Introduction
Myotonic dystrophy (MD) type 1 (MD1) is an autosomal
dominant multisystem disorder clinically characterized by skeletal
muscle weakness, myotonia, cardiac conduction abnormalities,
cataracts and other abnormalities (1). MD1 results from an expansion of a
cytosine-thymine-guanine (CTG) trinucleotide repeat in the
3'-untranslated region of the dystrophia myotonia protein kinase
(DMPK) gene (2). A study in
2021 based on the newborn screening program in New York State
reported DM1 prevalence of 4.76 per 10,000 births or 1 in 2100
births (3). The length of the CTG
repeat expansion is moderately associated with disease severity
(4). In the case that the number of
repeats is high, MD1 can be diagnosed based on characteristic
symptoms and a positive family history. The anesthetic management
of patients with MD1 warrants careful attention due to individual
differences in reactions to certain anesthetics and the multisystem
effects of the disease (5). However,
in the case that the number of repeats is low, MD1 may progress
unnoticed by patients and their families. General anesthesia in
patients with undiagnosed MD1 can cause post-anesthesia
complications.
Difficult-to-wean patients are those who fail
initial weaning and require up to three spontaneous breathing
trials (SBTs) or as long as 7 days from the first SBT to achieve
successful weaning (6). In the
surgery room, airway obstruction, inadequate ventilation and
oxygenation, and the clearance of secretions can present
difficulties in extubating patients. In a previous multicenter,
prospective study of patients in the intensive care unit (ICU), the
incidence rate of difficult-to-wean cases was found to be 39% of
the patients who received mechanical ventilation for at least 12 h
(7). Patients in this category
should be identified for etiologies, such as respiratory, cardiac,
psychological, ventilator circuit and nutritional issues, as well
as ICU-acquired weakness (8). This
workup process can sometimes be difficult if the disease etiology
is unknown.
The present study describes the case of a woman who
was diagnosed with MD1 for the first time following surgery that
was triggered by post-operative weaning failure from mechanical
ventilation.
Case report
A 42-year-old non-gravid Japanese woman visited
Nakatsu Gastrointestinal Hospital (Nakatsu, Japan) complaining of
sudden-onset lower abdominal pain. Contrast-enhanced computed
tomography revealed the torsion of the right ovarian tumor and
pulmonary discoid atelectasis (Fig.
1A and B). She was referred to
the gynecological department of Nakatsu Municipal Hospital and
scheduled for surgery. However, a pre-operative pulmonary function
test revealed severe restrictive respiratory failure (the vital
capacity was 30% and forced expiratory volume in the first second
was 103%). Arterial blood gas analysis revealed respiratory
acidosis (pH 7.25; partial pressure of carbon dioxide, 68.2 mmHg;
and bicarbonate, 28.9 mmol/l). Her chest radiograph,
electrocardiogram and laboratory test results did not reveal any
abnormalities. The patient was then transferred to Oita University
Hospital (Oita, Japan) for high-risk emergency surgery. Of note, 3
years prior, she had been diagnosed with obesity-related
restrictive lung disease by a primary care doctor and was advised
to lose weight (her body mass index was 33.6 kg/m2 at
the time of the present analysis). She had undergone a laparoscopic
myomectomy at 36 years of age and seven consecutive failures of
artificial inseminations. She underwent an abdominal right
salpingo-oophorectomy, which was converted from laparoscopy owing
to severe pelvic adhesions.
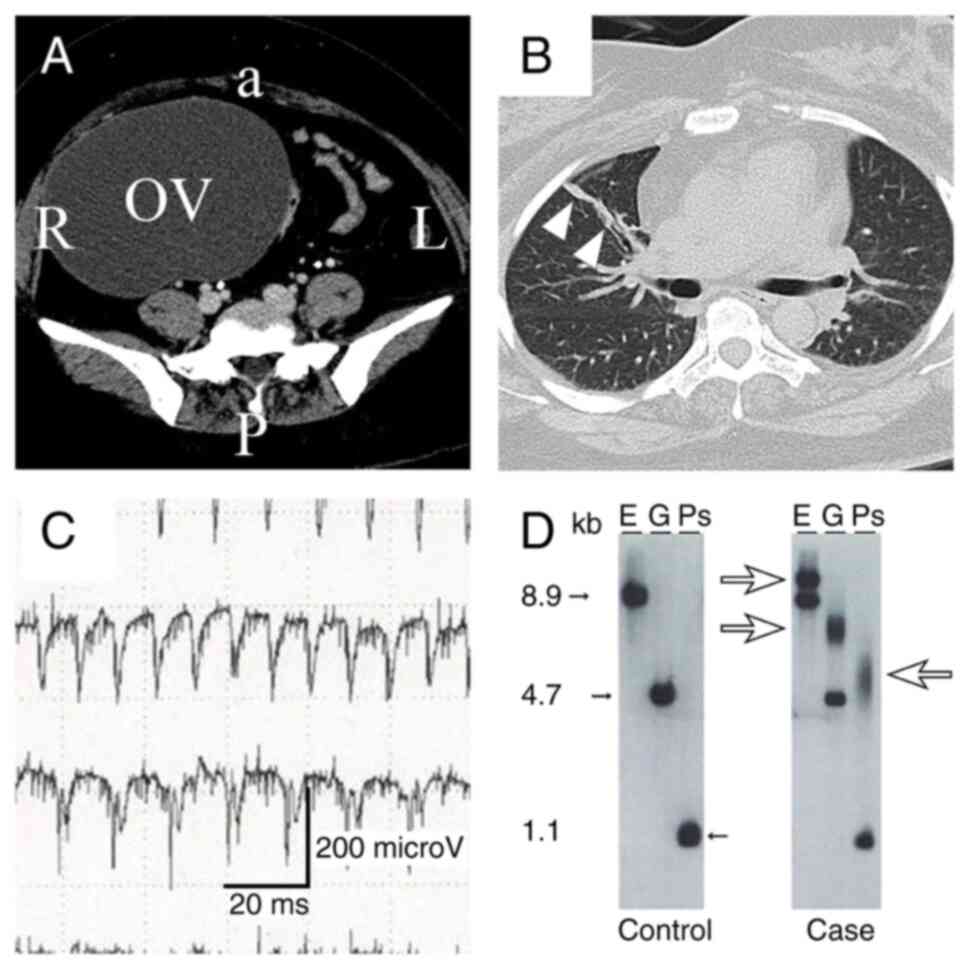 | Figure 1Results of pre-operative imaging and
tests for definitive diagnosis. (A) Pelvic computed tomography scan
illustrating a cystic right ovarian tumor (OV). a, anterior; P,
posterior; L, left; R, right. (B) Chest computed tomography scan
illustrating right pulmonary discoid atelectasis (arrowheads). (C)
Needle electromyography of the biceps indicating myotonic
discharge. (D) Southern blotting. The left panel demonstrates the
DMPK gene from a healthy control. The right panel
demonstrates the DMPK gene from the patient described
herein. Small arrows (closed) in lanes E, G and Ps indicate normal
repeat bands ranging from 5 to 34. Large arrows (open) in lanes E,
G and Ps indicate expansion repeat bands ranging from 700 to 1,100
kb. kb, kilobyte; E, EcoRI; G, BglI; Ps,
PstI. |
Anesthesia was induced with propofol (1.7 mg/kg) and
remifentanil [0.2 µg/kg/min, intravenously (i.v.)]. Tracheal
intubation was facilitated with rocuronium bromide (0.7 mg/kg,
i.v.), and anesthesia was maintained with 5% desflurane in 70:30
air: O2 and remifentanil infusion (0.25 µg/kg/min).
Ephedrine (4 mg/dose, i.v.) and phenylephrine (0.1 mg/dose, i.v.)
were used to reduce any hypotensive response during surgery. The
patient was mechanically ventilated to maintain the lower limit of
normal end-expiratory CO2 (36-37 mmHg). She was
extubated following surgery, but was promptly reintubated owing to
respiratory failure that was unresponsive to manual ventilation.
She was then transferred from the operating theater to the ICU for
post-operative respiratory management. On post-operative day
(POD)1, she was extubated again and supported by non-invasive
positive pressure ventilation. On POD2, her respiratory support was
gradually decreased from high nasal flow through a face mask to a
nasal cannula. On POD3, she was moved to the general ward. However,
owing to respiratory failure at 9 h after transfer, the patient was
reintubated for the second time and returned to the ICU. On POD5,
she was extubated again, and bi-level positive airway pressure
support was initiated following consultation with a pulmonologist
with the assessment of obesity-related respiratory failure. On
POD6, regardless of the cricothyroid membrane puncture-guided
tracheotomy, the sputum retention of the patient remained
uncontrolled, and she was intubated for the third time. She was
thus diagnosed as a difficult-to-wean patient and a tracheostomy
was performed on POD7, followed by further workup.
The consulting neurologist suspected MD based on the
patient's characteristic symptoms. Her facial and limb muscles were
weak (manual muscle test grade was 4/5) (9), her tongue was atrophied, and she was
obese. Percussion myotonia was observed. Her medical history
obtained through her written messages and from her family suggested
that her skeletal muscle weakness had gradually progressed from
adolescence onwards. At the age of 20, the patient could not
inflate a balloon, and at the age of 35, she could not open the lid
of a plastic bottle. At the most recent follow-up, she could not
walk for 100 m without resting. Her family history was not
definite; however, her mother had a similar facial appearance and
cognitive dysfunction. The patient's mother was a potential carrier
of an abnormal DMPK gene.
A needle electromyography revealed myotonic
discharge in the biceps (Fig. 1C).
Following genetic counseling, molecular genetic testing for the
DMPK gene was performed, which revealed a CTG repeat length
of 700-1,100 bp (Fig. 1D). The
genetic testing was performed by LSI Medicine Corporation
(Inspection code 02820, Tokyo, Japan). Finally, the patient was
diagnosed with MD1. She continued dysphagia rehabilitation for
weaning from the ventilator in hospital. Continuous dysphagia
rehabilitation resulted in recovery to ventilation with minimum
support [pressure support (PS)/continuous positive airway pressure;
fraction of inspired oxygen, 0.3; positive end-expiratory pressure,
6 mmHg; PS, 8 mmHg] on POD39. The patient was then transferred to
Nishibeppu Hospital (Beppu, Japan) on POD48.
Discussion
The present study describes a case of MD, with two
key clinical issues. First, MD1 may become clinically evident
following post-operative weaning difficulty in patients with a
history of uneventful general anesthesia. Second, MD1 may be
predicted much at an earlier stage based on a combination of
pre-operative respiratory failure and a gynecological history.
Several factors can cause an imbalance between
respiratory muscle strength and the workload of breathing,
resulting in weaning difficulties in the ICU. These factors
comprise respiratory or ventilatory, cardiac, psychological,
ventilator circuit-related and nutritional issues (8,10). A
summary of the reported cases of MD1 diagnosed following
post-operative weaning failure is presented in Table I (11-14).
The present study also conducted a literature review regarding
undiagnosed MD by searching PubMed/MEDLINE and Google Scholar. The
terms ‘respiratory failure’, ‘myotonic dystrophy’ and ‘undiagnosed
myotonic dystrophy’ were used to search the literature in English
without publication date filters. Patients from all four studies
required long-term post-operative ventilator management and
tracheostomy, resulting in a poor prognosis in some cases (11-14).
None of the patients were identified to have a family history of
MD1, although similar findings were later demonstrated in the
patients' families by consultant neurologists (11,12). It
should be noted that only the case described herein had a history
of uneventful general anesthesia. This is one of the reasons that a
certain amount of time was needed to reach a definite diagnosis of
MD1.
 | Table IReported cases of myotonic dystrophy 1
diagnosed after surgery triggered by post-operative weaning
failure. |
Table I
Reported cases of myotonic dystrophy 1
diagnosed after surgery triggered by post-operative weaning
failure.
| Study no. | Authors | Year of
publication | Age, years | Diagnosis | Type of surgery | Previous history of
surgery | DMPK
status | Symptoms and physical
signs | Outcome | (Refs.) |
|---|
| 1 | Fossen and
Gjerstad | 1986 | 34 | Atonic bleeding | Abdominal
hysterectomy | C-section (local
anesthesia) | N/A | Bilateral ptosis,
facial and distal limb muscle weakness, myotonic grip, percussion
myotonia | Recovery in the 6th
week | (11) |
| 2 | Gupta et
al | 2009 | 32 | Adnexal mass | Exploratory
laparotomy to rule out cancer | None | Expansion
(160,900) | Temporal flattening
and frontal balding, scoliotic deformity of dorsal spine, muscle
spasm | Dead of
cardio-pulmonary arrest in ICU on the 391st day | (12) |
| 3 | Gómez Hernández et
al | 2016 | 73 | Thymoma and thyroid
cancer | Cervicotomy and
partial sternotomy | Cataract surgery and
basal cell carcinoma surgery (local anesthesia) | Expansion | Baldness, dropping
eyelids, early cataracts, muscle weakness, swallowing
difficulties | Succumbed due to
asphyxia in the hospital in the 1st month | (13) |
| 4 | Ota et al | 2019 | 20 | Appendiceal
endometriosis | Laparoscopic biopsy
and appendectomy | N/A | Expansion (400 to
450) | Long and narrow
facies with atrophic temporalis | Alive and discharged
on the 69th day | (14) |
| 5 | Present study | 2023 | 42 | Adnexal torsion | Abdominal
salpingo-oophorectomy | Laparoscopic
myomectomy (general anesthesia) | Expansion (700 to
1,100) | Facial and limb
muscles weakness, tongue atrophy, percussion myotonia, obesity | Alive and transferred
on the 48th day | |
Previous studies have demonstrated that
tumorigenesis, including gynecological benign and malignant tumors,
is a key clinical manifestation of MD1. A previous meta-analysis
included five studies comprising 2,779 patients and revealed that
the standardized incidence ratio for endometrial cancer was 7.48
[95% confidence interval (CI), 4.72-11.8] and 5.56 (95% CI,
2.99-10.3) for ovarian cancer (15).
The UK Myotonic Dystrophy Patient Registry reported that the
incidence of benign gynecological tumors ranged from 6.1 to 37.6%
for uterine myoma and 3.5% for ovarian cysts (16). Furthermore, the female sex was
significantly associated with benign tumors in multiple organs
(16). Thus, patients with MD,
particularly females, have a higher chance of undergoing general
anesthesia surgery than the general population. Therefore,
clinicians should pay attention not only to common clinical
manifestations, such as musculoskeletal weakness, cardiac defects
and early cataracts, but to the tumorigenesis of MD1 as well.
In conclusion, the present study describes the case
of a woman with post-operative weaning difficulty with unrecognized
MD1. The risk of general anesthesia for patients with MD1 is widely
known. Therefore, many individuals with known MD1 carry a card to
alert authorities of their risk regarding anesthesia. Conversely, a
history of uneventful general anesthesia may be a pitfall in the
differential diagnosis of patients with weaning difficulties.
Furthermore, clinicians need to be more familiar with the variable
manifestations of MD1 in multiple organs other than neuromuscular
symptoms. In patients with certain risk factors (such as a history
of gynecological tumors or respiratory failure), timely
identification and evaluation of these risk factors would help to
ensure safer and more effective treatment outcomes. Furthermore,
based on the findings of the present case report, further
prospective studies are warranted to systematically evaluate the
risks and influencing factors that the majority of patients with
MD1 face regarding post-extubation difficulty, providing more
substantial evidence for clinical practice.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TY and KK contributed to the design of the study,
analysis of the patient's data and interpretation of the results,
and in the writing of the manuscript. NF performed the anesthesia
and intensive care management and was a major contributor to the
editing of the manuscript. MJ performed the neurological
examination and was a major contributor to the writing the
manuscript. SE and YKu contributed to the acquisition of the
patient's data and its curation and critically reviewed the
manuscript. YKa advised on patient treatment, supervised the study,
and critically reviewed the manuscript. TY and KK confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and Consent to
participate
The present study was conducted according to the
guidelines of the Declaration of Helsinki and approved by the
Ethics Committee of Oita University Hospital (approval no. 2492).
Written informed consent was obtained from the patient's husband
for her anonymized information to be included in the present case
report.
Patient consent for publication
Written informed consent was obtained from the
patient and the patient's husband for her anonymized information to
be published in the present case report.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Udd B and Krahe R: The myotonic
dystrophies: Molecular, clinical, and therapeutic challenges.
Lancet Neurol. 11:891–905. 2012.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Mahadevan M, Tsilfidis C, Sabourin L,
Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J,
O'Hoy K, et al: Myotonic dystrophy mutation: An unstable CTG repeat
in the 3' untranslated region of the gene. Science. 255:1253–1255.
1992.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Johnson NE, Butterfield RJ, Mayne K,
Newcomb T, Imburgia C, Dunn D, Duval B, Feldkamp ML and Weiss RB:
Population-Based prevalence of myotonic dystrophy type 1 using
genetic analysis of statewide blood screening program. Neurology.
96:e1045–e1053. 2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Groh WJ, Groh MR, Shen C, Monckton DG,
Bodkin CL and Pascuzzi RM: Survival and CTG repeat expansion in
adults with myotonic dystrophy type 1. Muscle Nerve. 43:648–651.
2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Mangla C, Bais K and Yarmush J: Myotonic
dystrophy and anesthetic challenges: A case report and review. Case
Rep Anesthesiol. 2019(4282305)2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Boles JM, Bion J, Connors A, Herridge M,
Marsh B, Melot C, Pearl R, Silverman H, Stanchina M,
Vieillard-Baron A and Welte T: Weaning from mechanical ventilation.
Eur Respir J. 29:1033–1056. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Penuelas O, Frutos-Vivar F, Fernandez C,
Anzueto A, Epstein SK, Apezteguia C, Gonzalez M, Nin N, Raymondos
K, Tomicic V, et al: Characteristics and outcomes of ventilated
patients according to time to liberation from mechanical
ventilation. Am J Respir Crit Care Med. 184:430–437.
2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Obata E, Kai K, Aso S, Tsukamoto N,
Hanaoka T, Nabeta Y and Kawano Y: Demons syndrome with pericardial
effusion followed by intensive care unit-acquired weakness: A case
report and literature review. SAGE Open Med Case Rep.
10(2050313X211069315)2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Medical Research Council: Memorandum No.
45: Aids to the Examination of the Peripheral Nervous System.
Medical Research Council, London, 1976.
|
|
10
|
Trudzinski FC, Neetz B, Bornitz F, Müller
M, Weis A, Kronsteiner D, Herth FJF, Sturm N, Gassmann V, Frerk T,
et al: Risk factors for prolonged mechanical ventilation and
weaning failure: A systematic review. Respiration. 101:959–969.
2022.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Fossen D and Gjerstad L: Obstetric
complications as the first sign of myotonic dystrophy. Acta Obstet
Gynecol Scand. 65:667–668. 1986.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Gupta N, N Saxena K, Kumar Panda A, Anand
R and Mishra A: Myotonic dystrophy: An anaesthetic dilemma. Indian
J Anaesth. 53:688–691. 2009.PubMed/NCBI
|
|
13
|
Gómez Hernández MT, Martin Posadas MT and
Garcia Sanchez Mdel C: Undiagnosed myotonic dystrophy type 1 in a
patient with synchronous thymoma and thyroid cancer. Arch
Bronconeumol. 52:393–394. 2016.PubMed/NCBI View Article : Google Scholar : (In English,
Spanish).
|
|
14
|
Ota K, Nakamura Y, Nakamura E, Takashima
S, Oka M, Ota K, Sakaue M, Sano Y and Takasu A: Massive abscess
with prolonged respiratory failure due to newly diagnosed myotonic
dystrophy: A case report. Medicine (Baltimore).
98(e15427)2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Emparanza JI, Lopez de Munain A, Greene
MH, Matheu A, Fernandez-Torron R and Gadalla SM: Cancer phenotype
in myotonic dystrophy patients: Results from a meta-analysis.
Muscle Nerve. 58:517–522. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Alsaggaf R, Wang Y, Marini-Bettolo C, Wood
L, Nikolenko N, Lochmuller H, Greene MH and Gadalla SM: Benign and
malignant tumors in the UK myotonic dystrophy patient registry.
Muscle Nerve. 57:316–320. 2018.PubMed/NCBI View Article : Google Scholar
|














