Introduction
Castleman disease (CD) describes a group of rare
heterogeneous lymphoproliferative disorders characterized by
enlarged hyperplastic lymph nodes. Based on the clinical features
and distribution of lymphadenopathy, CD can be classified into
unicentric CD (UCD) and multicentric CD (MCD) (1). Histopathologically, CD can be
classified into the hyaline-vascular (HV) variant, plasma cell (PC)
variant and mixed types. The HV type is more common in patients
with UCD, whereas the PC type is more commonly observed in MCD
(2). MCD is frequently observed in
patients with human immunodeficiency virus (HIV) infection
(3). Human herpes virus 8 (HHV-8)
infection is observed in all HIV-positive patients and in a few
HIV-negative patients with MCD (4).
Idiopathic MCD (iMCD) refers to those patients without HHV-8 and
HIV infection (5).
Patients with UCD often present with lymphadenopathy
and compressive symptoms. MCD is frequently associated with
systemic manifestations, including fever, fatigue, edema and weight
loss (2). Complete resection of the
involved lesion is the gold standard of treatment for UCD (6). MCD, and particularly iMCD, is
associated a poor prognosis and systemic treatment is preferred
(5). The present study examined the
demographic characteristics, clinical presentation, histology,
staging, treatment and outcomes of 11 patients diagnosed with CD at
the Regional Cancer Centre, Thiruvananthapuram, India.
Patients and methods
The present study was a retrospective study on 11
patients with CD, diagnosed and treated at Regional Cancer Centre
from 2017 to 2022. Data on demographic details including age and
sex, clinical presentation, laboratory investigations, radiological
findings, histopathological details and treatment received were
collected from the medical records. All patients underwent a tissue
biopsy for a histopathological diagnosis. They were classified as
HV, PC and mixed types based on the morphology and
immunohistochemistry. Lymph nodes were fixed in 10% neutral
buffered formalin at room temperature for a period of 24 to 48 h.
Sections were cut at a thickness of 3-4 µm. Immunohistochemistry
was performed using the automated Ventana Benchmark XT system. The
sections were incubated with primary antibody (CD138, dilution
1:50, Biocare Medical, LLC; clone M115) at 37˚C for 32 min and
secondary antibody (ultraview DAB detection kit, ready to use,
Ventana labx; Ventana Medical Systems, Inc.) at 37˚C for 12 min.
The sections were counterstained with hematoxylin (MilliporeSigma)
and visualized under a light microscope (Olympus Corporation).
Based on the anatomical distribution of CD, patients
were divided into UCD and MCD. The UCD group consisted of patients
who had histological evidence of CD in a single group of lymph
nodes without clinical or radiological evidence of adenopathy
elsewhere. Patients with MCD had histological evidence of CD in ≥1
group of lymph nodes and radiological or clinical evidence of
adenopathy elsewhere (1).
Histologically, CD was classified into the HV type and PC type
based on the histological criteria proposed by Keller et al
(7). Lymph nodes with
characteristics intermediate between HV and PC were categorized as
mixed type (1). The standard of care
for UCD is surgical excision and for multicentric CD, systemic
therapy is the mainstay of treatment. The response assessment was
performed based on the revised RECIST criteria (version 1.1)
(8). Follow-up details were noted
from the case file. The study was conducted after obtaining patient
consent and the approval of the Human Ethics Committee at Regional
Cancer Centre.
Results
The median age of the study population was 41 years
(range, 24 to 68 years). There were 8 males and 3 females. The
baseline clinical characteristics, treatment summary, outcomes and
survival are summarized in Table I.
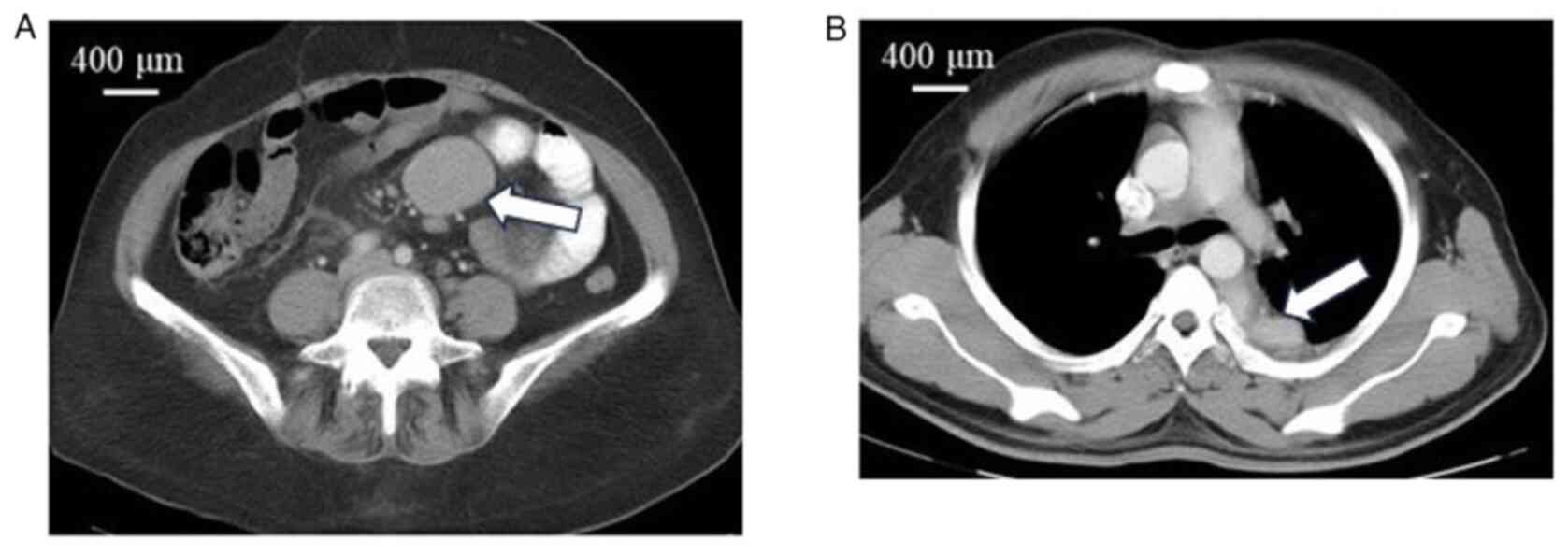
Posterior mediastinal mass, colonic mass and jejunal mass, which
are rare extranodal presentations, were each observed in 1 patient
(patient nos. 6, 7 and 10, respectively) (Fig. 1). The median duration of symptoms was
3 months (range, 1 to 10 months).
 | Table IClinical characteristics, treatment
summary, outcomes and survival of the study population. |
Table I
Clinical characteristics, treatment
summary, outcomes and survival of the study population.
| Patient no. | Age, years/sex | Clinical
presentation | Stage | Histology | Treatment | Status | Overall survival
(months) |
|---|
| 1 | 34/F | Retroperitoneal
mass | Unicentric | Hyaline vascular | Nephrectomy and en
bloc excision | Alive, complete
response | 48 |
| 2 | 24/M | Mesenteric mass POEMS
syndrome | Unicentric | Mixed | Excision | Alive, complete
response | 149 |
| 3 | 32/M | Retroperitoneal
mass | Multicentric | Hyaline vascular | Rituximab | Alive, partial
response | 48 |
| 4 | 54/F | Mesenteric mass | Unicentric | Plasma cell | Excision | Alive, complete
response | 48 |
| 5 | 63/M | Generalised
lymphadenopathy | Multicentric | Mixed | Rituximab | Alive, partial
response | 53 |
| 6 | 25/M | Posterior mediastinal
mass | Unicentric | Hyaline vascular | Debulking,
radiotherapy | Alive, partial
response | 40 |
| 7 | 60/M | Colonic mass | Unicentric | Not known | Right
hemicolectomy | Alive, complete
response | 60 |
| 8 | 45/M | Generalized
lymphadenopathy | Multicentric | Plasma cell | CHOP 1 cycle | Expired | 3 |
| 9 | 25/M | Generalized
lymphadenopathy | Multicentric | Hyaline vascular | RCHOP
*6 | Alive, complete
response | 14 |
| 10 | 68/F | Jejunal mass | Unicentric | Hyaline vascular | Bowel resection | Alive, complete
response | 20 |
| 11 | 41/M | External iliac and
inguinal adenopathy | Unicentric | Plasma cell | Rituximab | Alive, complete
response | 10 |
A total of 7 patients were diagnosed with UCD and 4
patients were diagnosed with MCD. A comparison of the clinical
profiles of the patients with UCD and MCD is presented in Table II. Systemic symptoms, including
fever and weight loss were observed in 5 patients, of whom 4
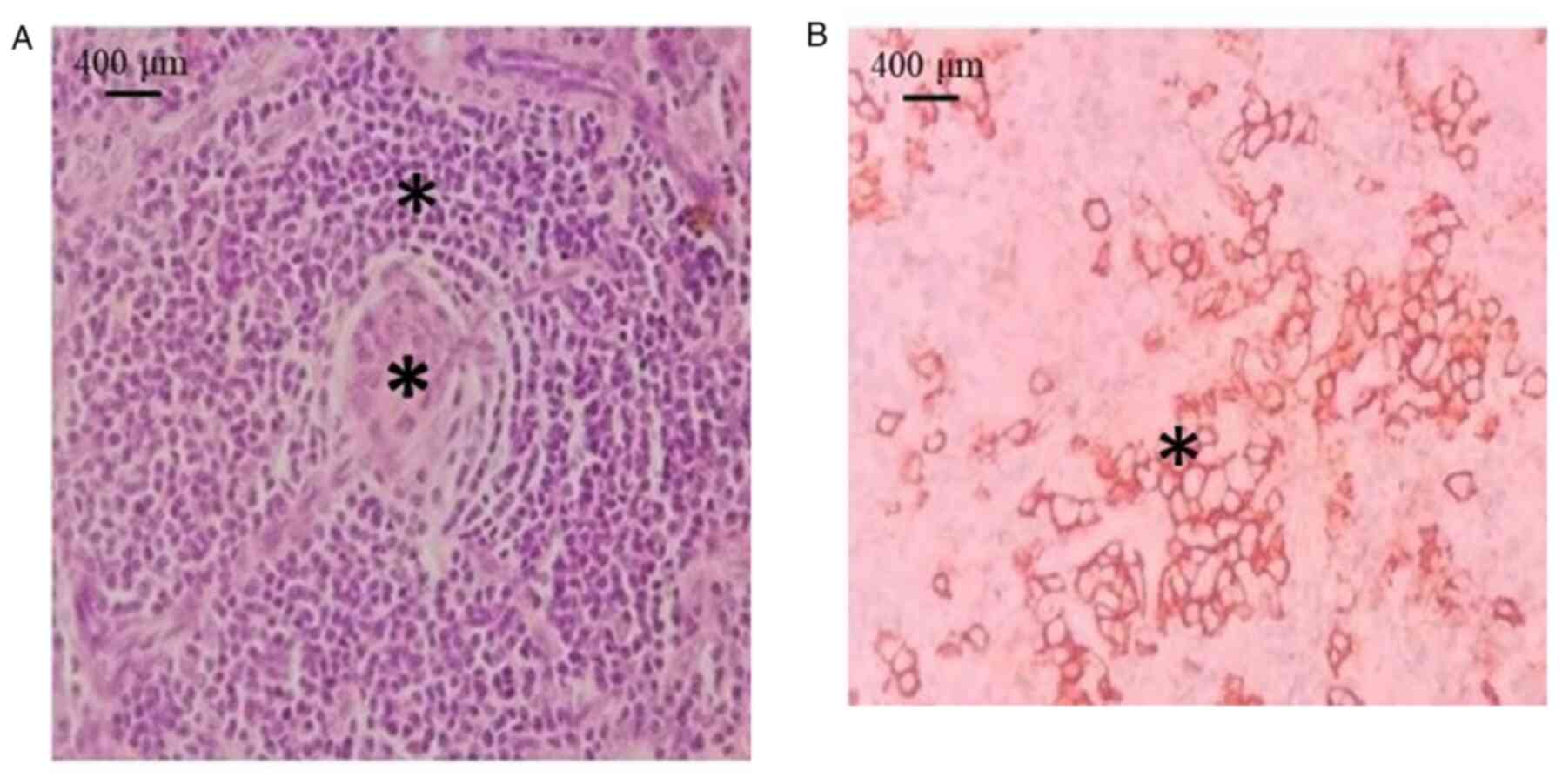
patients had UCD. Histopathologically, the HV variant exhibited
small lymphocytes with lymphocyte depletion in germinal centers,
penetrated by sclerotic blood vessels (lollipop lesions) and broad
mantle zones with concentric rings of small lymphocytes (onion skin
pattern) (Fig. 2A). The mixed
subtype exhibited clusters of plasma cells in the interfollicular
area with cytoplasmic membrane staining for CD138 (Fig. 2B). Among the 7 patients with UCD, 3
patients had the HV subtype, 2 patients had the PC subtype, 1
patient had a mixed histology and in 1 patient, the subtype was not
specified. Among the patients with MCD, 2 patients had the HV type
and 1 patient each had PC and a mixed histology. In total, 6
patients with UCD had visceral disease presentation. Furthermore, 3
patients with MCD presented with peripheral lymphadenopathy
(Table II). In addition, 1 patient
(patient no. 11) was positive for HHV-8 and 1 patient (patient no.
2) had associated polyneuropathy, organomegaly, endocrinopathy,
monoclonal plasma cell disorder, skin changes (POEMS) syndrome. All
patients were negative for HIV (Table
II).
| Table IIComparison of the clinical profiles of
patients with unicentric and multicentric Castleman disease. |
Table II
Comparison of the clinical profiles of
patients with unicentric and multicentric Castleman disease.
| Characteristic | Unicentric (n=7) | Multicentric
(n=4) |
|---|
| Age (years), median
(range) | 41 (24-68) | 38.5 (25-63) |
| Sex | | |
|
Male | 4 | 4 |
|
Female | 3 | - |
| Location | | |
|
Peripheral | 1 | 3 |
|
Visceral | 6 | 1 |
| Systemic
symptoms | 4 | 1 |
| Lymphadenopathy | 1 | 3 |
|
Hepatosplenomegaly | - | 2 |
| Anemia | 2 | 2 |
| Hypoalbuminemia | 2 | 2 |
| Pathological
subtype | | |
|
Hyaline-vascular | 3 | 2 |
|
Plasma
cell | 2 | 1 |
|
Mixed | 1 | 1 |
| Human herpes virus
8 | 1 | - |
| POEMS syndrome | 1 | - |
Among the 7 patients with UCD, 6 patients underwent
surgery, including 5 patients who underwent complete excision, 1
patient with debulking, and 1 patient (patient no. 11) received
rituximab. Six patients with UCD attained a complete response (CR)
and 1 patient had a partial response (PR). All patients with UCD
are alive and on follow-up. The patients with MCD received systemic
treatment with rituximab and/or chemotherapy with rituximab,
cyclophosphamide, adriamycin, vincristine and prednisolone
(R-CHOP). Of the patients with MCD, 1 patient (patient no. 9)
attained CR, 2 patients had a PR and are under follow-up. In
addition, 1 patient (patient no. 8) succumbed after 3 months. The
3-year survival rate for the study group was 91%.
Discussion
CD is a rare lymphoproliferative disorder first
described by Benjamin Castleman in 1956 in a series of 13 patients
with mediastinal mass resembling thymoma (9). In the present study, the authors
discuss their experience in treating this rare disease.
Patients with CD usually present in the fourth
decade of life (2). The HV variant
is commonly observed in UCD and the PC variant is more common in
MCD (10). In the study by Yu et
al (11), the abdomen was the
most common site of disease presentation (39.5%) in patients with
UCD followed by neck and mediastinum. In the present case series,
the median age was 41 years. UCD was the most common type observed
in >50% of patients, which is consistent with that reported in
previous studies (10,12). In the present study, HV was the most
common histology in both UCD and MCD. A total of 6 patients with
UCD in the present study had visceral disease and 1 patient had
posterior mediastinal mass.
Systemic symptoms and organomegaly are more frequent
in patients with MCD (13,14). However, in the present study,
systemic symptoms were more common in the UCD type. A total of 2
patients with MCD had hepatosplenomegaly, while none with UCD had
the same. The presence of POEMS syndrome is often associated with a
significant risk of mortality (14).
In the present series, 1 patient (patient no. 2) with POEMS
syndrome was alive with CR and on follow-up at 12 years.
Overall survival approaching 100% has been
documented for patients with UCD following surgery (15). Talat et al (6) studied 278 patients with UCD treated
with surgery and reported an overall survivalof 90% at a follow-up
of 10 years. In the study by Dispenzieri and Fajgenbaum (16), the 5-year survival rate was 91% for
patients with UCD treated surgically. Radiotherapy has a role in
patients with unresectable disease or in those who have an
incomplete resection (17). In the
present study, among the 6 patients with UCD who underwent surgery,
5 patients attained CR and are on follow-up. In addition, 1 patient
(patient no. 6) had a PR and is under follow-up with at 3
years.
Systemic therapy is the mainstay of treatment for
patients with MCD (13). The
monoclonal antibody against anti-CD20, rituximab, has been proven
to be effective in HIV-associated MCD. Bower et al (18) reported an overall survival rate of
95% at 2 years for patients with HIV-associated MCD treated with
rituximab. Rituximab-based therapies have led to a marked
improvement in the outcome of these patients with 5-year overall
survival rates reaching >90% (19). In patients with iMCD, a 78% overall
remission rate has been observed in those treated with
chemotherapy; however, many patients subsequently progress or
develop infectious complications (20). In the present study, among the 4
patients with MCD who received systemic treatment, 2 patients
progressed.
In conclusion, CD is a rare disease occurring in
immunodeficient patients. UCD is more common and is associated with
improved outcomes. Surgery is the mainstay of management in
unicentric disease, whereas MCD requires combination
chemotherapy.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used in the current study are available
from the corresponding author upon reasonable request.
Authors' contributions
SSA was involved in the literature search, in data
acquisition and analysis, in manuscript preparation, and in the
editing and reviewing of the manuscript. GN was involved in the
conception and design of the study, in the literature search, in
data acquisition, in manuscript preparation, and in the editing and
reviewing of the manuscript. SMT, JAV, DSJP, PNP, SGN and RN were
involved in data acquisition and in there viewing of the
manuscript. SSA and GN has confirmed the authenticity of all the
raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board of the Regional Cancer Centre (IRB no-09/2021/01) and
the Human Ethics Committee of the Regional Cancer Center, (HEC
no-24/22). The patients provided consent for their participation in
the study.
Patient consent for publication
The patients provided written informed consent for
their participation in the study and the publication of their data
and any related images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jiang JP, Shen XF, Du JF and Guan WX: A
retrospective study of 34 patients with unicentric and multicentric
Castleman's disease: Experience from a single institution. Oncol
Lett. 15:2407–2412. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Casper C: The aetiology and management of
Castleman disease at 50 years: Translating pathophysiology to
patient care. Br J Haematol. 129:3–17. 2005.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Krause JR, Robinson SD and Vance EA:
Multicentric Castleman's disease and HIV. Proc Bayl Univ Med Cent.
27:28–30. 2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Suda T, Katano H, Delsol G, Kakiuchi C,
Nakamura T, Shiota M, Sata T, Higashihara M and Mori S: HHV-8
infection status of AIDS-unrelated and AIDS-associated multicentric
Castleman's disease. Pathol Int. 51:671–679. 2001.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Fajgenbaum DC, van Rhee F and Nabel CS:
HHV-8-negative, idiopathic multicentric Castleman disease: Novel
insights into biology, pathogenesis, and therapy. Blood.
123:2924–2933. 2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Talat N, Belgaumkar AP and Schulte KM:
Surgery in Castleman's disease: A systematic review of 404
published cases. Ann Surg. 255:677–684. 2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Keller AR, Hochholzer L and Castleman B:
Hyaline-vascular and plasma-cell types of giant lymph node
hyperplasia of the mediastinum and other locations. Cancer.
29:670–683. 1972.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Eisenhauer EA, Therasse P, Bogaerts J,
Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S,
Mooney M, et al: New response evaluation criteria in solid tumours:
Revised RECIST guideline (version 1.1). Eur J Cancer. 45:228–247.
2009.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Castleman B, Iverson L and Menendez VP:
Localized mediastinal lymphnode hyperplasia resembling thymoma.
Cancer. 9:822–830. 1956.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Talat N and Schulte KM: Castleman's
disease: Systematic analysis of 416 patients from the literature.
Oncologist. 16:1316–1324. 2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Yu L, Tu M, Cortes J, Xu-Monette ZY,
Miranda RN, Zhang J, Orlowski RZ, Neelapu S, Boddu PC, Akosile MA,
et al: Clinical and pathological characteristics of HIV- and
HHV-8-negative Castleman disease. Blood. 129:1658–1668.
2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Ye B, Gao SG, Li W, Yang LH, Zhao SH, Ma
K, Zhu XL, Liu XY and Sun KL: A retrospective study of unicentric
and multicentric Castleman's disease: A report of 52 patients. Med
Oncol Northwood Lond Engl. 27:1171–1178. 2010.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Liu W, Cai Q, Yu T, Strati P, Hagemeister
FB, Zhai Q, Zhang M, Li L, Fang X, Li J, et al: Clinical
characteristics and outcomes of Castleman disease: A multicenter
Consortium study of 428 patients with 15-year follow-up. Am J
Cancer Res. 12:4227–4240. 2022.PubMed/NCBI
|
|
14
|
Dispenzieri A and Fajgenbaum DC: Overview
of Castleman disease. Blood. 135:1353–1364. 2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
van Rhee F, Oksenhendler E, Srkalovic G,
Voorhees P, Lim M, Dispenzieri A, Ide M, Parente S, Schey S,
Streetly M, et al: International evidence-based consensus
diagnostic and treatment guidelines for unicentric Castleman
disease. Blood Adv. 4:6039–6050. 2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Dispenzieri A, Armitage JO, Loe MJ, Geyer
SM, Allred J, Camoriano JK, Menke DM, Weisenburger DD, Ristow K,
Dogan A and Habermann TM: The clinical spectrum of Castleman's
disease. Am J Hematol. 87:997–1002. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Neuhof D and Debus J: Outcome and late
complications of radiotherapy in patients with unicentric Castleman
disease. Acta Oncol Stockh Swed. 45:1126–1131. 2006.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Bower M, Powles T, Williams S, Davis TN,
Atkins M, Montoto S, Orkin C, Webb A, Fisher M, Nelson M, et al:
Brief communication: Rituximab in HIV-associated multicentric
Castleman disease. Ann Intern Med. 147:836–839. 2007.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Pria AD, Pinato D, Roe J, Naresh K, Nelson
M and Bower M: Relapse of HHV-8-positive multicentric Castleman
disease following rituximab-based therapy in HIV-positive patients.
Blood. 129:2143–2147. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Van Rhee F, Voorhees P, Dispenzieri A,
Fosså A, Srkalovic G, Ide M, Munshi N, Schey S, Streetly M, Pierson
SK, et al: International, evidence-based consensus treatment
guidelines for idiopathic multicentric Castleman disease. Blood.
132:2115–2124. 2018.PubMed/NCBI View Article : Google Scholar
|