Introduction
Historically, cocaine was utilized as a topical
anesthetic and active ingredient in a variety of tonics, beverages
and cure-alls (1). At present, it is
a commonly abused illicit substance. The United Nations estimates
the global prevalence of cocaine use to be ~0.4% (2). Cocaine abuse results in several visits
to emergency departments and hospitalizations, and is responsible
for a variety of neurological and system complications. The present
study reports the case of a patient presenting with systemic
complications and significant neurological impairment secondary to
cocaine use, who made a good recovery.
Case report
A 23-year-old female presented with coma to a
tertiary hospital in Canada. She last been seen in a normal
condition 12-h prior and was subsequently found in bed. Collateral
history revealed no preceding headache, psychiatric nor cognitive
changes, focal neurological deficits, seizures, fever, neck
discomfort, or any other systemic symptoms. There was no history of
contact with any ill individuals, viral illness, travel, or
vaccination. Her medical history disclosed no comorbidities and no
prescribed medications. Her social history was significant for
polysubstance use and subsequent information revealed she had
recently attended a ‘pill party’.
Upon arrival to the emergency department, the
patient had a temperature of 36.5˚C, a heart rate of 151 beats/min,
a respiratory rate of 28 breaths/min, a blood pressure of 101/65, a
Glasgow Coma Scale score of 4 and a glucose level of 6 mmol/l.
Central painful stimuli produced extensor posturing, no verbal
response and her eyes remained closed. The eyes were mid-position
with no vertical eye movements on command, roving, bobbing/dipping,
or gaze deviation. Her pupils were 2-3 mm and were reactive
bilaterally with no hippus. A fundoscopy did not reveal any notable
findings and there were no signs indicating papilledema. The
remaining brainstem reflexes were intact. A more detailed motor
examination revealed a normal tone throughout, 4+ reflexes
symmetrically, bilateral clonus and an upgoing and equivocal
plantar response on the right and left respectively. A detailed
systemic examination revealed nuchal rigidity, normal otoscopy, no
atypical odours and tachypnea with no irregular pattern; the
respiratory examination yielded normal results; a cardiovascular
examination also revealed normal results, including no murmurs; an
abdominal examination did not reveal any notable findings and there
were no extrahepatic signs of liver disease; there were also no
concerning dermatological findings.
Simultaneous with the assessment were resuscitative
measures, including the administration of 2 mg naloxone with no
effect; rapid sequence induction and endotracheal intubation; fluid
resuscitation with normal saline; and the use of empiric acyclovir
(10 mg/kg IV q8h), ceftriaxone (2 g IV q12h) and vancomycin (20
mg/kg IV load followed by 15 mg/kg IV q12h). Investigations
(Table I) included a computed
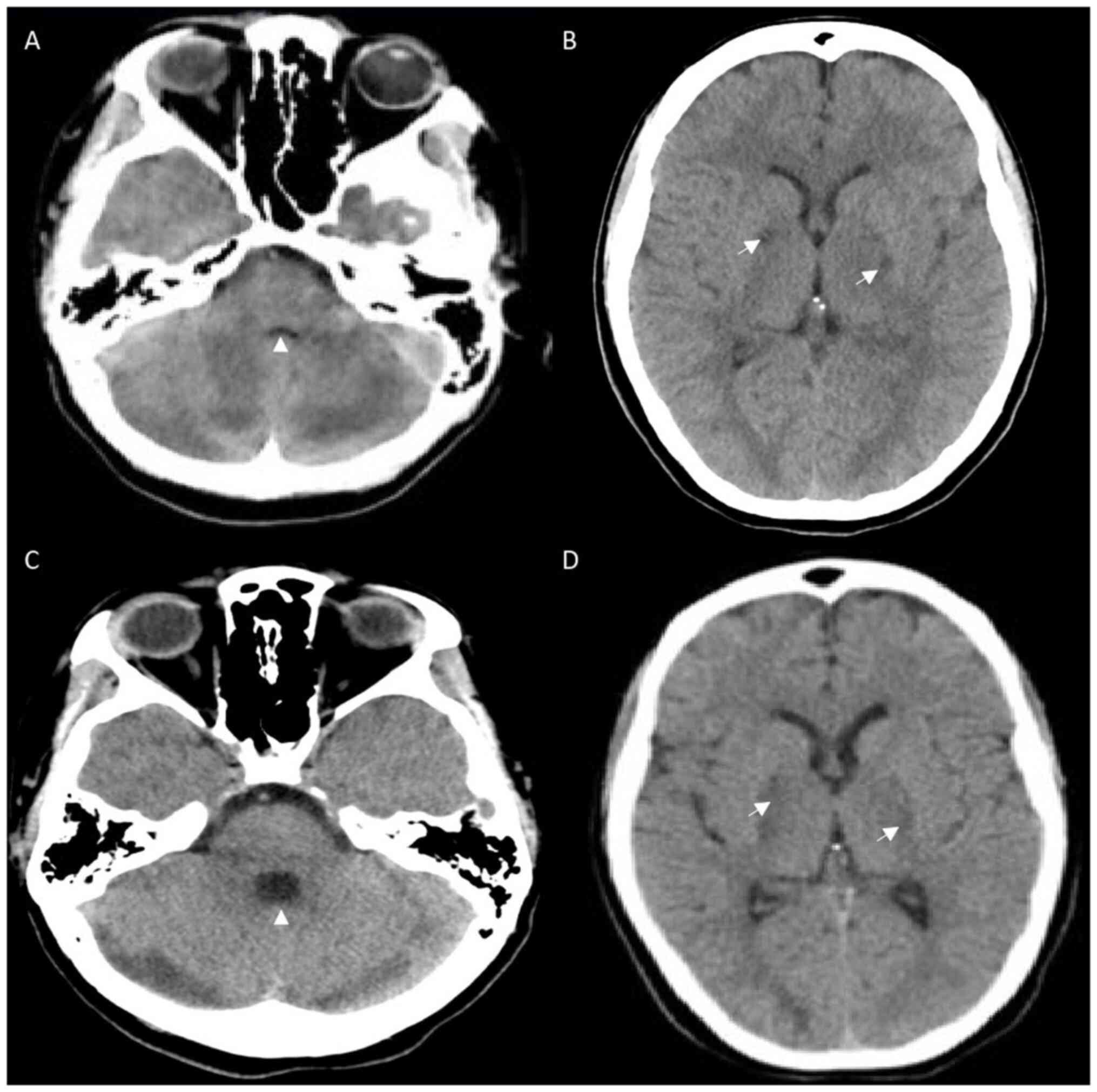
tomography (CT) scan of the head that revealed bilateral
hypodensities in the globus pallidus, and the loss of gray-white
differentiation in both cerebellar hemispheres (Fig. 1A and B). CT angiography revealed no venous or
arterial abnormalities. While optic nerve sheath diameters on point
of care ultrasound and CT head were <5 mm, the concern of mass
effect within the posterior fossa precluded a lumbar puncture.
Initial systemic investigations revealed acute kidney injury (AKI)
with myoglobinuria and granular casts, transaminitis with preserved
liver function, mild rhabdomyolysis and hypokinetic apex with
mildly reduced left ventricle function (Table I).
 | Table IInitial investigations performed for
the patient in the present case report. |
Table I
Initial investigations performed for
the patient in the present case report.
| Category | Investigations | Results |
|---|
| Neurological | Computed tomography
angiography- head and neck | Bilateral
hypodensities in globus pallidus and loss of gray-white matter
differentiation in cerebellar hemispheres. No venous or arterial
abnormalities. |
| | Magnetic resonance
imaging-brain | Edema involving
cerebellum, globus pallidus, hippocampus, peri-rolandic, centrum
semiovale, periventricular and occipital regions. Bilateral
cerebellar infarcts and diffusion restriction in gray matter
structures of cerebral hemispheres. Petechial hemorrhages in
occipital lobes and splenium of corpus callosum. |
| |
Electroencephalography | Diffuse alpha and
delta wave slowing. No rhythmic or periodic patterns. No
electrographic seizures. |
| Cardiac | Troponin | 421 ng/l |
| |
Electrocardiogram | Sinus tachycardia
with no other abnormalities |
| | Transthoracic
echocardiogram | Hypokinetic apex with
low-normal left ventricular systolic function (53.6%) |
| Respiratory | Venous blood gas | pH 7.33 |
| | | PaCO2 47
mmHg |
| Hepatic | Alanine
transaminase | 1562 U/l |
| | Aspartate
aminotransferase | 1037 U/l |
| |
Gamma-glutamyltransferase | 105 U/l |
| | Alkaline
phosphatase | Normal |
| | Bilirubin-total and
direct | Normal |
| | Abdominal
Ultrasound | Mild hepatomegaly and
splenomegaly, mild increase in liver echogenicity, edematous
gallbladder. Normal hepatic and portal vein blood flow via
doppler |
| | Ammonia | Normal |
| Pancreatic | Lipase | 173 U/l |
| Renal | Creatinine | Normal |
| | Blood urea
nitrogen | Normal |
| | Urinalysis | Positive for
myoglobinuria and granular casts |
| Metabolic | Sodium | Normal |
| | Potassium | |
| | Phosphorus | |
| | Magnesium | |
| | Calcium | |
| | Lactate
dehydrogenase | 2,500 U/l |
| | Glucose | Normal |
| | Thyroid stimulating
hormone | Normal |
| | Cortisol | Normal |
| | Vitamin B12 | Normal |
| Musculoskeletal | Creatinine
kinase | 1,162 U/l |
| Hematological | Hemoglobin; white
blood cells; platelets | Normal |
| | International
normalized ratio; partial thromboplastin time | Normal |
| | Lupus anticoagulant
testing; anti-cardiolipin antibodies; anti-β-2 glycoprotein
antibodies | Negative |
| Toxicological | Acetaminophen and
salicylate levels | Negative |
| | Ethanol level | Negative |
| | Anion gap/osmolar
gap | Negative-therefore no
toxic alcohol testing sent |
| | Toxicological
panel | Positive: Cocaine,
benzoylecgonine (782 ng/l), hydromorphone (1,166 ng/l), morphine
(144 ng/l), noroxycodone (61 ng/l), dextromethorphan, zopiclone,
trazadone, lidocaine. |
| | | Negative:
Amphetamine, 3,4-methylenedioxyamphetamine, methadone,
2-ethylidine-1,5-dimethyl-3,3-diphenylpyrrolidine, fentanyl,
norfentanyl, oxycodone, codeine, norcodeine, buprenorphine,
norbuprenorphine, hydrocodone, 6-monoacetylmorphine |
| Infectious | Blood cultures | Negative |
| | Sputum culture | Negative |
| | Viral swabs | Negative for
COVID-19, influenza A/B, respiratory syncytial virus A/B,
adenovirus, metapneumovirus, parainfluenza, enterovirus/rhinovirus,
coronavirus |
| | Mycoplasma Pneumonia
PCR | Negative |
| | Urine culture | Negative |
| | Legionella
antigen | Negative |
| | Hepatitis A, B, C,
E serology | Negative |
| | Human
immunodeficiency virus | Negative |
| | Anti-Epstein-Barr
virus viral capsid antigen IgM/IgG; Anti-Epstein-Barr virus nuclear
antigen 1 IgG | Negative |
| | Cytomegalovirus
IgM | Negative |
| Inflammatory and
demyelinating | C-reactive
protein | Normal |
| | Anti-nuclear
antibodies | Negative |
| | Anti-neutrophilic
cytoplasmic anti-bodies | Negative |
| | Extractable nuclear
antigen anti-bodies | Negative |
| | Immunoglobulin
levels-M, G, A | Normal |
| | Complement
levels-C3, C4 | Normal |
| | Anti- | Negative |
| |
N-methyl-D
aspartate (NMDA) Voltage gated potassium channel (VGKC) | |
| |
Glutamic
acid decarboxylase 65 (GAD65) | |
| |
GABA-aminobutyric
acid (GABA) Myelin oligodendrocyte glyco-protein | |
| | (MOG) | |
| |
Aquaporin 4
(AQP4) | |
| |
Glomerular
basement membrane antibody (GBM) | |
| |
Myelin-associated
glycoprotein neuropathy (MAG) | |
The patient was admitted to the intensive care unit.
For her systemic issues, fluid resuscitation continued, and the use
of N-acetylcysteine was initiated with a bolus of 150 mg/kg
followed by an infusion (50 mg/kg IV over 4 h followed by 100 mg/kg
over 16 h). Ampicillin (2 g IV q4h) was added for possible
rhombencephalitis. As dexamethasone was not administered prior to
or with antibiotics, it was not initiated. High-dose thiamine (500
mg IV q8h) was initiated. Neuroprotective measures were maintained,
including fever avoidance; mean arterial pressure augmentation with
norepinephrine to 80 mmHg given the posterior fossa mass effect;
the avoidance of hyponatremia; the maintenance of euglycemia; and
the maintenance of the partial pressure of oxygen and carbon
dioxide at 80-120 mmHg and 35-40 mmHg, respectively. An
electroencephalography (EEG) revealed no seizures.
Once stabilized, efforts were directed at
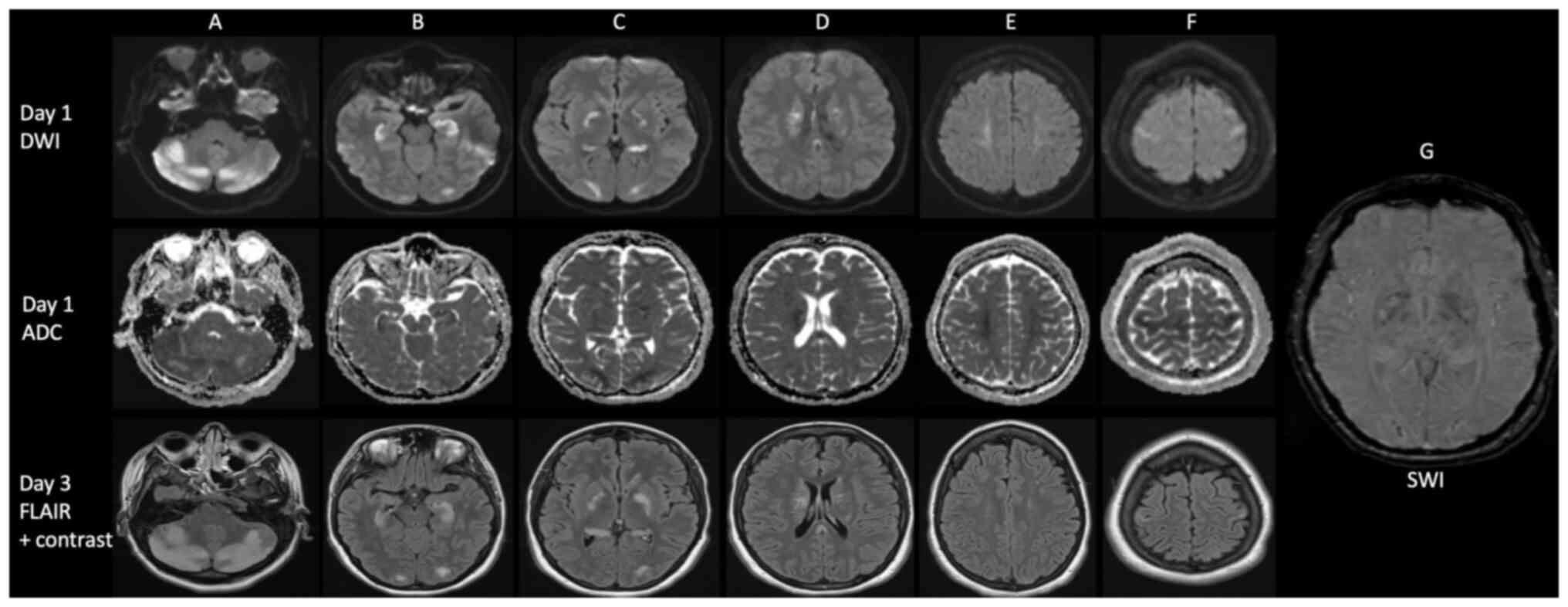
identifying an underlying etiology. An enhanced MRI of the brain at
day 1 demonstrated restricted diffusion involving bilateral
cerebellar hemispheres with posterior fossa mass effect and
distended optic nerve sheaths, as well as restricted diffusion
involving the bilateral hippocampus, globus pallidus, caudate
nuclei, occipital lobes, periventricular region, the splenium of
the corpus callosum, centrum semiovale and perirolandic area.
Petechial hemorrhage was noted in the occipital lobes and globus
pallidus (Fig. 2).
The urine toxicology was positive for cocaine,
dextromethorphan, trazadone and zopiclone. Liquid
chromatography-tandem mass spectrometry yielded positive results
for: Benzoylecgonine, noroxycodone, morphine and hydromorphone.
Tests for other substances, including levamisole, heroin,
amphetamines, barbiturates, acetaminophen and salicylate yielded
negative results. The ethanol level was negative and no osmolar or
anion gaps were observed to suggest a toxic alcohol ingestion.
While a lumbar puncture was not able to be
performed, given the high suspicion for a toxic-metabolic
encephalopathy and unremarkable systemic work-up for concerning
infectious etiologies (Table I), a
central nervous system (CNS) infection was considered unlikely, and
antivirals and antibiotics were discontinued with cautious
monitoring. This also precluded cerebrospinal fluid analysis for
possible inflammatory and autoimmune disorders, as well as systemic
tests, the results of which were reassuring (Table I).
A follow-up enhanced MRI of the brain and cervical
spine on day 3 revealed the complete regression of the restricted
diffusion, although increasing FLAIR hyperintensities and subtle
enhancement in the previously affected regions were observed with
no new lesions within the brain or cervical spine (Fig. 2).
As a result of the aforementioned clinical
assessment and investigations, cocaine toxicity was considered to
explain both her neurological presentation and associated
rhabdomyolysis, AKI, hepatitis and takotsubo cardiomyopathy.
With ongoing care, the systemic disturbances of the
patient rapidly improved. Neurologically, the condition of the
patient remained unaltered for 10 days until she began to
spontaneously open her eyes and weakly withdraw. A follow-up CT
scan of the head on day 13 revealed expected evolution (Fig. 1C and D). She began to localize with questionable
tracking of visual stimuli on day 14 and obeyed simple motor
commands shortly thereafter. Extensive discussions and shared
decision-making with family members were performed regarding her
diagnosis, uncertain prognosis and ongoing care. Due to ongoing
somnolence, weakness and a resulting ineffective cough, a
tracheostomy was required for safe liberation from mechanical
ventilation to facilitate more time for possible recovery. At the
time of transfer from the intensive care unit on day 16, she could
obey simple commands, but had profound spasticity in all four
extremities and left hemiparesis. More detailed assessments while
recovering in the ward revealed severe cognitive impairment and
aphasia.
She underwent intensive rehabilitation with
occupational therapy and speech language pathology focused. Her
spasticity was managed with baclofen and botox, and she underwent
physical therapy to regain her strength. At 3 months following her
presentation, a weakness of the left iliopsoas, quadricep and
tibialis anterior muscles was observed, in addition to sensory
disturbances involving the anteromedial thigh. Nerve conduction
analyses and electromyography testing indicated a left lumbar
plexus injury with extensive denervation. After a period of 3
months, she underwent surgery to reinnervate the left femoral neve
using the obturator nerve, followed by progressive improvement in
strength and mobility.
The patient returned home with support at 38 weeks
following her presentation and continued with outpatient
rehabilitation. At 1 year from her presentation, she was able to
return to university part-time to work toward an engineering
degree.
Discussion
As regards the presentation of patients with coma
(absent arousal and awareness), coma is the most severe syndrome on
the disorders of consciousness spectrum and when presenting acutely
must be regarded as a neurological emergency. Patients often lack
overt signs of ongoing secondary injury and neurological decline
that may only be detected with nuanced neurological exams or
specialized non-invasive and invasive monitoring. This is in
contrast to other medical emergencies and may give care providers a
false sense of security. The approach to coma should be efficient,
yet systematic and comprehensive. There are three parallel streams
of thought and corresponding actions are required, including: i)
Stabilization and supportive care; ii) diagnosis; and iii)
management (Fig. 3).
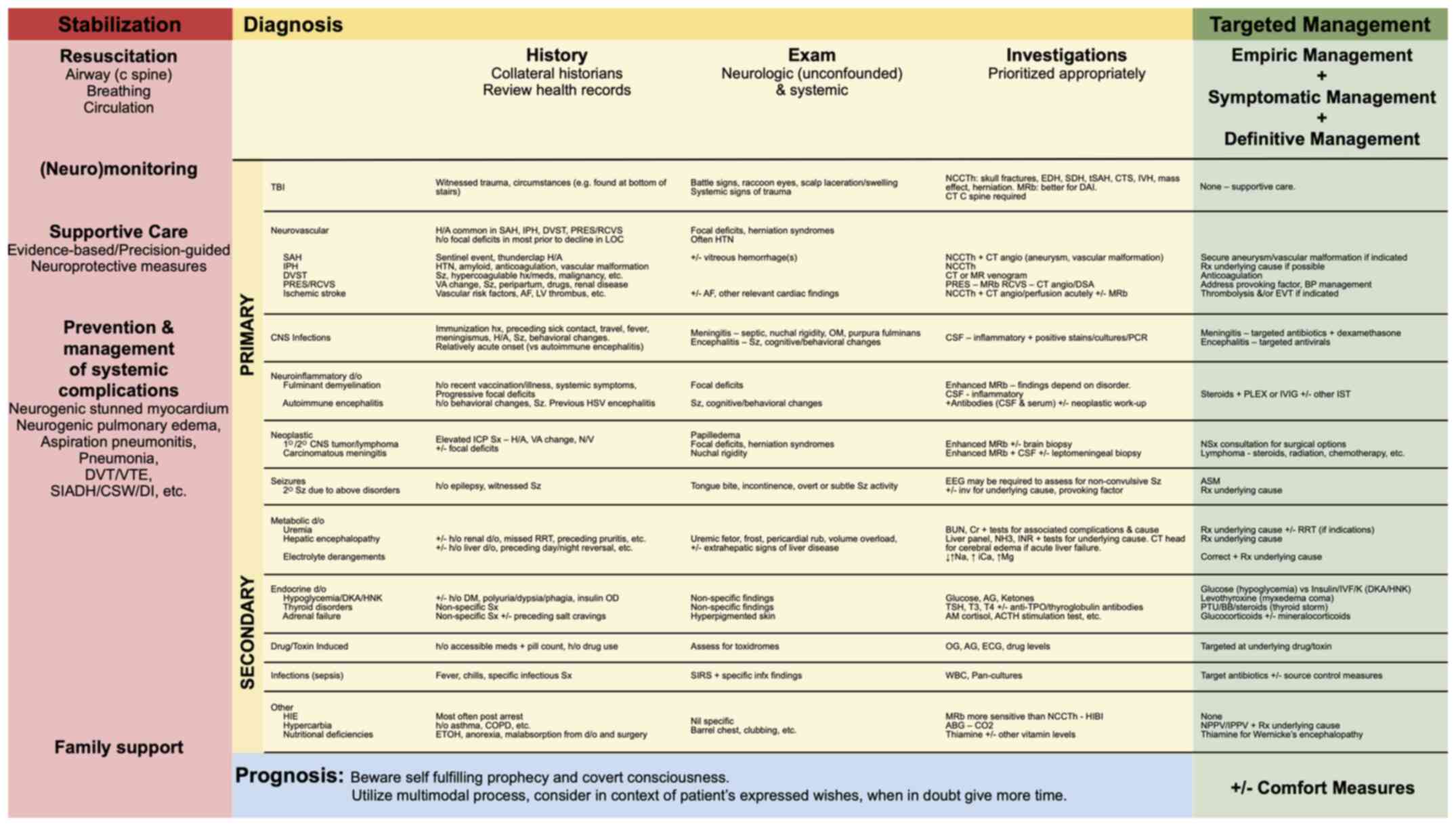 | Figure 3Clinical approach to the comatose
patient: Stabilization, diagnosis (history, examination,
investigations) and targeted management. Other considerations
pertaining to the approach to an altered level of consciousness
have been previously reported (3,4). ACTH,
adrenocorticotropic hormone; AF, atrial fibrillation; AG, anion
gap; ASM, anti-seizure medication; BB, beta blocker; BP, blood
pressure; BUN, blood urea nitrogen; CNS, central nervous system;
COPD, chronic obstructive pulmonary disease; CSF, cerebrospinal
fluid; CT, computed tomography; CTS, contusion; DAI, diffuse axonal
injury; DKA, diabetic ketoacidosis; d/o, diagnosis; DVST, dural
venous sinus thrombosis; DVT, deep vein thrombosis; ECG,
electrocardiography; EEG, electroencephalography; EDH, epidural
hematoma; EtOH, ethanol; EV, endovascular thrombectomy; H/A,
headache; HIBI, hypoxic ischemic brain injury; HIE, hepatic
ischemic encephalopathy; HNK, hyperosmolar hyperglycemic
non-ketotic coma; h/o, history of; HTN, hypertension; iCa, ionized
calcium; infx, infectious; IPH, intraparenchymal hemorrhage; IPPV,
intermittent positive-pressure ventilation; IST, immunosuppressive
therapy; IVF, intravenous fluids; IVH, intraventricular hemorrhage;
IVIG, intravenous immunoglobulin; LOC, level of consciousness; LV,
left ventricle; MRb, MR brain; NCCTh, non-enhanced CT head; NPPV,
non-invasive positive pressure ventilation; NSx, neurosurgical;
N/V, nausea/vomiting; OG, osmolar gap; OM, otitis media; PLEX,
plasma exchange; PNA, pneumonia; PRES, posterior reversible
encephalopathic syndrome; PTU, propylthiouracil; RCVS, reversible
cerebral vasoconstriction syndrome; RRT, renal placement therapy;
Rx, treatment; SAH, subarachnoid hemorrhage; SDH, subdural
hematoma; SIADH, syndrome of inappropriate antiduretic hormone
secretion; SIRS, systemic inflammatory response syndrome; Sx,
symptoms; Sz, seizure; tSAH, traumatic subarachnoid hemorrhage; T3,
triiodothyronine; T4, thyroxine; TBI, traumatic brain injury; TPO,
thyroid peroxidase; TSH, thyroid stimulating hormone; v/a, visual
acuity; VTE, venous thromboembolism; WBC, white blood cells. |
Initial resuscitation should be focused on securing
the airway (3). Cervical spine
precautions should be maintained when trauma is a concern. When no
anatomic airway difficulties are predicted, rapid sequence
induction is preferred to avoid negative effects of the procedure
on intracranial pressure. When time permits, a focused neurological
examination should be performed prior, as it will be temporarily
confounded by sedatives and paralytics. Hemodynamic stability must
be ensured throughout intubation and thereafter to ensure adequate
cerebral perfusion, while avoiding excessive hypertension. As
demonstrated by the case described herein, patients may have
concomitant cardiovascular issues potentially due to neurogenic
stunned myocardium, a direct result of the causative disorder, or
an unrelated comorbidity. Specific blood pressure parameters will
depend on the clinical circumstances. Neuroprotective measures are
a crucial and sometimes overlooked element of supportive care that
are necessary to minimize secondary injury (4).
The differential diagnosis for causes of coma is
broad (Fig. 3). These can be
categorized into structural/primary neurological disorders and
systemic/secondary neurological disorders and suggest that the
former will often have focal signs, while the latter will not.
There are exceptions to this rule, however. Patients with
systemic/secondary neurological disorders may have focal deficits
from prior diagnoses. Other systemic disorders have been reported
to cause focal deficits (e.g., hypoglycemia). Given the relative
limitations of the neurologic examination in coma, one may not
detect subtle findings suggestive of structural disorders. This may
also be confounded by medications utilized during resuscitation. As
such, urgent neuroimaging should be obtained, unless there is an
overt systemic cause.
Once neuroimaging excludes an acute vascular cause,
empiric management for meningoencephalitis with acyclovir,
antibiotics and dexamethasone may be prudent. Thiamine
administration is also reasonable with a history of polysubstance
abuse when little is known regarding the alcohol consumption of the
patient. Antiseizure medications should only be administered in the
case that seizures are confirmed either clinically or via EEG.
Following stabilization, extensive investigations
and ongoing empiric and supportive care, the presumed cause for the
coma in the patient in the present study was cocaine-induced CNS
injury. Cocaine induced CNS injury occurs via three categories of
pathophysiologic mechanisms. Vascular-mediated CNS damage occurs
due to vasospasms, vasculitis, platelet aggregation and thrombus
formation, cardioembolism and/or hypertension (5,6).
Metabolic damage due to mitochondrial dysfunction causes
demyelination, vacuolar degeneration, and axonal injury (5). Immune-mediated responses occur with or
without levamisole. While levamisole can cause inflammation via the
activation of dendritic cells, cocaine disrupts the endothelium,
permitting migration of immune cells into the CNS and promotes
increased levels of inflammatory cytokines (7).
Cocaine-induced CNS injury presents via a variety of
mechanisms. With intoxication, patients can present with mydriasis,
tics, fasciculations, vertigo, nausea and vomiting (8). Furthermore, the excessive stimulation
of adrenergic and dopaminergic pathways can contribute to other
neuropsychiatric sequelae, such as agitation, akathisia,
formication and other hallucinations (8). Depending on the severity of symptoms,
antipsychotic treatment may be indicated. Via vascular mechanisms,
patients can experience isolated thunderclap headaches, reversible
cerebral vasoconstriction syndrome, posterior reversible
encephalopathy syndrome (PRES), and seizures and/or focal deficits
in isolation (8,9). Cocaine-induced leukoencephalopathy
resulting from metabolic and immune-mediated mechanisms often
presents with cognitive impairments, and an altered level of
consciousness including coma, impaired vision, and spasticity
(5,10,11). The
patient described herein presented predominantly with a
leukoencephalopathy phenotype comprised of an altered level of
consciousness, subsequent cognitive deficits and spasticity. While
the initial coma could have been confounded by other medications
(e.g., zopiclone, trazadone, narcotics) the lack of response to
naloxone, neuroimaging, and delayed awakening suggests
otherwise.
Imaging modalities are helpful in excluding other
potential causes, assessing the degree of involvement, and for
provide insight into the underlying pathophysiological mechanisms.
Vascular mediated damage will result in infarctions, hemorrhage,
arterial inflammation, occlusions and/or segmental
vasoconstriction, in addition to patterns associated with PRES.
Metabolic injury causes bihemispheric white matter (FLAIR and T2)
hyperintensities, often with absent restricted diffusion, and
absent gadolinium enhancement on MR brain (5). Subcortical U-fibers, the brainstem and
cerebellum are often spared (5). MR
spectroscopy demonstrates increased lactate and/or decreased
N-acetylaspartate peaks (5).
Immune-mediated pathology presents with subcortical and
periventricular white matter (T2/FLAIR) hyperintensities, although
with variable, diffusion-weighted signal abnormality, gadolinium
enhancement and surrounding edema. However, case reports of
brainstem, cerebellar and globus pallidus involvement have been
noted (12,13). In the present study, the CNS injury
of the patient appeared to be driven by all three pathophysiologic
mechanisms, including immune mediated/inflammatory despite the
absence of levamisole. This was evidenced by the following: The
petechial hemorrhages in the occipital lobe; both vasogenic and
cytotoxic edema; the involvement of regions highly susceptible to
metabolic injury including the corpus callosum and globus pallidus;
in addition to the subtle gadolinium enhancement.
In addition to the CNS effects, cocaine has been
shown to be associated with a multitude of systemic complications
(Table II) (4,14-17).
The treating physician should consider the importance of managing
the systemic issues of a patient, as further systemic decline will
only predispose to worsening secondary neurological injury and
outcomes.
| Table IIComplications of cocaine
toxicity. |
Table II
Complications of cocaine
toxicity.
| System | Mechanism | Clinical
manifestations | (Refs.) |
|---|
| CNS | Vascular Mediated:
Vasospasm, vasculitis, platelet aggregation, thrombus formation,
cardio-embolism, hypertension Metabolic Damage: Mitochondrial
dysfunction, vacuolar degeneration, demyelination, axonal injury
Immune-Mediated: Activation of dendritic cells, disruption of
endothelium permitting immune cell migration into CNS, increased
levels of inflammatory cytokines | Acute | (5,8-11) |
| | |
Reversible
cerebral vasoconstrictive syndrome (RCVS) | |
| | |
Posterior
reversible encephalopathy syndrome (PRES) | |
| | |
Hemorrhagic
and ischemic strokes | |
| | |
Leukoencephalopathy | |
| | |
Resulting
in: | |
| | |
Altered
level of consciousness | |
| | |
Seizures | |
| | |
Focal
neurological deficits | |
| | |
Spasticity | |
| | |
Headache | |
| | | Chronic | |
| | |
Movement
Disorders-Tourette's syndrome, dystonia, tardive dyskinesia,
chorea, akathisia | |
| | |
Cognitive
deficits | |
| | |
Spasticity,
focal neurological deficits | |
| Cardiovascular | ↑ Sympathetic
tone/circulating catecholamines | Myocardial
infarction | (14) |
| | ↑ Oxygen demand via
↑ inotropy, chronotropy and afterload | Aortic
dissection | |
| | Coronary
vasoconstriction, platelet adherence and thrombus formation | Infective
endocarditis | |
| | Conduction
abnormalities (↑PR, QRS, QTc intervals) | Reduced systolic
and diastolic dysfunction | |
| | | Arrhythmias | |
| Respiratory | Pulmonary
vasoconstriction | Pulmonary
hypertension and right heart failure | (15) |
| | Other vascular
mediated effects including vasculitis, platelet aggregation,
thrombus formation, etc. | Acute respiratory
distress syndrome (ARDS) | |
| | | Diffuse alveolar
hemorrhage | |
| | | Pneumothorax,
pneumomediastinum | |
| | | Organizing
pneumonias | |
| |
Bronchoconstriction | Pulmonary
edema | |
| | Immune-mediated
effects | Pneumonia, lung
abscess, empyema | |
| | Introduction of
infections | | |
|
Gastrointestinal | Mesenteric ischemia
due to vasospasm, vasculitis, platelet aggregation, thrombus
formation, and/or cardio-embolism | Ischemic bowel | (15) |
| | | Intestinal
perforations | |
| | | Gastric
ulcerations | |
| | | Retroperitoneal
fibrosis | |
| Hepatic | Hepatic ischemia
and/or necrosis from vasospasm, vasculitis, platelet aggregation,
and thrombus formation | Transaminitis,
varying degrees of liver failure | (15) |
| Renal | Rhabdomyolysis,
hypertension, vasoconstriction, thrombosis, infarctions, and
vasculitis | Acute kidney
injury, renal failure | (15) |
| PNS | Direct muscle
toxicity, seizures and muscle ischemia from arterial
vasoconstriction and compression from prolonged downtime Arterial
vasoconstriction, direct toxicity and/or compression | Rhabdomyolysis | (9,16,17) |
| | | Peripheral
neuropathies | |
| Dermatological | Vasospasm,
vasculitis, platelet aggregation, thrombus formation and immune
activation | Blackened
hyperkeratotic palms (‘crack hands’) | (6) |
| | | Acute multifocal
skin necrosis | |
| | | Acute generalized
exanthematous pustulosis | |
| | | Cutaneous
fibrosis | |
| | | Chronic skin
ulcers | |
| | | Scleroderma | |
| | | Cocaine-related
bullous disease | |
| | | Buerger
disease | |
| | |
Pseudovasculitis | |
| | | Urticarial
vasculitis | |
| | | Eosinophilic
granulomatosis polyangitis | |
| | | IgA vasculitis | |
| | | Necrotizing
granulomatous vasculitis and necrotizing vasculitis | |
| | | Steven-Johnson
syndrome | |
| Head and neck | Nasal and palatal
ischemia and necrosis | Nasal septum
perforation | (15) |
| | | Nasal bone
osteomyelitis | |
| | | Nasal solid tumors
or lymphoma | |
| | | Dental carries | |
| | | Palatal necrosis
and/or perforation | |
| Psychiatric | Facilitation of
dopamine neurotra-nsmission involving D-1, D-2 and D-3
receptors | Euphoria,
psychosis, agitation, panic, paranoia | (15) |
| | | Crash phase lasting
several hours-anxiety, depression, drug craving, exhaustion,
hypersomnolence | |
| Other | Increased motor
activity, increased heat production, reduced heat dissipation (due
to vasoconstriction) | Hyperthermia | (15) |
Cocaine causes increased sympathetic tone and
circulating catecholamines. This simultaneously increases oxygen
demand via increased chronotropy, inotropy and afterload, while
decreasing supply via coronary vasoconstriction, platelet adherence
and thrombus formation. Local anesthetic effects and reduced sodium
transport causes conduction abnormalities and reduced ventricular
function (14). A number of these
mechanisms may have been in effect in the patient described herein
and/or neurogenic stunned myocardium. While the patient did
demonstrate evidence of a type II myocardial infarction, therapies
incorporated into proposed treatment algorithms (14), such as antiplatelets/anticoagulation
and nitrates were not employed due to concerns regarding
intracranial hemorrhage and posterior fossa mass effect and
potential intracranial hypertension respectively.
The most common hepatic presentation is hepatic
necrosis, accompanied by elevated serum aminotransferase and
lactate dehydrogenase levels, as was managed in this patient. AKI
most often results from rhabdomyolysis-induced acute tubular
necrosis, but can also occur due to hypertension, vasoconstriction,
thrombosis, infarctions and vasculitis (15). The AKI of the patient in the present
study likely resulted from several of these mechanisms as her
rhabdomyolysis was mild with no myoglobinuria and a downtime of
12-h without hyperthermia would not cause significant
hypovolemia.
Rhabdomyolysis results from direct muscle toxicity,
seizures and muscle ischemia due to arterial vasoconstriction, and
compression in situations involving prolonged downtime. When
severe, one must be vigilant to monitor for resulting
complications, including compartment syndrome, hyperkalemia,
hyperphosphatemia, hypocalcaemia in addition to AKI. Cocaine can
also cause peripheral mononeuropathies, as became apparent in the
course of the patient described herein. Mechanisms for this include
arterial vasoconstriction, direct toxicity and/or compression
(16,17).
There are also several direct and indirect drug-drug
interactions with possible clinical implications in cocaine-users
that physicians need to be aware of. These can occur due to changes
in pharmacokinetics or pharmacodynamics, or due to genetic or
epigenetic factors (18). The most
important of which is the potential interaction with β-blockers.
Cocaine promotes the release of norepinephrine and epinephrine. Τhe
subsequent stimulation of β-1 receptors increases the heart rate
and cardiac contractility, and β-2 receptors promote smooth muscle
relaxation. α1 receptors, on the other hand, induce
vasoconstriction. It has been suggested that in the context of
stimulant use, β-blockers may lead to unopposed α-receptor
stimulation, which can result in unopposed vasoconstriction, which
in turn could cause coronary ischemia, hypertension and subsequent
cardiovascular complications (18).
However, this association has been recently called into question
(19). Cocaine also induces an
increase in serotonin synaptic activity, which may lead to the
development of serotonin syndrome in the event that other
serotonergic drugs are administered concurrently (e.g., fentanyl,
linezolid, serotonin reuptake inhibitors). While cocaine is largely
metabolized by serum esterases to inactive metabolites
benzoylecgonine and ecgonine, a small portion undergoes hepatic
N-demethylation by CYP3A4 to the active metabolite norcocaine,
which is responsible for some of the toxic effects of cocaine.
Several commonly prescribed medications are known inducers of
CYP3A4 (e.g., phenytoin, carbamazepine) and may lead to increased
levels of the toxic metabolite when used concurrently with cocaine
(18). Additionally, the use of
cocaine with acetylcholinesterase inhibitors, may lead to reduction
of serum esterases and shunt cocaine metabolism toward the hepatic
pathway, thus increasing norcocaine formation (18).
There is no consensus available to date on the
treatment for cocaine-induced CNS injury. Treatment with steroids,
intravenous immunoglobulin and plasmapheresis has been reported
(5,11). The response to these therapies has
been inconsistent. The patient in the present study received
supportive care with early attention focused on preventing
secondary brain injury and the management of complicating systemic
factors. Later in the course, the focus shifted toward symptomatic
management and rehabilitation.
Disorders of consciousness comprise a spectrum
including delirium, minimally conscious state, unresponsive
wakefulness syndrome and coma. While bedside clinical examinations
are crucial, in patients with impaired consciousness, these are
relatively basic, lack sensitivity and may have inter-examiner
differences. Even careful standardized neurological assessments may
misclassify conscious patients as unresponsive. Research regarding
the detection of covert consciousness is emerging (20).
Impaired levels of consciousness may influence the
decision to withdraw life-sustaining therapies in patients with
acute brain injury, such as this when prognosis is uncertain. The
potential to augment the accuracy of prognostication by various
tests has been researched over the years and is perhaps best
established within patients post-arrest. Often, however, evidence
is limited in less common disorders (21) and research regarding covert
consciousness that identify patients with better prognoses has yet
to be translated into clinical practice (20). It is imperative that healthcare teams
remain humble regarding their own knowledge; open regarding
limitations in evidence; avoid personal biases that may cause
inappropriate nihilism or optimism; and be vigilant of the
self-fulfilling prophecy for the neuroprognostication of patients
(22). Quality of life is subjective
and multifactorial and shared decision-making with families is
imperative. The patient in the present study subsequently obtained
a good functional neurological outcome despite the profound acute
presentation.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZMH and JAK contributed to the conception and design
of the present case report, as well as to the acquisition and
interpretation of the patient's data, and in the drafting and
critical revision of the manuscript for intellectual content. ZMH
and JAK confirm the authenticity of all the raw data. Both authors
have read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The patient provided informed consent to participate
in the present study.
Patient consent for publication
The patient provided informed consent for the
publication of the present case report and any related images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Middleton RM and Kirkpatrick MB: Clinical
use of cocaine. A review of the risks and benefits. Drug Saf.
9:212–217. 1993.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Annual Reports Questionnaire: Annual
prevalence of drug use: Amphetamines (Internet). https://public.tableau.com/views/Prevalence-general/Prevalence-general-heatmap?:showVizHome=no.
Accessed August 23, 2024.
|
|
3
|
Rajajee V, Riggs B and Seder DB: Emergency
neurological life support: Airway, ventilation, and sedation.
Neurocrit Care. 27 (Suppl 1):S4–S28. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Stocchetti N, Taccone FS, Citerio G, Pepe
PE, Le Roux PD, Oddo M, Polderman KH, Stevens RD, Barsan W, Maas
AI, et al: Neuroprotection in acute brain injury: An up-to-date
review. Crit Care. 19(186)2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Vosoughi R and Schmidt BJ: Multifocal
leukoencephalopathy in cocaine users: A report of two cases and
review of the literature. BMC Neurol. 15(208)2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Brewer JD, Meves A, Bostwick JM, Hamacher
KL and Pittelkow MR: Cocaine abuse: Dermatologic manifestations and
therapeutic approaches. J Am Acad Dermatol. 59:483–487.
2008.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Núñez MJ, Balboa J, Rey-Méndez M, Brenlla
J, González-Peteiro M, Rodrigo E and Freire-Garabal M: Effects of
amphetamine and cocaine on the development of acute experimental
allergic encephalomyelitis in Lewis rats. Hum Exp Toxicol.
26:637–643. 2007.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Richards JR and Le JK: Cocaine toxicity.
In: StatPearls. StatPearls Publishing, Treasure Island, FL, 2024.
https://www.ncbi.nlm.nih.gov/books/NBK430976/. Updated
June 8, 2023.
|
|
9
|
Farooque U, Okorie N, Kataria S, Shah SF
and Bollampally VC: Cocaine-induced headache: A review of
pathogenesis, presentation, diagnosis, and management. Cureus.
12(e10128)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kondziella D, Danielsen ER and
Arlien-Soeborg P: Fatal encephalopathy after an isolated overdose
of cocaine. J Neurol Neurosurg Psychiatry. 78:437–438.
2007.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Vitt JR, Brown EG, Chow DS and Josephson
SA: Confirmed case of levamisole-associated multifocal inflammatory
leukoencephalopathy in a cocaine user. J Neuroimmunol. 305:128–130.
2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Cisneros O, Garcia de de Jesus K, Then EO
and Rehmani R: Bilateral basal ganglia infarction after intranasal
use of cocaine: A case report. Cureus. 11(e4405)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Renard D, Brunel H and Gaillard N:
Bilateral haemorrhagic infarction of the globus pallidus after
cocaine and alcohol intoxication. Acta Neurol Belg. 109:159–161.
2009.PubMed/NCBI
|
|
14
|
Schwartz BG, Rezkalla S and Kloner RA:
Cardiovascular effects of cocaine. Circulation. 122:2558–2569.
2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Glauser J and Queen JR: An overview of
non-cardiac cocaine toxicity. J Emerg Med. 32:181–186.
2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
de Souza A, Desai PK and de Souza RJ:
Acute multifocal neuropathy following cocaine inhalation. J Clin
Neurosci. 36:134–136. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Beniczky S, Tfelt-Hansen P, Fabricius M
and Andersen KV: Multiple mononeuropathy following cocaine abuse.
BMJ Case Rep. 2009(bcr07.2008.0446)2009.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Gallelli L, Gratteri S, Siniscalchi A,
Cione E, Sirico S, Seminara P, Caroleo MC and De Sarro G: Drug-drug
interactions in cocaine-users and their clinical implications. Curr
Drug Abuse Rev. 10:25–30. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wilson T, Pitcher I and Bach P: Avoidance
of β-blockers in patients who use stimulants is not supported by
good evidence. CMAJ. 194:E127–E128. 2022.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Rohaut B, Eliseyev A and Claassen J:
Uncovering consciousness in unresponsive ICU patients: Technical,
medical and ethical considerations. Crit Care.
23(78)2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wijdicks EFM: Predicting the outcome of a
comatose patient at the bedside. Pract Neurol. 20:26–33.
2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Wijdicks EFM and Hwang DY: Predicting coma
trajectories: The impact of bias and noise on shared decisions.
Neurocritical Care. 35:291–296. 2021.PubMed/NCBI View Article : Google Scholar
|