Introduction
Pancreatic neuroendocrine tumors (PanNETs) are a
group of malignant neoplasms that arise exclusively from the
neuroendocrine tissue of the pancreas. They are typically
slow-growing and exhibit the strong expression of
immunohistochemical markers, such as synaptophysin and chromogranin
A (1,2). PanNETs account for ~2 to 7% of all
pancreatic tumors (3) and include
entities, such as insulinoma, gastrinoma, VIPoma, ACTHoma, PPoma
and glucagonoma, among others. These neoplasms may originate from
either differentiated pancreatic cells or stem cells located within
the islets of Langerhans (4).
The incidence of PanNETs has exhibited a steady
increase worldwide, with notable regional and national variations.
The average reported incidence is ~1 case per 100,000 inhabitants
(5,6). According to the World Health
Organization (WHO), PanNETs are classified based on histological
differentiation and molecular expression profiles (7). Well-differentiated PanNETs exhibit a
low proliferative activity, whereas poorly differentiated tumors
are associated with high Ki-67 indices and demonstrate a more
aggressive clinical behavior (8).
Regardless of grade, these neoplasms often present with
eosinophilic cytology, hyperchromatic nuclei, and a fibrotic stroma
typically devoid of necrosis (9).
Approximately 30% of PanNETs secrete functional
hormones, which may facilitate clinical recognition through
symptoms related to ectopic hormone production (10,11).
However, the majority of PanNETs are clinically silent or
non-specific, necessitating a comprehensive diagnostic approach
involving biochemical markers, molecular analysis and advanced
imaging modalities (12,13). In this context, genetic screening
aimed at detecting specific pathogenic mutations has also been
proposed (14). The treatment of
PanNETs is guided by their malignant potential and functional
status. Poorly differentiated tumors usually require aggressive
treatment strategies due to their unfavorable prognosis (15). By contrast, well-differentiated
functional tumors may be initially managed with active
surveillance, with surgical resection and lymphadenectomy indicated
upon evidence of disease progression (16,17).
Early detection, close clinical monitoring and
timely surgical intervention are associated with improved survival
outcomes of patients with PanNETs (18). Nevertheless, despite the increasing
global incidence, the majority of cases continue to be diagnosed at
advanced stages, thus often being associated with a poor prognosis
(19).
Considering the above, the present study describes
the clinical case of a patient with a confirmed diagnosis of a
functional PanNET whose initial presentation was atypical. Given
the rarity of these tumors and their often non-specific
manifestations, the present case report aimed to highlight key
clinical features that may support early diagnostic suspicion in
patients presenting with compatible symptoms, thereby contributing
to the prompt recognition of these uncommon neoplasms.
Case report
A 52-year-old female patient presented at Hospital
General Regional No. 1, Unidad Morelos of the Instituto Mexicano
del Seguro Social (IMSS), Chihuahua, Mexico, with a family history
of malignancies, including colorectal, breast, pancreatic cancer,
and an unspecified lymphoma. She had no history of chronic diseases
or substance abuse. Her surgical history included an appendectomy
at age 21, a myomectomy at age 42 and a hysterectomy at age 45.
She was admitted to the emergency department at
Hospital General Regional No. 1, Unidad Morelos of the Instituto
Mexicano del Seguro Social (IMSS) following an episode of syncope,
with no other associated symptoms. During a clinical evaluation,
she reported the onset of symptoms with unintentional weight loss
of ~25% of her usual body weight over a 6-month period (from 78 to
58 kg), accompanied by fatigue, generalized weakness, and hyporexia
secondary to cheilitis and glossitis. This was followed by
intermittent colicky abdominal pain localized to the right upper
quadrant and mesogastric region, with no radiation or identifiable
triggering or relieving factors. More recently, she reported the
worsening of her clinical condition with progressive dyspnea
initially upon moderate exertion and later minimal exertion, as
well as orthopnea and predominantly daytime lower extremity
edema.
Upon a physical examination, altered vital signs
were recorded: A blood pressure of 96/58 mmHg, a heart rate of 115
beats per minute and a respiratory rate of 25 breaths per minute.
Laboratory analyses revealed normocytic normochromic anemia with a
hemoglobin level of 6.8 g/dl (reference range, 12-15.5 g/dl), a
mean corpuscular volume of 80.4 fl (reference range, 80-96 fl),
mean corpuscular hemoglobin level of 25.2 pg (reference range,
27-33 pg) and a mean corpuscular hemoglobin concentration of 31.3
g/dl (reference range, 32-36 g/dl). The patient was subsequently
admitted to the internal medicine ward for further diagnostic
workup.
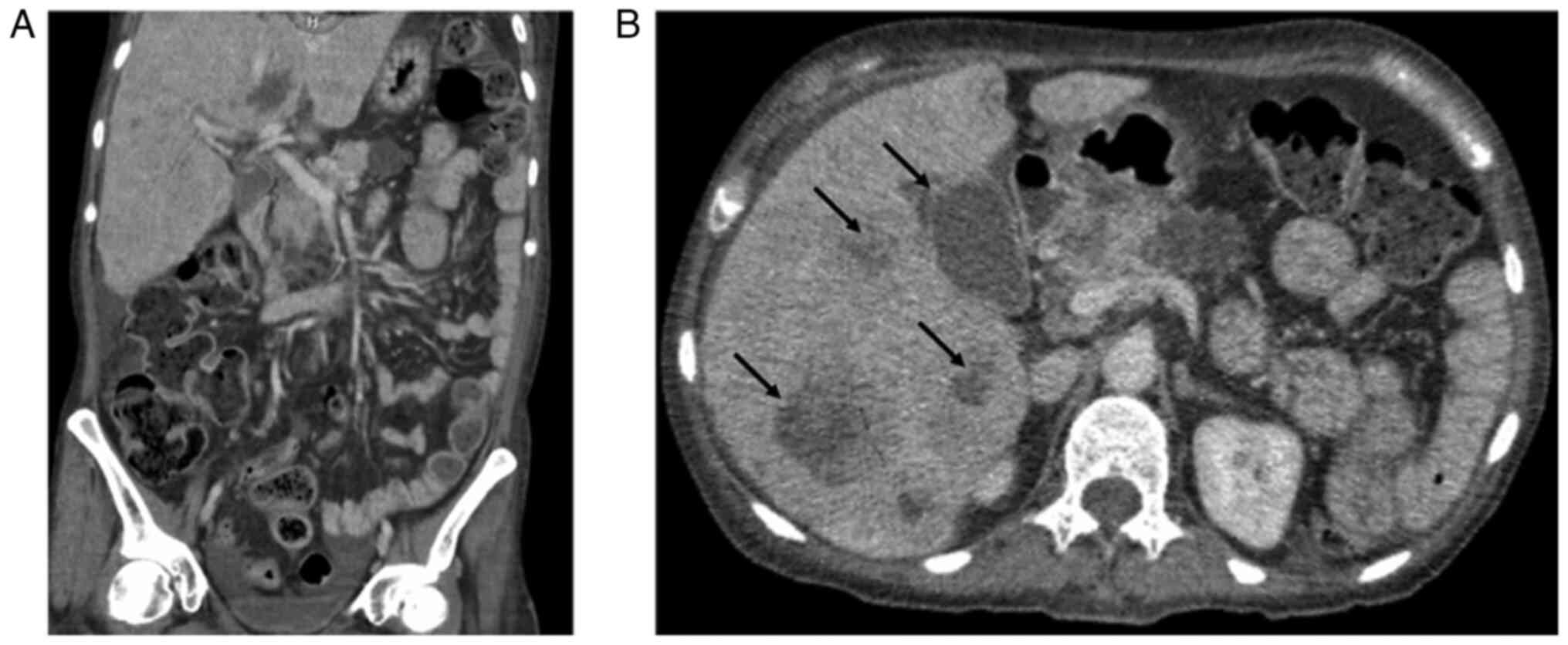
A contrast-enhanced computed tomography (CT) scan of
the abdomen and pelvis revealed multiple hepatic and pancreatic
lesions of varying lengths and isodense characteristics, suggestive
of metastatic disease, as well as hydrocholecystis (Fig. 1). Given these findings, a CT-guided
liver biopsy was performed. A histopathological examination was
performed using hematoxylin and eosin (H&E) staining (Merck
KGaA). The surgical specimens were fixed in 10% neutral-buffered
formalin at room temperature for 24 h, embedded in paraffin, and
sectioned at a thickness of 4 µm. The sections were subsequently
deparaffinized, rehydrated, and stained with hematoxylin for 5 min,
followed by eosin counterstaining for 2 min. The slides were
mounted with a synthetic resin and examined under a light
microscope (Olympus CX43, Olympus Corporation) at x40
magnification.
The histopathological examination revealed a
malignant epithelial neoplasm with focal areas of both recent and
chronic coagulative necrosis. The lesion exhibited fibrotic areas
and was composed of cellular clusters with cylindrical or cuboidal
morphology, nuclear enlargement, oval to irregular or elongated
nuclei, inconspicuous nucleoli and scant cytoplasm.
Immunohistochemical analysis was performed on paraffin-embedded
tissue blocks using the avidin-biotin immunoenzymatic method at the
Pathology and Immunohistochemistry Laboratory of the Hospital
General Regional No. 1, Unidad Morelos of the Instituto Mexicano
del Seguro Social (IMSS). Sections of 4 µm thickness were obtained
from formalin-fixed paraffin-embedded tissue. For intracellular
antigens, permeabilization was carried out with 0.1% Triton X-100
(Merck KGaA) for 10 min at room temperature. Endogenous peroxidase
activity was blocked with 3% hydrogen peroxide for 10 min, followed
by blocking with 5% normal goat serum (MilliporeSigma) for 30 min
at room temperature. The following primary antibodies were used:
CK7 (1:100, cat. no. M7018, Dako, Agilent Technologies, Inc.), CK20
(1:100, cat. no. M7019, Dako, Agilent Technologies, Inc.), CK19
(1:200, cat. no. ab15463, Abcam), MUC5AC (1:100, cat. no. ab3649,
Abcam), CA19-9 (1:100, cat. no. ab15146, Abcam), chromogranin A
(1:200, cat. no. M0869, Dako, Agilent Technologies, Inc.),
synaptophysin (1:200, cat. no. M0776, Dako), and Ki-67 (1:200, cat.
no. M7240, Dako, Agilent Technologies, Inc.). Incubation with
primary antibodies was performed overnight at 4˚C. Sections were
then incubated with secondary antibodies conjugated to horseradish
peroxidase (HRP) (EnVision+ System-HRP, Dako, Agilent Technologies,
Inc.) for 30 mins at room temperature. Immunoreactivity was
visualized using 3,3'-diaminobenzidine (DAB; Dako, Agilent
Technologies, Inc.) as a chromogen, followed by counterstaining
with hematoxylin for 1 min at room temperature. Slides were mounted
with resin and examined using a light microscope (Olympus CX43,
Olympus Corporation) at x40 magnification. Immunohistochemical
analyses confirmed the diagnosis of a grade II neuroendocrine tumor
(Fig. 2). The patient was evaluated
by the medical oncology team, who recommended initiating treatment
with intramuscular octreotide at a dose of 20 mg every 21 days. She
was discharged under palliative care with close outpatient
follow-up.
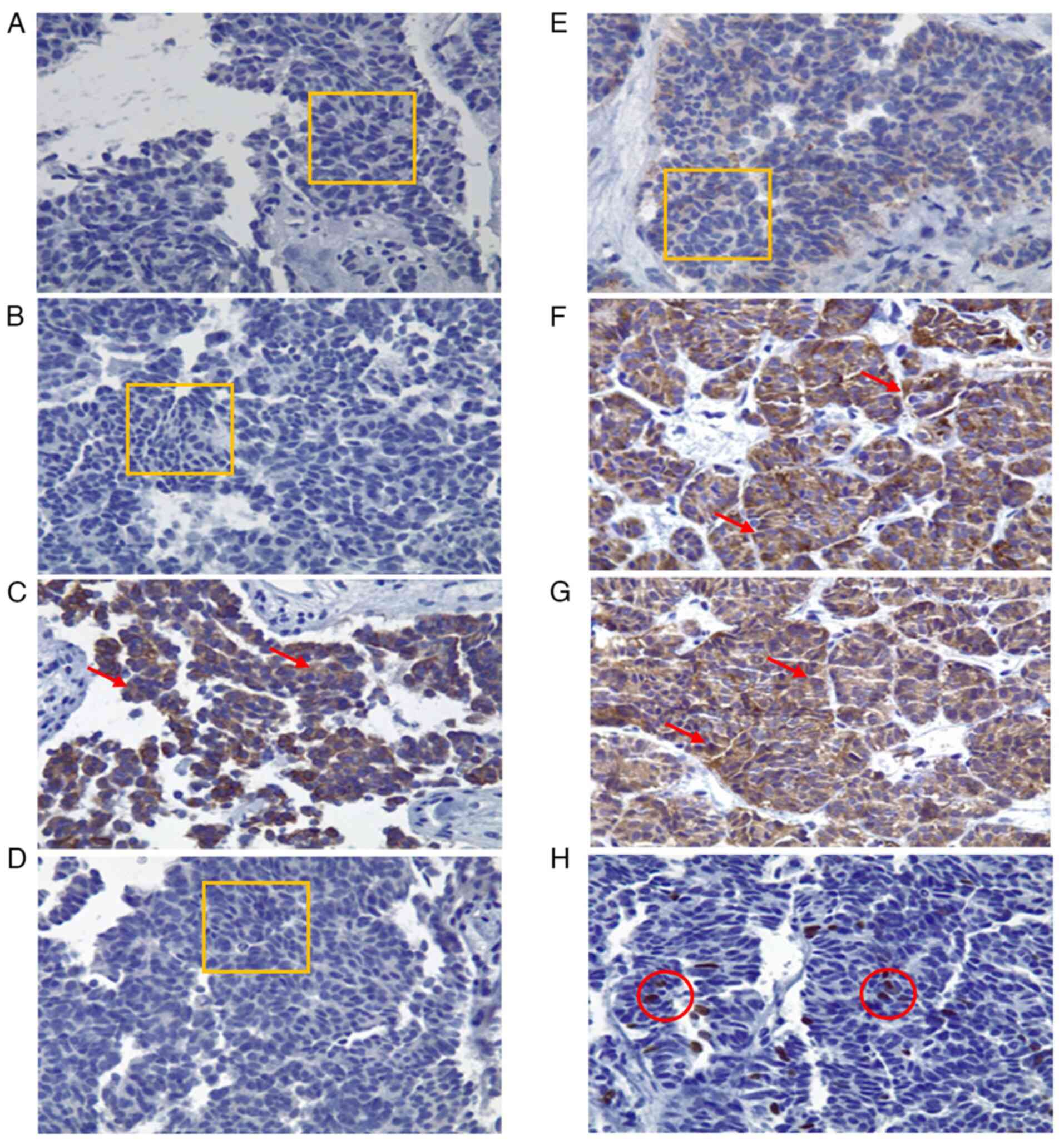 | Figure 2Immunohistochemical analysis of the
biopsied tissue (magnification, x40). Histological specimens
illustrating neoplastic cells with cylindrical and cuboidal
morphology, irregular nuclear enlargement, inconspicuous nucleoli
and scant cytoplasm. Three mitotic figures per high-power field
(40X objective) are identified. Findings are consistent with
moderately differentiated adenocarcinoma associated with extensive
desmoplasia. (A) CK7 immunohistochemistry: 0% expression, negative
intensity. (B) CK20: 0% expression, negative intensity. (C) CK19:
90% of cells positive, 80% intensity, cytoplasmic pattern. (D)
MUC5AC: 0% expression, negative intensity. (E) CA 19-9: 0%
expression, negative intensity. (F) Chromogranin A: 100%
expression, 90% intensity, cytoplasmic pattern. (G) Synaptophysin:
100% expression, 90% intensity, cytoplasmic pattern. (H) Ki-67:
proliferation index of 8%, 100% intensity, nuclear pattern. (A, B,
D and E) The thin orange rectangle outlines the evaluated area with
no staining (negative expression). C, F and G) Red arrows indicate
cells with brown cytoplasmic staining (positive cytoplasmic
pattern). (H) Thin red circles indicate nuclei with brown nuclear
staining (positive nuclear pattern, 8%). |
At her first outpatient visit on January 24, 2025,
the laboratory results revealed hemoglobin levels at 11.3 g/dl, a
platelet count of 662x109/l and a serum albumin level of
2.5 g/dl. Despite initial clinical stability and an ECOG
performance status of 1, the patient began experiencing persistent
vomiting and progressive weight loss. By March 7, 2025, laboratory
tests revealed a decline in hemoglobin levels to 9.3 g/dl and a
platelet count of 412x109/l. She was hospitalized again
on April 9, 2025, due to a worsening functional status, but was
discharged the same day under symptomatic management. At the final
follow-up, at 6 months after discharge, the patient remained alive
under palliative care, with no major complications other than
general functional decline.
Discussion
The present study describes the clinical case of a
middle-aged female patient who, following a prolonged course of
non-specific constitutional symptoms, was diagnosed with a grade
II, moderately differentiated, metastatic PanNET. The case
described herein underscores the clinical complexity and frequently
indolent nature of these neoplasms, which often leads to delayed
diagnosis despite evidence of systemic progression.
Previous case reports have described PanNETs with
atypical clinical presentations similar to the present case, in
which non-specific constitutional symptoms preceded definitive
diagnosis by several months (20).
In particular, incidental findings or vague symptoms, such as
weight loss, fatigue, or syncope have led to the eventual discovery
of PanNETs, emphasizing the need for a more thorough evaluation in
patients with persistent unexplained symptoms (21). The clinical value of this case lies
in highlighting how the absence of classic signs can delay the
diagnosis of potentially treatable neoplasms, particularly in
individuals with a significant family history of malignancy
(22). It also underscores the
importance of including PanNETs in the differential diagnosis in
high-risk individuals, even in the absence of functional signs
(23). Furthermore, the present case
points to the need for future research evaluating the role of
genetic screening in predisposed populations and the use of
molecular biomarkers as tools for early detection and non-invasive
monitoring of neuroendocrine tumors.
Globally, neuroendocrine neoplasms account for only
0.5% of all tumors, with ~70% of them located in the
gastrointestinal tract and pancreas (24). The pancreas is a dual-function organ
in which endocrine and exocrine components interact through shared
molecular mechanisms under both physiological and pathological
conditions, including tumorigenesis (25). In the case of PanNETs, their clinical
progression is often slower and less symptomatic compared to other
pancreatic neoplasms, rendering early detection more challenging
(26).
In this event, the absence of classic clinical signs
such as jaundice, severe abdominal pain, or symptoms related to
hormonal hypersecretion suggests a non-functional PanNET. These
tumors are often diagnosed incidentally or when they cause
compressive or vague systemic symptoms (27). In the case described herein, it was a
syncopal episode that prompted medical attention and ultimately led
to the diagnosis, despite the presence of prior symptoms consistent
with constitutional syndrome.
Several studies have reported an increase in
incidental diagnoses of PanNETs in individuals without an apparent
oncological history, particularly in those presenting with
long-standing digestive symptoms (28). In the case in the present study,
diagnostic confirmation was achieved through liver biopsy and
immunohistochemistry, demonstating positivity for characteristic
neuroendocrine tumor markers and a Ki-67 index consistent with a
grade II neoplasm, according to the current WHO classification
(29,30).
The prognosis of metastatic PanNETs is variable,
with 3-year survival rates ranging from 13 to 54% (31). This variability is closely related to
tumor burden, functional status and the availability of therapeutic
interventions. In the patient in the present study, treatment was
initiated with a somatostatin analog (octreotide), which is
considered first-line therapy for intermediate-grade PanNETs,
although the efficacy of this approach in advanced stages remains
limited (32).
Moreover, the significant family history of
malignancy of the patient raises the possibility of an underlying
genetic predisposition. This highlights the importance of
implementing targeted screening strategies in individuals with a
family history of PanNETs or other endocrine neoplasms. Recent
research has proposed the use of molecular biomarkers and liquid
biopsy as promising tools for early detection and non-invasive
monitoring of these tumors in high-risk populations (33).
Currently, new molecular targets are under
investigation for the treatment of PanNETs resistant to
conventional therapies. Among these, FOXM1 (implicated in multiple
oncogenic processes) has been identified (34), along with CYR61, a tumor-promoting
gene, and the proteins, PAK4 and NAMPT, whose overexpression in
patient biopsies suggests their potential as therapeutic targets in
advanced disease stages (35). These
research pathways may lead to the timely development of more
effectively targeted therapies, even in patients with tumor
recurrence (36).
The atypical presentation in the patient in the
present study suggests the need for the closer monitoring of
individuals with a strong familial history of cancer who present
mild or non-specific symptoms, including consideration of proactive
screening in selected cases. However, as the present case report
focuses on a single patient, it does not allow for the
generalization regarding the clinical presentation or treatment
response of grade II PanNETs. Furthermore, no genetic or molecular
profiling was performed in this case, which would have provided
greater insight into the etiology and potential hereditary variants
involved. Therefore, targeted familial screening is recommended in
similar cases.
In conclusion, the present case report highlights
the importance of considering PanNETs in the differential diagnosis
of persistent and non-specific constitutional symptoms,
particularly in patients with a significant family history of
malignancy. Timely diagnosis, supported by histopathological and
immunohistochemical techniques, along with access to targeted
therapies, is essential to improve the prognosis of patients with
these neoplasms, which often present with variable clinical
behavior. The future integration of molecular screening methods may
transform the management of PanNETs, enabling earlier and more
personalized interventions.
Acknowledgements
The authors would like to thank their host
institution Hospital General Regional No. 1, Unidad Morelos of the
Instituto Mexicano del Seguro Social (IMSS), Chihuahua, Mexico, for
its logistical support in providing access to the patient's
clinical information during the preparation of this manuscript.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
LFMB, HARL, AVU and EIMR participated equally in the
preparation of this manuscript, both in the medical care process
and during data collection, literature search, information
synthesis, and writing of this manuscript. LFMB and HARL confirm
that the information provided here is accurate. LFMB and HARL
confirm the authenticity of all the raw data. All authors have read
and approved the final manuscript.
Ethics approval and consent for
participation
The present study was performed in accordance with
the ethical standards of the Declaration of Helsinki, 1964.
Informed consent was obtained from the patient for inclusion in the
study. Ethics approval was waived by the local committee as no
personal data was used.
Patient consent for publication
Written informed consent was obtained from the
patient for the publication of the present case report and any
related images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lloyd RV, Osamura RY, Klöppel G and Rosai
J (eds): WHO Classification of Tumours of Endocrine Organs. Vol 10.
4th edition. IARC Press, Lyon, 2017.
|
|
2
|
Rizen EN and Phan AT: Neuroendocrine
tumors: A relevant clinical update. Curr Oncol Rep. 24:703–714.
2022.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Jensen RT, Bodei L, Capdevila J, Couvelard
A, Falconi M, Glasberg S, Kloppel G, Lamberts S, Peeters M, Rindi
G, et al: Unmet needs in functional and nonfunctional pancreatic
neuroendocrine neoplasms. Neuroendocrinology. 108:26–36.
2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Yang M, Tian B, Zhang Y, Su A, Yue P, Xu S
and Wang L: Epidemiology, diagnosis, surgical treatment and
prognosis of the pancreatic neuroendocrine tumors: Report of 125
patients from one single center. Indian J Cancer. 52:343–349.
2015.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Das S and Dasari A: Epidemiology,
incidence, and prevalence of neuroendocrine neoplasms: Are there
global differences? Curr Oncol Rep. 23(43)2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Öberg K: Management of functional
neuroendocrine tumors of the pancreas. Gland Surg. 7:20–27.
2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kasajima A and Klöppel G: Neuroendocrine
neoplasms of lung, pancreas and gut: A morphology-based comparison.
Endocr Relat Cancer. 27:R417–R432. 2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Gill A, Klimstra D and Lam A (eds): WHO
Classification of Tumours: Digestive System Tumours. 5th edition.
International Agency for Research on Cancer, Lyon, 20190.
|
|
9
|
Konukiewitz B, Jesinghaus M, Kasajima A
and Klöppel G: Neuroendocrine neoplasms of the pancreas: Diagnosis
and pitfalls. Virchows Arch. 480:247–257. 2022.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Reid MD, Bagci P, Ohike N, Saka B, Erbarut
Seven I, Dursun N, Balci S, Gucer H, Jang KT, Tajiri T, et al:
Calculation of the Ki67 index in pancreatic neuroendocrine tumors:
A comparative analysis of four counting methodologies. Mod Pathol.
28:686–694. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Crona J, Norlen O, Antonodimitrakis P,
Welin S, Stalberg P and Eriksson B: Multiple and secondary hormone
secretion in patients with metastatic pancreatic neuroendocrine
tumours. J Clin Endocri nol Metab. 101:445–452. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Hofland J, Falconi M, Christ E, Castaño
JP, Faggiano A, Lamarca A, Perren A, Petrucci S, Prasad V,
Ruszniewski P, et al: European Neuroendocrine Tumor Society 2023
guidance paper for functioning pancreatic neuroendocrine tumour
syndromes. J Neuroendocrinol. 35(e13318)2023.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Zilli A, Arcidiacono PG, Conte D and
Massironi S: Clinical impact of endoscopic ultrasonography on the
management of neuroendocrine tumors: Lights and shadows. Dig Liver
Dis. 50:6–14. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Alshareefy Y, Cummins S, Mazzoleni A,
Sharma V, Guggilapu S, Leong AWY and Wireko AA: A review of
functional pancreatic neuroendocrine tumors: Exploring the
molecular pathogenesis, diagnosis and treatment. Medicine
(Baltimore). 102(e36094)2023.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Ito T, Masui T, Komoto I, Doi R, Osamura
RY, Sakurai A, Ikeda M, Takano K, Igarashi H, Shimatsu A, et al:
JNETS clinical practice guidelines for gastroenteropancreatic
neuroendocrine neoplasms: Diagnosis, treatment, and follow-up: A
synopsis. J Gastroenterol. 56:1033–1044. 2021.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Titan AL, Norton JA, Fisher AT, Foster DS,
Harris EJ, Worhunsky DJ, Worth PJ, Dua MM, Visser BC, Poultsides
GA, et al: Evaluation of outcomes following surgery for locally
advanced pancreatic neuroendocrine tumors. JAMA Netw Open.
3(e2024318)2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Rinke A, Ambrosini V, Dromain C,
Garcia-Carbonero R, Haji A, Koumarianou A, van Dijkum EN, O'Toole
D, Rindi G, Scoazec JY and Ramage J: European Neuroendocrine Tumor
Society (ENETS) 2023 guidance paper for colorectal neuroendocrine
tumours. J Neuroendocrinol. 35(e13309)2023.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Wu Z, Wang W, Zhang K, Fan M and Lin R:
The impact of surgery and survival prediction in patients with
gastroenteropancreatic neuroendocrine tumors: A population-based
cohort study. Int J Surg. 109:1629–1638. 2023.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Loosen SH, Kostev K, Eschrich J, Krieg S,
Krieg A, Luedde T, Jann H and Roderburg C: Clinical characteristics
of 662 patients with pancreatic neuroendocrine tumors receiving
antitumoral therapy. Medicine (Baltimore).
101(e32044)2022.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Rikhraj N, Fernandez CJ, Ganakumar V and
Pappachan JM: Pancreatic neuroendocrine tumors: A case-based
evidence review. World J Gastrointest Pathophysiol.
16(107265)2025.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Popa SG, Golli AL, Matei CF, Sonei AN,
Vere C, Cimpeanu R, Munteanu M and Munteanu A: Pancreatic
neuroendocrine tumors-diagnostic pitfalls of non-diabetic severe
hypoglycemia: Literature review and case report. Diagnostics
(Basel). 15(337)2025.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Danek E, Kavnoudias H, McLean C,
Gerstenmaier JF and Di Muzio B: Radiological variability in
pancreatic neuroendocrine neoplasms: A 10-year single-center study
on atypical presentations and diagnostic challenges. Biomedicines.
13(496)2025.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Yalcin S and Öberg K (eds): Neuroendocrine
Tumours: Diagnosis and Management. 2nd edition. Springer
International Publishing, Cham, 2024.
|
|
24
|
Klöppel G: Neuroendocrine neoplasms:
Dichotomy, origin and classifications. Visc Med. 33:324–330.
2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Hu C, Chen Y, Yin X, Xu R, Yin C, Wang C
and Zhao Y: Pancreatic endocrine and exocrine signaling and
crosstalk in physiological and pathological status. Signal
Transduct Target Ther. 10(39)2025.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhu X, Xiao Z, Liu H, Zhang P, Deng S,
Ding L, Feng J, Luo J, Ni Q, Luo G and Yu X: Pancreatic Cancer: An
Exocrine Tumor With Endocrine Characteristics. Ann Surg.
280:e17–e25. 2024.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Fang JM, Li J and Shi J: An update on the
diagnosis of gastroenteropancreatic neuroendocrine neoplasms. World
J Gastroenterol. 28:1009–1023. 2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Sonbol MB and Halfdanarson TR: Management
of well-differentiated high-grade (G3) neuroendocrine tumors. Curr
Treat Options Oncol. 20(74)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ma ZY, Gong YF, Zhuang HK, Zhou ZX, Huang
SZ, Zou YP, Huang BW, Sun ZH, Zhang CZ, Tang YQ and Hou BH:
Pancreatic neuroendocrine tumors: A review of serum biomarkers,
staging, and management. World J Gastroenterol. 26:2305–2322.
2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Oronsky B, Ma PC, Morgensztern D and
Carter CA: Nothing but NET: A review of neuroendocrine tumors and
carcinomas. Neoplasia. 19:991–1002. 2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Fairweather M, Swanson R, Wang J, Brais
LK, Dutton T, Kulke MH and Clancy TE: Management of neuroendocrine
tumor liver metastases: Long-term outcomes and prognostic factors
from a large prospective database. Ann Surg Oncol. 24:2319–2325.
2017.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Falconi M, Eriksson B, Kaltsas G, Bartsch
DK, Capdevila J, Caplin M, Kos-Kudla B, Kwekkeboom D, Rindi G,
Klöppel G, et al: ENETS consensus guidelines update for the
management of patients with functional pancreatic neuroendocrine
tumors and non-functional pancreatic neuroendocrine tumors.
Neuroendocrinology. 103:153–171. 2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ahmed M: Gastrointestinal neuroendocrine
tumors in 2020. World J Gastrointest Oncol. 12:791–807.
2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Ambrosini V, Kunikowska J, Baudin E, Bodei
L, Bouvier C, Capdevila J, Cremonesi M, de Herder WW, Dromain C,
Falconi M, et al: Consensus on molecular imaging and theranostics
in neuroendocrine neoplasms. Eur J Cancer. 146:56–73.
2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Mpilla GB, Philip PA, El-Rayes B and Azmi
AS: Pancreatic neuroendocrine tumors: Therapeutic challenges and
research limitations. World J Gastroenterol. 26:4036–4054.
2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Pulvirenti A, Javed AA, Michelakos T,
Sekigami Y, Zheng J, Kalvin HL, McIntyre CA, Nebbia M, Chou JF,
Gonen M, et al: Recurring pancreatic neuroendocrine tumor: Timing
and pattern of recurrence and current treatment. Ann Surg.
278:e1063–e1067. 2023.PubMed/NCBI View Article : Google Scholar
|