesarean section at term, with a birth
weight of 3,250 g, a body length of 50 cm, a normal Apgar score and
timely vaccination. The child had a history of convulsions,
long-term thrombocytopenia, hyperCKemia and repeated skin petechiae
and ecchymoses from the age of 1 month. The girl was admitted to
the Children's Hospital of Soochow University due to petechiae and
ecchymoses that were scattered across the skin of the whole body. A
physical examination revealed a temperature of 36.5˚C, a pulse of
112 bpm, a respiratory rate of 26 breaths per minute, a body length
of 82 cm and a weight of 11.2 kg, and petechiae and ecchymoses were
noted over the skin of the whole body. Eye movements were normal.
Chest, cardiac and abdominal examinations did not reveal any
abnormalities. No marked muscular weakness was noted in her
extremities, and Kernig's and Brudzinski's signs were negative.
Family history
In the family, only the patient had thrombocytopenia
and long-term hyperCKemia. The parents were not inbred, and there
was one sibling sister, who was in good health. There was not
familial history of the disorder.
Laboratory tests
Blood tests revealed that the patient's white blood
cell count was high (11.2x109/l; reference range (RR),
4.0-10.0x109/l). She had low platelet counts
(16x109/l; RR, 101-320x109/l), anemia
(hemoglobin, 90 g/l; RR, 110-140 g/l), normal calcium serum levels
(2.27 mmol/l; RR, 2.25-2.67 mmol/l) and very high levels of
creatine kinase (CK; hyperCKemia; 498.6 U/l; RR, 25-225 U/l).
Antinuclear antibody tests revealed positive results for anti-SSA,
anti-AMA-M2 and anti-Ro-52 antibodies. Alanine aminotransferase,
aspartate aminotransferase and serum creatinine levels were normal.
Fibrinogen, prothrombin time and activated partial thromboplastin
time were normal. Trace element serum levels, ammonia serum levels,
ceruloplasmin serum levels, fibrinogen, prothrombin time, activated
partial thromboplastin time, tandem mass spectrometry and
cerebrospinal cultures did not reveal any obvious anomalies. In the
bone marrow assessment, nucleated cell hyperplasia was markedly
active, megakaryocytes were visible and platelets were rare. An
abdominal computed tomography scan revealed normal structures of
her liver, gallbladder, pancreas and spleen. Brain magnetic
resonance imaging and a 24-h electroencephalogram revealed no
abnormalities. Blood and urine tandem mass spectrometry and
hemolysis examinations were normal.
Whole-exome sequencing and mutation
detection
Genomic DNA was extracted from 200 µl peripheral
blood, using a Qiagen DNA Blood Midi/Mini kit (Qiagen GmbH). The
extracted DNA was fragmented with DNase and purified via the
magnetic bead method, followed by ligation of the adaptor sequence
and PCR amplification. The PCR products were validated using an
Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.). PCR was
performed using Takara PrimeSTARMax DNA Polymerase (Takara
Biotechnology, Co., Ltd.) under the following thermocycling
conditions: An initial denaturization at 95˚C for 5 min, followed
by 35 cycles of denaturation at 95˚C for 3 sec, annealing at 60˚C
for 30 sec and extension at 72˚C for 30 sec. A final extension was
performed at 72˚C for 5 min. Sequencing of the genomic DNA of the
family was performed using the Novaseq6000 platform (Illumina,
Inc.). The original data were obtained following sequencing of all
exons. The Burrows Wheeler Aligner algorithm was used to compare
the reference sequence (UCSChg19), and GATK version 3.7.0
(https://software.broadinstitute.org/gatk/) and
VarScanversion 2 software (http://dkoboldt.github.io/varscan/) were used to
identify mutations, e.g., single nucleotide polymorphisms (SNPs)
and InDels, including detection, annotation and statistical
analysis. The pathogenicity of variants was analyzed according to
the American College of Medical Genetics and Genomics standards and
guidelines (10).
Construction
The STIM1 domain was searched on UCSC (http://genome.ucsc.edu/). The STIM1 bioconservative
analysis view was structured using T-Coffee (https://www.ebi.ac.uk/Tools/msa/tcoffee/). The 3D
structures of STIM1 and mutated proteins were constructed in
SWISS-MODEL (https://swissmodel.expasy.org/).
Expression analysis
The bottom blood cells were obtained by the
centrifugation of peripheral blood (centrifugation conditions, 4
ml, 4˚C, 1,509.3 x g). The blood cells were frozen and dissolved,
and treated using Red Blood Cell Lysis Buffer (Shanghai Acmec
Biochemical Co., Ltd.). The products were washed with
phosphate-buffered saline (PBS) buffer (ABclonal Biotech Co., Ltd.)
and treated with RIPA lysis buffer (ABclonal Biotech Co., Ltd.).
The protein concentration was measured using a BCA Protein
Quantification kit [Yeasen Biotechnology (Shanghai) Co., Ltd.]. The
samples (10 µg protein/lane) were resolved by 10% SDS-PAGE and
electroblotted onto Immobilon-P transfer membranes (ABclonal
Biotech Co., Ltd.). The membrane was blocked at 4˚C overnight with
5% skim milk, and incubated with primary antibodies at room
temperature for 1 h, and then incubated with secondary antibodies
at room temperature for 1 h. STIM1 rabbit polyclonal antibody
(diluted 1:1,000, cat. no. A19894, ABclonal Biotech Co., Ltd.) was
used as a primary antibody. Anti-rabbitIgG-HRP (diluted 1:1,000,
cat. no. AS080, ABclonal Technology, Wuhan, Hubei, China) was used
as a secondary antibody. GAPDH rabbit polyclonal antibody (cat. no.
AC027, ABclonal Technology, Wuhan, Hubei, China) as the loading
control. The membranes were again washed three times with TBS and
the color reaction was carried out by incubating the membranes with
chromogenic substrate for peroxidase, 4-chloro-1-naphtol and
H2O2. Protein bands were visualized using an
Enhanced Chemiluminescence Detection kit (Beyotime Institute of
Biotechnology), and images were captured using Image Lab software
version 3.0 (Bio-Rad Laboratories, Inc.).
Literature search
The literature review was based on a thorough
retrieval of studies from PubMed, China National Knowledge
Infrastructure (CNKI; https://www.cnki.net/) and Wanfang Standards Database
(WFSD; http://www.wanfangdata.com.cn/). The
key terms used in the search included Stormorken, STIM1 and
TAM. Clinical, laboratory, therapeutic and outcome data were
collected.
Treatment
After admission, the patient was administered
intravenous immunoglobulin 500 mg/(kg/day) for supportive
treatment, and methylprednisolone was added at 2 mg/(kg/day) for 5
days; carbazochrome sodium sulfonate 20 mg/(once/day) was
administered to prevent bleeding, vitamin C was administered to
improve vascular permeability, and other comprehensive treatments
were applied. No new skin bleeding points occurred in the child,
and her platelet count was progressively maintained
>60x109/l after two consecutive reexaminations. The
patient was discharged after 8 days with improved symptoms. At the
last follow-up, the patient's platelet count was
68x109/l, CK levels were 501.4 U/l and no related
manifestations of muscle weakness were found. Comprehensive
language and exercise training was conducted at home, with regular
outpatient follow-up visits at the Department of Child Health care
and Rehabilitation, Children's Hospital of Soochow University. The
quality of life of the child was the same as that of a healthy
individual during the non-pathogenic period, specifically, the
motor function is normal, and can communicate and play normally
with children of the same age.
Molecular genetics analysis
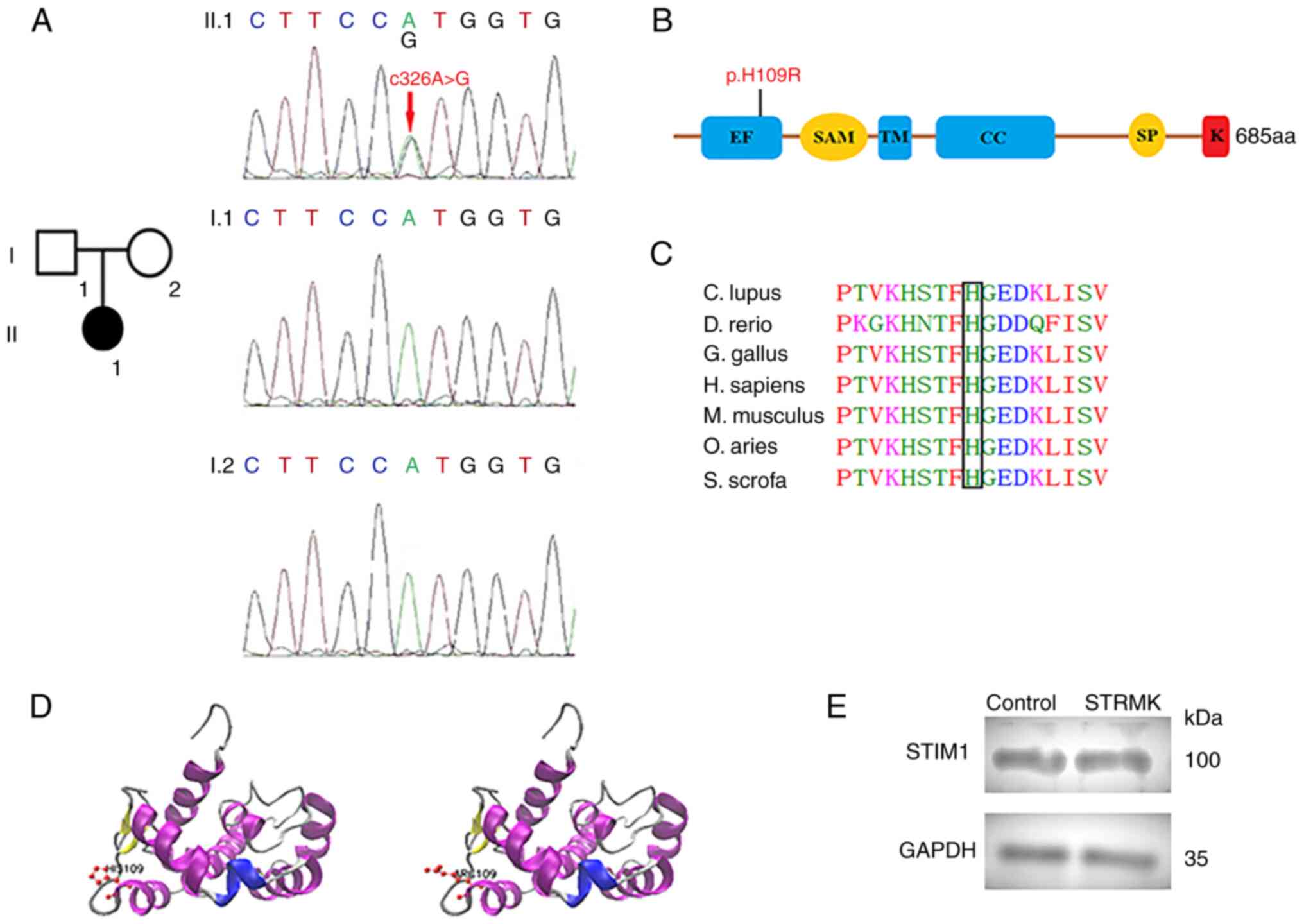
To identify the disease, exome sequencing was
performed on the patient and her parents, and the c.326A>G
transition in an STIM1 allele (p.H109R) was identified
(Fig. 1A). The mutation was present
in the affected patient and absent in the unaffected parents,
indicating that it was a spontaneous STIM1 mutation in the
patient. The mutation was predicted to be harmful by protein
function prediction software (http://www.mutationtaster.org/). Mutations associated
with TAM/STRMK were found upon consulting the HGMD database
(http://www.hgmd.org/). The variant located in the
EF-hand (Fig. 1B) supposedly led to
constitutive STIM1 unfolding and oligomerization (11). The affected amino acids in this
residue was evolutionarily conserved (Fig. 1C), which indicated an important
functional role. The 3D structures of STIM1 and mutated proteins
were constructed in SWISS-MODEL (https://swissmodel.expasy.org/) (Fig. 1D). No differences in STIM1 expression
levels were observed between the case in the present study and the
healthy control (the healthy control was selected from a 2-year-old
healthy Chinese girl without clinical manifestations of STRMK)
(Fig. 1E); thus, the present study
reached similar conclusions as those of a previous study (9).
Literature review
The literature search revealed only a few reported
cases of suspected TAM/STRMK worldwide, comprising 52 families and
87 cases in total. Among these, 30 families (46 cases) had STIM1
EF-hand mutations, 14 families (21 cases) had STIM1 CC1 mutations,
and 8 families (20 cases) had ORAI1 mutations (1,4,8,9,11-24).
TAM/STRMK was mainly induced after STIM1 EF-hand, STIM1 CC1 and
ORAI1 mutations. The mutation sites identified in previous studies
(including the present case) are summarized in Table I. A total of 7 patients had the same
mutation site as the patient described herein, and the phenotypic
and histopathological data of the patients with STRMK with
c.326A>G (p.H109R) mutations were collated (Table II) (9,15,22,24).
 | Table ISummary of mutation sites reported in
previous studies (1,4,8,9,11-24). |
Table I
Summary of mutation sites reported in
previous studies (1,4,8,9,11-24).
| STIM1 EF-hand
(N=46) | STIM1 CC1
(N=21) | ORAI1
(N=20) |
|---|
| c.216C>A(p.H72Q)
(10) | c.910C>T(p.R304W)
(18) | c.290C>G(p.S97C)
(3) |
| c.239A>C(p.N80T)
(2) | c.911 G>A(p.R304Q)
(3) | c.292G>A(p.G98S)
(6) |
| c.242G>A(p.G81D)
(3) | | c.319G>A(pV107M)
(7) |
| c.251A>G(p.D84G)
(3) | | c.412C>T(p.L138F)
(1) |
| c.252T>A(p.D84E)
(2) | | c.551C>T(p.T184M)
(1) |
| c.262A>G(p.S88G)
(1) | |
c.734C>T(p.P245L) (2) |
| c.274C>G(p.L92V)
(1) | | |
| c.286C>G(p.L96V)
(1) | | |
| c.293A>G(p.Y98C)
(1) | | |
|
c.322T>A(p.F108I) (2) | | |
|
c.322T>C(p.F108L) (1) | | |
|
c.325C>A(p.H109N) (4) | | |
|
c.326A>G(p.H109R) (8) | | |
|
c.343A>T(p.I115F) (5) | | |
|
c.1095G>C(p.K365N) (1) | | |
|
c.312A>T(p.K104N) (1) | | |
| Table IIPhenotypic and histopathological data
of patients with STRMK with the c.326A>G (p.H109R) mutation
(9,15,22,24). |
Table II
Phenotypic and histopathological data
of patients with STRMK with the c.326A>G (p.H109R) mutation
(9,15,22,24).
| | Family 2 | | Family 1 | |
|---|
| Period of
onset | Patient 1 (father)
Childhood | Patient 2 (son)
Childhood | Patient 3
Childhood | Patient 4
Childhood | Patient 5 (son)
Childhood | Patient 6 (mother)
Childhood | Patient 7
Childhood | Patient 8 (present
case) Childhood |
|---|
| Myasthenia | Upper and lower
limbs, predominantly proximal | Lower limbs
proximal, upper limbs mild and proximal | Ligature band
muscle, lower limbs | Upper and lower
limbs | Lower limbs
proximal | Lower limbs
proximal | Limb-girdle muscle
weakness | - |
| Eye movement
defects | Ophthalmoplegia,
upwards gaze paresis | Ophthalmoplegia,
upwards gaze paresis | - | Ophthalmoplegia,
mild bilateral upper eyelid ptosis | - | - | - | - |
| Contractures | Neck, elbows,
wrists, fingers | Neck, heels | Ankle joint | - | - | - | Elbows, wrists,
heels | - |
| CK level | 5x198 U/l | 12x198 U/l | 6x198 U/l | 7x198 U/l | 4.1x198 U/l | 9.1x198 U/l | 4x198 U/l | 2.5x198 U/l |
| TA
distribution | Mainly in type I
fibers | In type I and type
II fibers | Mainly in type I
fibers | In type I and type
II fibers | - | In type I and type
II fibers | - | - |
| Special
histological characteristics | Type I fiber
predominance, type II fiber atrophy | Type I fiber
predominance, type II fiber atrophy | Type I fiber
predominance, type II fiber atrophy | Type I fiber
predominance | - | Type I fiber
predominance, type II fiber atrophy | - | - |
| Thrombocytopenia/
bleeding tendency | - | - | - | - | + | + | - | + |
| Prognosis | Intermittent
ventilation treatment at night, not walking normally | Walking normally
due to long-term exercise training | Progressive limb
weakness, unable to run and climb up from the floor | Systemic muscle
atrophy and weakness | Difficulty in
climbing the stairs | Lower limb muscle
weakness progressive aggravation, walking restriction | Normal walking | Normal walking |
Discussion
The case in the present study had thrombocytopenia
and hyperCKemia, and the dominant missense variant, p.H109R in
STIM1 was identified in this patient. The mutation was
present in the affected patient and absent in the unaffected
parents, indicating that it was a spontaneous STIM1 mutation
in the patient. The level of STIM1 was not decreased in the case
described herein. Previous studies have demonstrated that the
variant probably results in constitutive STIM1 unfolding and
oligomerization, further impairing SOCE, and further changing
resting cell Ca2+ levels, which first affects skeletal
muscle as it is susceptible to changes in Ca2+ levels
(4,25). The mechanism may be related to the
fact that TAM/STRMK patients due to STIM1 mutations mostly
present with muscular involvement.
Shahrizaila et al (17) first reported 4 patients with STRMK
from one British family in 2004. To date, a total of 8 patients
with TAM/STRMK with c.326A> G (p.H109R) have been identified,
including the present case. In total, 7 patients with c.326A> G
(p.H109R) were all found in childhood. The other 7 patients had a
typical TAM-related phenotype, apart from the present patient. By
contrast, the patient in the present study only had hyperCKemia,
which was also the main reason for the absence of muscle biopsy.
Compared with other patients, patient 3 (Table II) had an obvious muscle phenotype.
However, the patient did not have obvious abnormalities in the
early stages, and the clinical symptoms gradually worsened as the
age of the last follow-up increased. Other cases have also
exhibited this phenomenon. Patient 1 (Table II) was a 53-year-old male who
required intermittent ventilation treatment at night and could not
walk normally. The patient described herein was <3 years of age
at the last follow-up and only exhibited hyperCKemia. This
phenomenon suggests that the severity of TAM/STRMK clinical
manifestations is associated with age. The patient in the present
study was diagnosed earlier than the other 7 patients, mainly due
to its recurrent thrombocytopenia/bleeding tendency. Platelet
analyses of the patient with heterozygous STIM1 R429C
mutation revealed a reduced Ca2+ store content, a
partially impaired SOCE and a secretion defect, which further
caused decreased platelet cohesion and platelet dysfunction
(19,26). This may account for the bleeding
tendency in children with STRMK. Three patients (including the
patient in the present study) with c.326A>G (p.H109R) had
thrombocytopenia/bleeding tendency. The other 2 patients (patient 5
and 6) (Table II) began to develop
muscle weakness at 3 years and >10 years of age. The case
described herein lacked the TAM-relevant performance and was <3
years old at the last follow-up, which may be related to her
long-term and medium-intensity exercise training. Further follow-up
observations are required.
STRMK has a variable degree of multisystemic signs,
including muscle weakness, miosis, thrombocytopenia, hyposplenism,
ichthyosis, a short stature and dyslexia (4). A literature review suggested that the
type and site of TAM/STRMK mutations do not determine the clinical
presentation and that the genotype-phenotype correlation remains
loose. The case in the present study first manifested
thrombocytopenia/bleeding tendency and convulsion, and was
misdiagnosed with thrombocytopenia or epilepsy. Following treatment
for thrombocytopenia, there was no significant improvement. The
case was diagnosed with TAM/STRMK by combining the previous
clinical history of the child, the clinical characteristics,
laboratory examinations and exome sequencing. Currently, the
diagnosis of TAM/STRMK is dependent on clinical features,
hyperCKemia, routine blood examination, muscle biopsies, muscle
histochemical analysis, gene sequencing and other related
assessments (11).
Some patients with TAM/STRMK develop the disease in
early life, and patients can develop motor dysfunction as the
disease progresses; however, the life expectancy of patients is not
markedly reduced. The child described in the present study had an
early age of onset and now has no significant motor dysfunction and
adverse prognosis. In patients with the p.H109R mutation identified
in the literature, patient 2 had a milder clinical manifestation
than his father (patient 1) at the same age (Table II), which may be related to the
long-term moderate-intensity exercise training of patient 2. By
contrast, patient 3 (Table II) was
unable to perform physical training activities due to a fracture of
the left greater trochanter of the femur, resulting in weight gain,
which rapidly caused secondary muscle atrophy and weakness
throughout the body. The phenomenon suggests that patients with
combined muscular system involvement may be more effectively
treated with timely exercise training of appropriate intensity.
TAM, bleeding tendency and thrombocytopenia are factors that affect
the quality of life of patients. Following the symptomatic
treatment of the aforementioned conditions, they can live normally,
or the disease may not affect their normal life. Long-term exercise
cannot only reduce the average number of tubular aggregates (TAs)
per fiber, the number of fibers containing TAs, and the average
size of the few remaining TAs, but can also promote colocalization
of STIM1 with ORAI1, significantly reduce the formation of TAs,
increase Ca2+ influx via SOCE, and enhance muscle
function and resistance to fatigue (27,28).
Markello et al (16)
hypothesized that compounds targeting the Ca(2+)-selective
release-activated Ca(2+) channel may be beneficial for patients
with TAM/STRMK. The case in the present study is now regularly
followed up in outpatient clinics, and comprehensive long-term
exercise and language training is performed at home. The patient
has not yet exhibited typical TAM-related clinical
manifestations.
TAM/STRMK is very rare and there is currently no
effective cure. Patients with TAM/STRMK have multisystemic signs
that are not easily distinguishable from other diseases. Overall,
when encountering patients with thrombocytopenia of unknown a
etiology, bleeding tendency and convulsions, whose causes are not
readily apparent by relevant tests and whose therapies are not very
useful, first, a careful assessment of the possibility of the
development of TAM/STRMK should be conducted. Second, genetic
testing must be obtained to assist in the diagnosis. At present,
there is no effective radical cure for TAM/STRMK. Patients with
TAM-related phenotypes should commence long-term exercise training
as soon as possible. In addition, the further exploration of the
pathogenesis of TAM/STRMK is warranted, and relevant targeted drugs
need to be developed as early as possible.
Acknowledgements
Not applicable.
Funding
Funding: The present study was financially supported by the
following grants: The National Natural Science Foundation of China
(nos. 81771626 and 81971423), the Jiangsu Provincial Maternal and
Child Health Key Talents Project (no. FRC201731), the Jiangsu
Provincial Key Social Development Project (no. BE2020658), and the
Suzhou Livelihood Science and Technology Project (no.
SYS2020030).
Availability of data and materials
The data that support the findings of this study are
openly available at ‘Mendeley Data’ (http://dx.doi.org/10.17632/rjmn9x7fb9.3).
Authors' contributions
WS participated in the study design and in the
writing of the manuscript. JHu and MLi participated in the
collection of clinical data. JHuo performed the interpretation of
the data. XZ participated in the data analysis, interpretation of
the data and in the writing of the manuscript. WS and XZ confirm
the authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was reviewed and approved by the
Ethics Committee of the Children's Hospital of Soochow University
(Suzhou, China; Approval no. 2020CS059). All clinical, laboratory
and molecular biological examinations and treatment programs were
performed after obtaining informed consent from the parents of the
children.
Patient consent for publication
All clinical, laboratory and molecular biological
examinations and treatment programs were performed after obtaining
informed consent from the parents of the children.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Böhm J, Chevessier F, Koch C, Peche GA,
Mora M, Morandi L, Pasanisi B, Moroni I, Tasca G, Fattori F, et al:
Clinical, histological and genetic characterisation of patients
with tubular aggregate myopathy caused by mutations in STIM1. J Med
Genet. 51:824–833. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Lacruz RS and Feske S: Diseases caused by
mutations in ORAI1 and STIM1. Ann N Y Acad Sci. 1356:45–79.
2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bohm J and Laporte J: Gain-of-function
mutations in STIM1 and ORAI1 causing tubular aggregate myopathy and
Stormorken syndrome. Cell Calcium. 76:1–9. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Misceo D, Holmgren A, Louch WE, Holme PA,
Mizobuchi M, Morales RJ, De Paula AM, Stray-Pedersen A, Lyle R,
Dalhus B, et al: A dominant STIM1 mutation causes Stormorken
syndrome. Hum Mutat. 35:556–564. 2014.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Dynes JL, Yeromin AV and Cahalan MD:
Cell-wide mapping of Orai1 channel activity reveals functional
heterogeneity in STIM1-Orai1 puncta. J Gen Physiol.
152(e201812239)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lee KW, Maeng JS, Choi JY, Lee YR, Hwang
CY, Park SS, Park HK, Chung BH, Lee SG, Kim YS, et al: Role of
Junctin protein interactions in cellular dynamics of calsequestrin
polymer upon calcium perturbation. J Biol Chem. 287:1679–1687.
2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Peche GA, Spiegelhalter C, Silva-Rojas R,
Laporte J and Böhm J: Functional analyses of STIM1 mutations reveal
a common pathomechanism for tubular aggregate myopathy and
Stormorken syndrome. Neuropathology. 40:559–569. 2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Böhm J, Bulla M, Urquhart JE, Malfatti E,
Williams SG, O'Sullivan J, Szlauer A, Koch C, Baranello G, Mora M,
et al: ORAI1 mutations with distinct channel gating defects in
tubular aggregate myopathy. Hum Mutat. 38:426–438. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Bohm J, Chevessier F, Maues De Paula A,
Koch C, Attarian S, Feger C, Hantaï D, Laforêt P, Ghorab K, Vallat
JM, et al: Constitutive activation of the calcium sensor STIM1
causes tubular-aggregate myopathy. Am J Hum Genet. 92:271–278.
2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Morin G, Biancalana V, Echaniz-Laguna A,
Noury JB, Lornage X, Moggio M, Ripolone M, Violano R, Marcorelles
P, Maréchal D, et al: Tubular aggregate myopathy and Stormorken
syndrome: Mutation spectrum and genotype/phenotype correlation. Hum
Mutat. 41:17–37. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Walter MC, Rossius M, Zitzelsberger M,
Vorgerd M, Müller-Felber W, Ertl-Wagner B, Zhang Y, Brinkmeier H,
Senderek J and Schoser B: 50 years to diagnosis: Autosomal dominant
tubular aggregate myopathy caused by a novel STIM1 mutation.
Neuromuscul Disord. 25:577–584. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Harris E, Burki U, Marini-Bettolo C, Neri
M, Scotton C, Hudson J, Bertoli M, Evangelista T, Vroling B,
Polvikoski T, et al: Complex phenotypes associated with STIM1
mutations in both coiled coil and EF-hand domains. Neuromuscul
Disord. 27:861–872. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Noury JB, Bohm J, Peche GA,
Guyant-Marechal L, Bedat-Millet AL, Chiche L, Carlier RY, Malfatti
E, Romero NB and Stojkovic T: . Tubular aggregate myopathy with
features of Stormorken disease due to a new STIM1 mutation.
Neuromuscul Disord. 27:78–82. 2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hedberg C, Niceta M, Fattori F, Lindvall
B, Ciolfi A, D'Amico A, Tasca G, Petrini S, Tulinius M, Tartaglia
M, et al: Childhood onset tubular aggregate myopathy associated
with de novo STIM1 mutations. J Neurol. 261:870–876.
2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Markello T, Chen D, Kwan JY,
Horkayne-Szakaly I, Morrison A, Simakova O, Maric I, Lozier J,
Cullinane AR, Kilo T, et al: York platelet syndrome is a CRAC
channelopathy due to gain-of-function mutations in STIM1. Mol Genet
Metab. 114:474–482. 2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Shahrizaila N, Lowe J and Wills A:
Familial myopathy with tubular aggregates associated with abnormal
pupils. Neurology. 63:1111–1113. 2004.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Nesin V, Wiley G, Kousi M, Ong EC, Lehmann
T, Nicholl DJ, Suri M, Shahrizaila N, Katsanis N, Gaffney PM, et
al: Activating mutations in STIM1 and ORAI1 cause overlapping
syndromes of tubular myopathy and congenital miosis. Proc Natl Acad
Sci USA. 111:4197–4202. 2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Morin G, Bruechle NO, Singh AR, Knopp C,
Jedraszak G, Elbracht M, Brémond-Gignac D, Hartmann K, Sevestre H,
Deutz P, et al: Gain-of-Function mutation in STIM1 (P.R304W) is
associated with Stormorken syndrome. Hum Mutat. 35:1221–1232.
2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Okuma H, Saito F, Mitsui J, Hara Y,
Hatanaka Y, Ikeda M, Shimizu T, Matsumura K, Shimizu J, Tsuji S and
Sonoo M: Tubular aggregate myopathy caused by a novel mutation in
the cytoplasmic domain of STIM1. Neurol Genet.
2(e50)2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Endo Y, Noguchi S, Hara Y, Hayashi YK,
Motomura K, Miyatake S, Murakami N, Tanaka S, Yamashita S, Kizu R,
et al: Dominant mutations in ORAI1 cause tubular aggregate myopathy
with hypocalcemia via constitutive activation of store-operated
Ca2+ channels. Hum Mol Genet. 24:637–648.
2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Li A, Kang X, Edelman F and Waclawik AJ:
Stormorken syndrome: A rare cause of myopathy with tubular
aggregates and dystrophic features. J Child Neurol. 34:321–324.
2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Jiang LJ, Zhao X, Dou ZY, Su QX and Rong
ZH: Stormorken syndrome caused by a novel STIM1 mutation: A case
report. Front Neurol. 12(522513)2021.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ticci C, Cassandrini D, Rubegni A, Riva B,
Vattemi G, Matà S, Ricci G, Baldacci J, Guglielmi V, Di Muzio A, et
al: Expanding the clinical and genetic spectrum of pathogenic
variants in STIM1. Muscle Nerve. 64:567–575. 2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Schiaffino S and Reggiani C: Fiber types
in mammalian skeletal muscles. Physiol Rev. 91:1447–1531.
2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Nakamura L, Sandrock-Lang K, Speckmann C,
Vraetz T, Bührlen M, Ehl S, Heemskerk JW and Zieger B: Platelet
secretion defect in a patient with stromal interaction molecule 1
deficiency. Blood. 122:3696–3698. 2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Boncompagni S, Pecorai C, Michelucci A,
Pietrangelo L and Protasi F: Long-term exercise reduces formation
of tubular aggregates and promotes maintenance of Ca2+
entry units in aged muscle. Front Physiol.
11(601057)2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Protasi F, Pietrangelo L and Boncompagni
S: Calcium entry units (CEUs): Perspectives in skeletal muscle
function and disease. J Muscle Res Cell Motil. 42:233–249.
2021.PubMed/NCBI View Article : Google Scholar
|