Introduction
Congenital muscular dystrophies (CMD) are a group of
neuromuscular disorders characterized by severe muscle hypotonia at
birth or within the first years of life. Symptoms include delayed
motor milestones, respiratory and swallowing difficulties as well
as generalized muscle weakness (1). The major CMD subtypes include
merosin-deficient CMD, integrin-deficient CMD, fukuyama CMD, CMD
with rigid spine syndrome, muscle-eye-brain disease, Ullrich CMD
and Walker-Warburg syndrome (2).
Merosin is absent in approximately half of the patients with
classical CMD and frequently in the most severe cases. Therefore,
classical CMDs are classified into two groups: Merosin-negative and
merosin-positive CMD (3).
Merosin-deficient CMD is characterized by the absence of the
laminin α2 chain around muscle fibres as well as elevated serum
creatine kinase (CK) levels, particularly in the early months of
life. The majority of patients with merosin-deficient CMD have
normal intelligence but certain individuals may exhibit moderate
mental retardation and epilepsy (4–6).
LAMA2-associated muscular dystrophy, which is
also known as merosin-deficient CMD type 1A (MDC1A), is inherited
in an autosomal recessive manner (7). The prevalence of CMDs was estimated
to be between 0.7/100,000 (8) and
2.5/100,000 (9), although the
precise prevalence of MDC1A in China has remained unknown. The
LAMA2 gene spans 260 kb, encompassing 65 exons and
transcribing a messenger RNA transcript of 9.5 kb (NM_000426.3)
(7). LAMA2 encodes a
400-kDa protein that is post-translationally cleaved into two
subunits (300- and 80-kDa) associated with disulfide bonds
(7). In skeletal muscle,
LAMA2 has been suggested to mediate the migration,
organization and attachment of cells during embryonic development
via interactions with components of the extracellular matrix
(10). A potential hypothesis for
the mechanism underlying the pathogenesis of
LAMA2-associated muscular dystrophy was suggested following
studies in zebrafish, in which the sarcolemma was destabilized due
to cellular damage and subsequent apoptosis (11).
At present, next-generation sequencing (NGS) is
becoming more widely recognized as a genetic analysis tool
(12,13). Unlike traditional direct sequence
analyses, NGS provides thousands of reads at single nucleotide
resolution. The high-throughput sequencing data generated by such
large-scale parallel sequencing enables more accurate
quantification of sequence changes. In order to improve efficiency
and reduce costs, it is often necessary to select specific regions
of interests and enrich these regions prior to sequencing. Targeted
NGS, which is based on chip capturing and large-scale parallel
sequencing, may present an efficient strategy to circumvent this
problem. At present, targeted NGS technology is frequently used to
search for alleles underlying Mendelian disorders (14–17).
The present study aimed to demonstrate the utility
of targeted NGS by efficiently identifying novel intragenic
deletions of the LAMA2 gene in two patients with CMD.
Materials and methods
Patients
Study participants provided informed written
consent, and the study was approved by the Ethics Committee of the
Beijing Genomics Institute at Shenzhen (Shenzen, China). Samples
were taken at Wuhan Medical and Health Center for Women and
Children in November 2012. DNA was isolated from peripheral blood
using QIAamp DNA BloodMiNi kit (Qiagen, Hilden, Germany).
The proband was a six-year-old boy who exhibited
limb weakness that resulted in difficulties jumping, running and
walking. CMD symptoms were identified at birth, indicated by poor
spontaneous movements, a weak cry and feeding difficulties. No
intellectual deficiencies were identified. Neurological examination
revealed that the proximal end of the lower limb muscle strength
was graded III, distal muscle strength was graded level III+ and
lower limb muscle strength was graded IV (18). Electromyography (EMG) using Nicolet
VikingQuest (Thermo Nicolet, Madison, WI, USA) identified a
potential myogenic lesion. Magnetic resonance imaging (MRI) of the
brain using a Siemens Tro 3.0T superconductive MRI scanner (Siemens
Healthcare, Erlangen, Germany). The regular gradient recalled echo
sagittal T1-weighted sequence was imaged under the parameters:
TR/TE = 400/2.5 ms; matrix, 256×320; excitation number, two; slice
thickness, 5 mm; slice gap, 1.5 mm. The T1-weighted axial image was
taken under the parameters: TR/TE = 200/2.5 ms; matrix, 256×320;
excitation number, two; slice thickness, 5 mm; slice gap, 2 mm. In
addition, axial fluid attenuation inversion recovery sequence
(FLAIR) was scanned with TR/TI/TE = 8500/2440/93 ms; matrix,
464×512; excitation number, two. Axial FLAIR and T1-weighted axial
image were corresponding, supplemented by coronal FLAIR scanning,
which was performed following an identical protocol apart from the
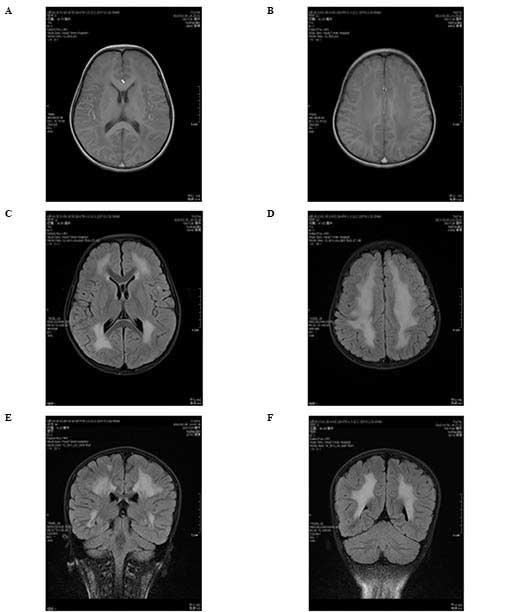
direction of imaging. The results revealed abnormal signaling in
bilateral symmetry of the white matter (Fig. 1). Concentrations of CK and
CK-muscle/brain (MB) isoenzyme were 491 and 28 U/l, respectively.
Concentrations of L-lactate dehydrogenase (LDH-L) and lactate
dehydrogenase isoenzyme 1 (LDH-1) were 352 and 79 U/l,
respectively. The proband tested negative for metabolic
enzymes.
Patient two, four months old at the time of
examination, was the younger brother of the proband. Patient two
was delivered via cesarean section at full-term without
asphyxiating. At the time of birth, the parents of patient two
identified limb weakness, weak cry and feeding difficulties. Aged
sixty days, patient two was able to raise his head, unstably. It
took three months for patient two to exhibit a laughing facial
expression. Following rehabilitation treatment for ~one month,
patient two appeared alert and responded normally in the physical
examination. Head circumference was 40 cm, limb muscle strength was
graded IV and muscular tension was normal. The bilateral patellar
reflex was active, although the Achilles tendon reflex was not
elicited. The cranial nerve examination was normal. Patient two was
able to raise his head when he was pulled up in supine position,
but not in prone position. A brain MRI indicated bilateral lateral
fissure, and the longitudinal and frontal brain extracellular
spaces were wider. Electroencephalography results were normal. The
results of EMG analysis indicated that patient two may exhibit
myogenic damage. The Gesell test (19) demonstrated that the general
developmental level was ten weeks when patient two was 15 weeks of
age. Concentrations of CK and CK-MB isoenzyme were 5,354 and 197
U/l, respectively. Concentrations of LDH-L and LDH-1 were 755 and
113 U/l, respectively.
Chip capturing, library construction and
next-generation sequencing
Total DNA was fragmented into the range of 200–250
bp using an ultrasonoscope (Covaris S2; Covaris, Inc., Woburn, MA,
USA). The DNA library was constructed according to standard
Illumina protocols (Illumina, Inc., San Diego, CA, USA) (20). Following adaptor ligation, the
library was amplified using four-cycle PCR with PCR primers
containing a custom-synthesized barcode sequence (8 bp) as a sample
index signature, and a high-fidelity Pfx DNA polymerase (Invitrogen
Life Technologies, Calsbad, CA, USA). Reactions were conducted in
the GeneAmp PCR system 9700 (Applied Biosystems Life Technologies,
Foster City, CA, USA). The first phase was 2 min at 94°C, followed
by the second phase of 15 sec at 94°C, 30 sec at 62°C and 30 sec at
72°C for four cycles. The third phase was 5 min at 72°C, followed
by 4°C. A custom-designed capture array (Roche NimbleGen, Inc.,
Madison, WI, USA) was used to capture neuromuscular genes. The
custom-made array targeted all exons plus the flanking 10 bp of the
adjacent introns of the target gene. The final PCR products were
pooled and hybridized to the capture array for 72 h. The washing,
elution and additional amplification steps were performed according
to NimbleGen protocols (Roche NimbleGen, Inc.). The captured
samples were subsequently analyzed using the Agilent 2100
Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA) and
the ABI StepOne (Applied Biosystems Life Technologies, Foster City,
CA, USA) for DNA fragment length measurement and estimation of
enrichment magnitude, respectively (21,22).
The captured libraries were sequenced on the Illumina genome
analyzer (Illumina HiSeq2000 Analyzer) as 90-bp paired-end reads,
according to the manufacturer’s instructions. Image analysis and
base calling were performed using Illumina Pipeline (version
1,3,4).
Data filtering and analysis pipeline
Following completion of the entire run, filter
criteria were used to remove unqualified sequences from the primary
data. Unqualified sequences were defined as reads that contained
>10% no signal detected in the read length, adapter sequences
including indexed sequence and 50% reads with a quality value of
<5 as well as an average quality <10 (23). Qualified sequences were designated
as clean reads for further analysis. Clean reads with a length of
90 bp were aligned against the reference human genome (hg19) using
the Burrows Wheeler Aligner Multi-Vision software package
(http://bio-bwa.sourceforge.net/)
(24). The single-nucleotide
variants (SNVs) and insertions and deletions (indels) were
identified using SOAPsnp software (25) and the SAMtools (26), respectively. Identified SNVs were
annotated using four databases, including the Single Nucleotide
Polymorphism database (www.ncbi.nlm.nih.gov/projects/SNP/), the Hapmap
project (hapmap.ncbi.nlm.nih.gov/), the 1000
Genomes Project (www.1000genomes.org/) and the 124
healthy reference samples sequenced. These healthy reference
samples were obtained from members of staff at BGI-Central China
who passed a health screen. Known disease-causing mutations were
identified from the Leiden Open Variation Database (www.lovd.nl/).
Sanger sequencing
PCR products containing potential variants were
analyzed using Sanger sequencing in order to ascertain the accuracy
of variant identification by NGS. The specific primers were
designed by Primer 6.0 software (Premier Biosoft, Palo Alto, CA,
USA). The Sanger sequencing was performed using a 3730xl DNA
Analyzer (Applied Biosystems Life Technologies) according to a
previously described protocol (23).
Copy number of exons in LAMA2
To determine the copy number (CN) of exons in the
gene in a given sample, intra- and inter-sample normalization were
performed. For each sample subjected to intra-sample analysis, the
relative depth count of an exon was defined as the ratio of the
depth count of an exon over the total depth count of exons in the
gene to which the exons belong. Inter-sample normalization was
subsequently performed to divide the relative depth count of each
exon by the corresponding relative depth count of the control
sample in the same experiment. In short:
CN=(depthi/depthall)test/(depthc-i/depthc-all)ref,
where depthi=depth of i exon; depthall=depth
of all exons in one gene; depthc-i=depth of i exon in
control sample; depthc-all=depth of all exons in one
gene in control sample; test=test individual and ref=reference
individual (27,28).
qPCR analysis
To further quantify the intragenic deletions in the
specimens from patients with CMD, qPCR with SYBR Green (Takara Bio,
Inc., Otsu, Japan) was performed in the StepOne™ Real-Time PCR
System (Applied Biosystems Life Technologies) according to the
manufacturer’s instructions. All primers were designed using Primer
6.0 and tested for specificity using National Center for
Biotechnology Information Basic Local Alignment Search Tool
software (blast.ncbi.nlm.nih.gov/Blast.cgi). The housekeeping
gene, GAPDH, was used as an endogenous control. Measurements were
repeated ≥three times to verify the reproducibility of the results.
The cycling conditions were as follows: 10 min at 95°C, 40 cycles
at 95°C for 15 sec and 60°C for 30 sec. Following PCR
amplification, a melting curve was generated for each PCR product
to confirm reaction specificity.
Statistical analysis
Statistical analyses were performed using Microsoft
Office Excel 2007 (Albuquerque, NM, USA). Values are expressed as
the mean ± standard error.
Results
Identification of candidate mutations in
the LAMA2 gene
To identify the most probable pathogenic mutations,
all SNVs previously reported in the four databases described were
excluded. The common variants (indel and substitution) were
filtered out if their frequency was >0.05 in any of the four
databases. Only mutations in the coding DNA sequence regions were
selected. One compound heterozygous mutation was identified in the
LAMA2 gene in the proband (c. 2045_2046delAG). A pair of
primers was designed for each specific site to test four family
members, including the proband and patient two (data not shown),
LAMA-F-1 (5′-AGTCTATTGAGGGTGGAGGATACA-3′) and LAMA-R-1
(5′-GGCTCCGTTCTTATCTGCTCTT-3′), and the PCR products were validated
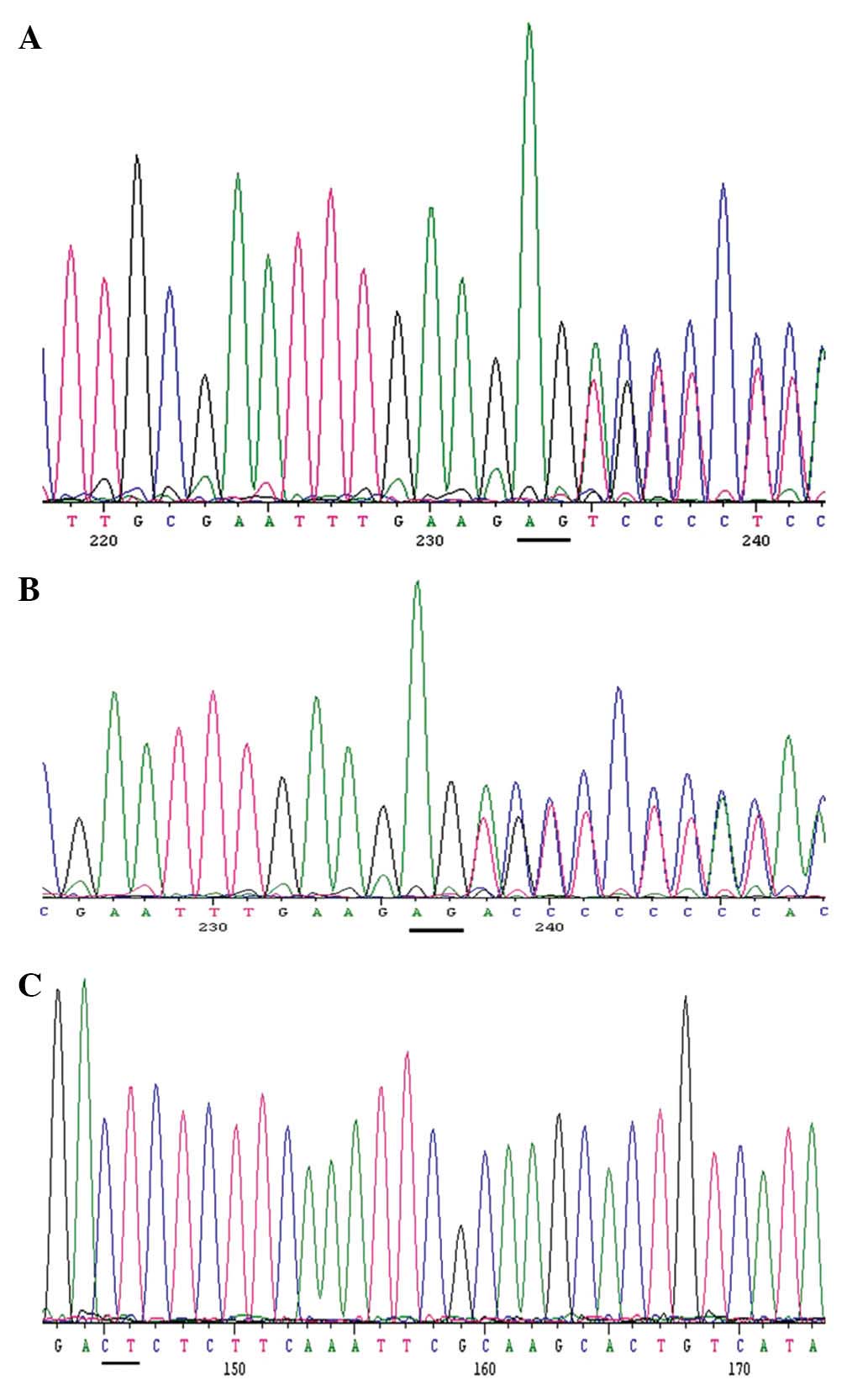
by Sanger sequencing. A small deletion in LAMA2 was
confirmed in the proband (Fig.
2A). Concurrently, the potential site was validated by PCR and
Sanger sequencing in the proband’s father (Fig. 2B), but not in the mother (Fig. 2C). The (c. 2045_2046delAG) mutation
causes a frameshift and introduces a premature stop codon that may
shorten the protein transcribed (29). Read mapping was used to analyze the
two base pair deletion in the proband (Fig. 3).
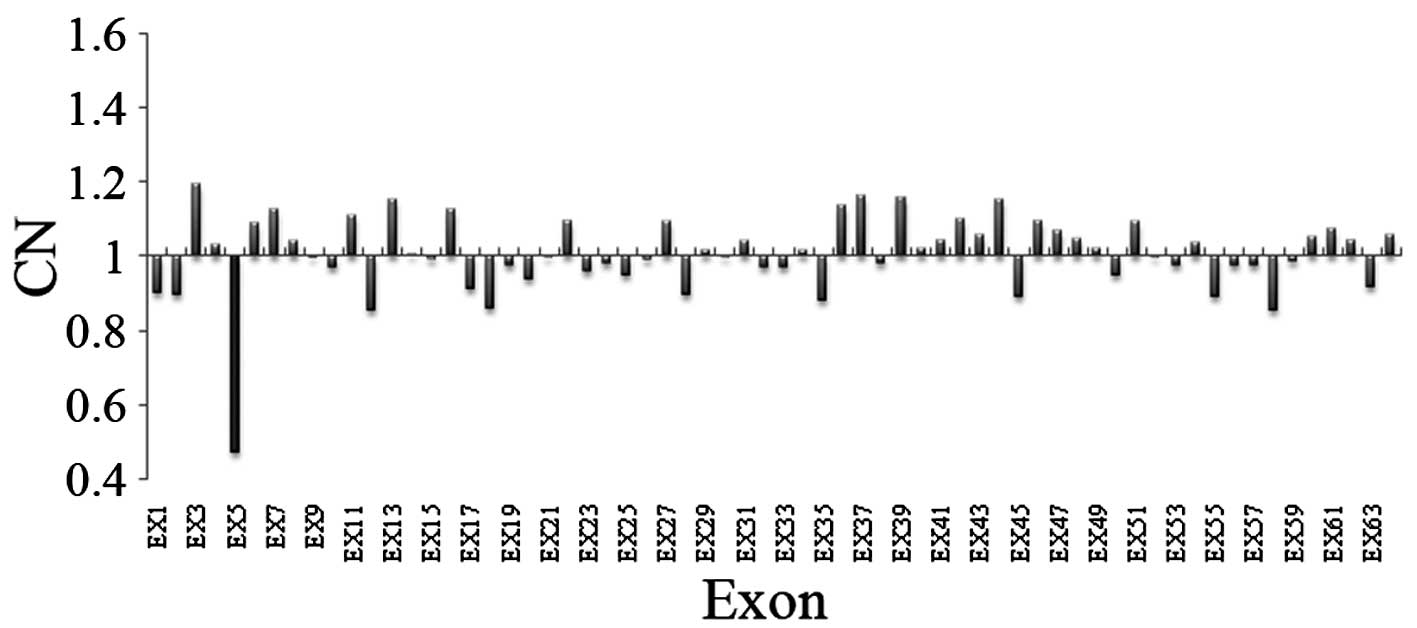
Identification of a deletion of exon five
of LAMA2 (Exon5del) in the proband
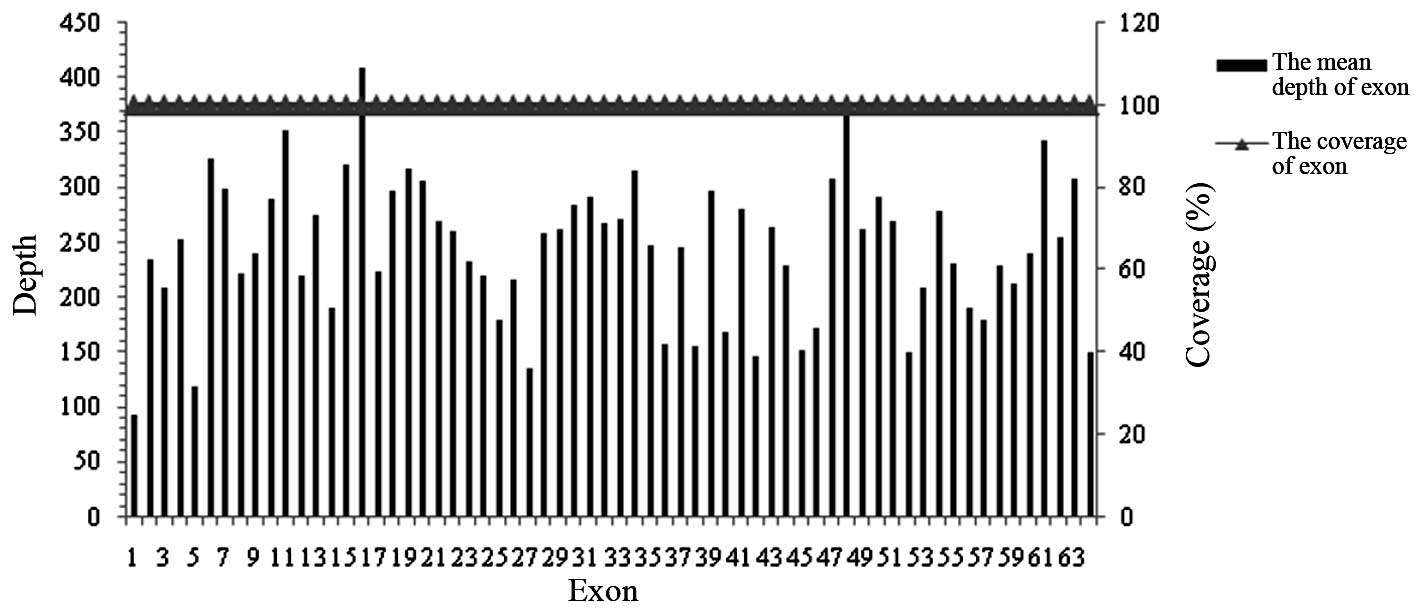
In the present study, the average sequencing depth
of all exons of the LAMA2 gene were analyzed in the proband
and controls using targeted NGS. The mean sequencing depth of the
target region was >227-fold in each sample and on average, 100%
of the targeted region was covered in each sample, indicating that
the method was reliable at detecting DNA variants (Figs. 4 and 5). In addition to specific and accurate
variant detection, simultaneous CN variation analysis within the
same targeted NGS experiment was significant for diagnostic
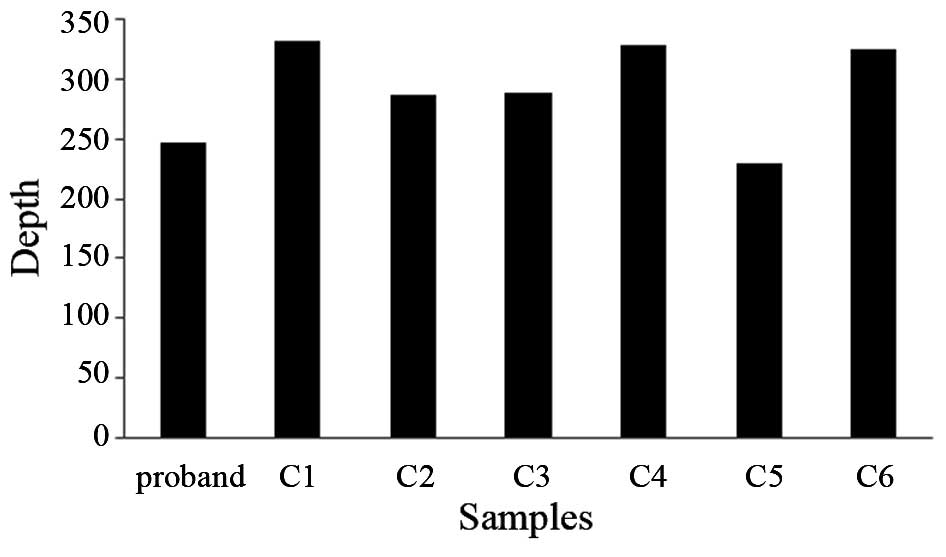
purposes. The average relative depth count of six normal samples
was used as a reference. Each mean depth of exons of LAMA2
from the proband was divided by the corresponding average depth of
exons from the reference sample. Based on the observations
presented in Fig. 6, exon five was
selected for subsequent screening.
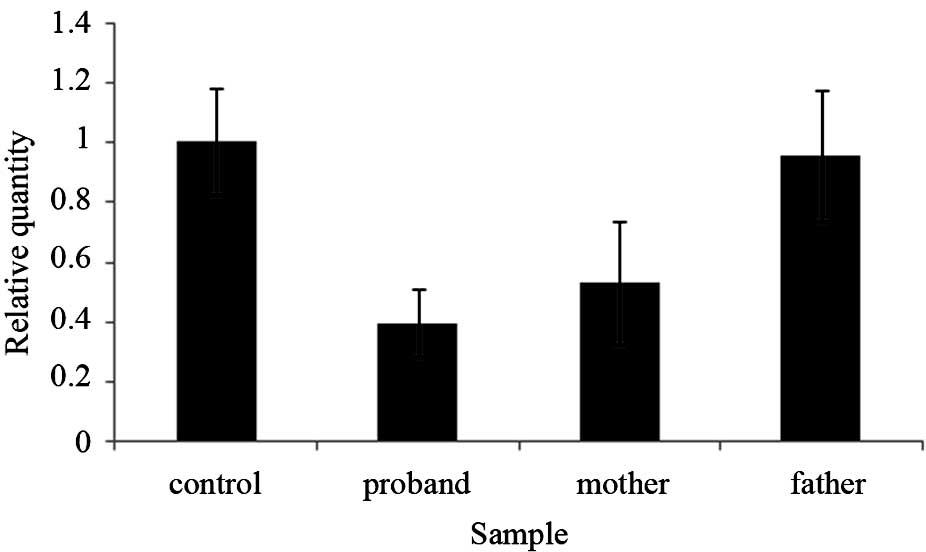
The Exon5del mutation is on the maternal
allele
To confirm the deletion of exon five of the
LAMA2 gene in the proband, an exon quantification strategy
based on PCR was utilized. qPCR was used to detect the DNA copy
number of the exons in the proband, the proband’s mother and
father, patient two (data not shown) and a normal control, where
the genomic GAPDH gene was used as a loading control. The
primers were as follows: Forward, 5′-GACCAAGTGCCGATGATCCTTC-3′ and
reverse, 5′-GTCAGCATTCAGTGTGCGGAT-3′. The results indicated that
the quantity of exon five detected in the DNA sample of the proband
was ~50% of that of his father and the control samples, and almost
100% of that of his mother’s sample (Fig. 7). These results suggested that
there was a heterozygous deletion of exon five in the LAMA2
gene in the proband and the proband’s mother (30). It was therefore concluded that the
deletion of LAMA2 exon five occurred on the maternal allele,
and that this heterozygous mutation was inherited by the
proband.
Discussion
The comprehensive study of single-gene disorders and
the corresponding advances in technologies available for disease
detection has meant that increasing numbers of monogenic inherited
diseases may be identified by clinical molecular diagnosis
(31). There are numerous
drawbacks of routine molecular diagnosis, including genetic and
clinical disease heterogeneity, unidentified genes, non-specific
clinical features and the involvement of large and/or multiple
genes requiring complementary methods, which result in an increased
diagnosis time and delays in the subsequent molecular validation of
the diagnosis (32). Second
generation sequencing technology and directional capture tools have
facilitated the development of novel clinical molecular diagnostic
techniques to detect single gene disorders (33). The advantage of this approach is
that the techniques are relatively low-cost, produce data that are
easier to interpret and allow corrected diagnosis, which confirms
the genetic heterogeneity of diseases (34).
The clinical manifestations observed in the proband
and patient two suggested that they may have had congenital
muscular dystrophy. The disease was subsequently diagnosed and
classified accurately via genetic testing, as indicated by the
results of the present study. Using a combination of targeted-gene
enrichment and NGS technology, an intragenic deletion on the
maternal allele and a two base pair deletion on the paternal allele
were identified in the proband. To confirm the candidate variants,
genomic qPCR and Sanger sequencing were used. If a conventional
sequencing method had been used it would have been unlikely to
detect the large intragenic LAMA2 gene deletion identified
in the present study.
In LAMA2, which comprises 64 exons, mutations
are widely distributed and there is no clear hot-spot amongst
identified mutations (35–37). The small deletion (p.
Lys682LysfsX22) potentially causes a frame shift, resulting in
premature translational termination at amino acid 682 - a mutation
previously confirmed to be disease-causing (29) - yielding a non-functional protein
product. The entire deletion of exon five was a novel mutation
identified in a CMD patient in the present study. The protein
containing the intragenic deletion mutation was damaged as exon
five comprised part of domain IV of laminin α-2. Domain IV
participates in calcium ion-dependent intermolecular interactions
and integrin binding (38), and is
highly conserved in the laminin isoforms of numerous human and
non-human species. The two identified mutations were additionally
confirmed in patient two (data not shown). Genetic testing has an
increasingly significant role in the diagnosis of patients with
elusive muscular dystrophy phenotypes, particularly in the absence
of known family history.
In conclusion, the present study indicated that an
integrated approach, combining exon-capture sequencing and clinical
molecular diagnosis, was a fast, efficient and a reliable
diagnostic tool for congenital myopathies. To the best of our
knowledge, the present study was the first to report the deletion
of exon five in LAMA2 in a Chinese family.
Acknowledgements
The authors would like to thank the blood donors for
their contribution to the present study. The present study received
financial support from the National High-Tech Research and
Development Program of China (863 Program; no. 2012AA02A201), the
Guangdong Enterprise Key Laboratory of Human Disease Genomics
(Shenzen, China), Shenzhen Key Laboratory of Transomics
Biotechnologies (Shenzen, China; no. CXB201108250096A), Shenzhen
Engineering Laboratory for Clinical Molecular Diagnostics (Shenzen,
China) and the China National GeneBank (Shenzen, China).
References
|
1
|
Allamand V and Guicheney P:
Merosin-deficient congenital muscular dystrophy, autosomal
recessive (MDC1A, MIM#156225, LAMA2 gene coding for alpha2 chain of
laminin). Eur J Hum Genet. 10:91–94. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sparks S, Quijano-Roy S, Harper A, et al:
Congenital Muscular Dystrophy Overview, 2012.
GeneReviews® [Internet]. Pagon RA, Bird TD, Dolan CR, et
al: University of Washington; Seattle, WA: 1993–2014
|
|
3
|
Kobayashi O, Hayashi Y, Arahata K, Ozawa E
and Nonaka I: Congenital muscular dystrophy: Clinical and
pathologic study of 50 patients with the classical (Occidental)
merosin-positive form. Neurology. 46:815–818. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Louhichi N, Triki C, Quijano-Roy S,
Richard P, Makri S, Méziou M, Estournet B, Mrad S, Romero NB, Ayadi
H, et al: New FKRP mutations causing congenital muscular dystrophy
associated with mental retardation and central nervous system
abnormalities. Identification of a founder mutation in Tunisian
families. Neurogenetics. 5:27–34. 2004. View Article : Google Scholar
|
|
5
|
Voit T, Cirak S, Abraham S, Karakesisoglou
I, Parano E, Pavone P, Falsaperla R, Amthor H, Schroeder J, Mutoni
F, et al: Congenital muscular dystrophy with adducted thumbs,
mental retardation, cerebellar hypoplasia and cataracts is caused
by mutation of Enaptin (Nesprin-1): The third nuclear envelopathy
with muscular dystrophy. In: Proceedings of the 12th International
Congress of the World-Muscle-Society Italy Neuromuscular Disorders;
Pergamon-Elsevier Science Ltd; Oxford, UK: pp. 833–834. 2007
|
|
6
|
Messina S, Tortorella G, Concolino D,
Spanò M, D’Amico A, Bruno C, Santorelli FM, Mercuri E and Bertini
E: Congenital muscular dystrophy with defective alpha-dystroglycan,
cerebellar hypoplasia, and epilepsy. Neurology. 73:1599–601. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Quijano-Roy S, Sparks S and Rutkowski A:
LAMA 2-Related Muscular Dystrophy, 2012. GeneReviews®
[Internet]. Pagon RA, Bird TD, Dolan CR, et al: University of
Washington; Seattle, WA: 1993–2014
|
|
8
|
Mostacciuolo ML, Miorin M, Martinello F,
Angelini C, Perini P and Trevisan CP: Genetic epidemiology of
congenital muscular dystrophy in a sample from north east Italy.
Hum Genet. 97:277–279. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Darin N and Tulinius M: Neuromuscular
disorders in childhood: a descriptive epidemiological study from
western Sweden. Neuromuscul Disord. 10:1–9. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Suzuki N, Yokoyama F and Nomizu M:
Functional sites in the laminin alpha chains. Connect Tissue Res.
46:142–152. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hall TE, Bryson-Richardson RJ, Berger S,
Jacoby AS, Cole NJ, Hollway GE, Berger J and Currie PD: The
zebrafish candyfloss mutant implicates extracellular matrix
adhesion failure in laminin alpha2-deficient congenital muscular
dystrophy. Proc Natl Acad Sci USA. 104:7092–7097. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang Y, Muzny DM, Reid JG, et al: Clinical
whole-exome sequencing for the diagnosis of mendelian disorders. N
Engl J Med. 369:1502–1511. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Biesecker LG, Burke W, Kohane I, et al:
Next-generation sequencing in the clinic: are we ready? Nat Rev
Genet. 13:818–824. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Iqbal Z, Neveling K, Razzaq A, Shahzad M,
Zahoor MY, Qasim M, Gilissen C, Wieskamp N, Kwint MP, Gijsen S, et
al: Targeted next generation sequencing reveals a novel intragenic
deletion of the TPO gene in a family with intellectual disability.
Arch Med Res. 43:312–316. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wei X, Jin F, Ye Y, Xu C, Qu N, Ju X and
Yi X: A novel mutation of IDS gene in a Chinese patient with
mucopolysaccharidosis II by next-generation sequencing. Clin Chim
Acta. 412:2340–2342. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wei X, Sun Y, Xie J, Shi Q, Qu N, Yang G,
Cai J, Yang Y, Liang Y, Wang W and Yi X: Next-generation sequencing
identifies a novel compound heterozygous mutation in MYO7A in a
Chinese patient with Usher Syndrome 1B. Clin Chim Acta.
413:1866–1871. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jones MA, Bhide S, Chin E, Ng BG,
Rhodenizer D, Zhang VW, Sun JJ, Tanner A, Freeze HH and Hegde MR:
Targeted polymerase chain reaction-based enrichment and next
generation sequencing for diagnostic testing of congenital
disorders of glycosylation. Genet Med. 13:921–932. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mendell JR and Florence J: Manual muscle
testing. Muscle Nerve. 13(Suppl): S16–S20. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ames LB, Gillespie BS, Haines J and Ilg
FL: The Gesell Institute’s child from one to six: Evaluating the
behavior of the preschool child. Harper & Row; New York:
1979
|
|
20
|
Illumina Protocol for Whole Genome
Sequencing using SBS Technology. BioTechniques Protocol Guide.
Biotechniques; New York, NY: 2006
|
|
21
|
Chilamakuri CS, Lorenz S, Madoui MA, Vodák
D, Sun J, Hovig E, Myklebost O and Meza-Zepeda LA: Performance
comparison of four exome capture systems for deep sequencing. BMC
Genomics. 15:4492014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guo G, Sun X, Chen C, Wu S, et al:
Whole-genome and whole-exome sequencing of bladder cancer
identifies frequent alterations in genes involved in sister
chromatid cohesion and segregation. Nat Genet. 45:1459–1463. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wei X, Ju X, Yi X, Zhu Q, Qu N, et al:
Identification of sequence variants in genetic disease-causing
genes using targeted next-generation sequencing. PLoS One.
6:e295002011. View Article : Google Scholar
|
|
24
|
Li H and Durbin R: Fast and accurate
long-read alignment with Burrows-Wheeler transform. Bioinformatics.
26:589–595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li R, Li Y, Fang X, Yang H and Wang J,
Kristiansen K and Wang J: SNP detection for massively parallel
whole-genome resequencing. Genome Res. 19:1124–1132. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R: 1000 Genome
Project Data Processing Subgroup: The Sequence alignment/map (SAM)
format and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Goossens D, Moens LN, Nelis E, Lenaerts
AS, Glassee W, Kalbe A, Frey B, Kopal G, De Jonghe P, De Rijk P and
Del-Favero J: Simultaneous mutation and copy number variation (CNV)
detection by multiplex PCR-based GS-FLX sequencing. Hum Mutat.
30:472–476. 2009. View Article : Google Scholar
|
|
28
|
Kumps C, Van Roy N, Heyrman L, Goossens D,
Del-Favero J, Noguera R, Vandesompele J, Speleman F and De Preter
K: Multiplex Amplicon Quantification (MAQ), a fast and efficient
method for the simultaneous detection of copy number alterations in
neuroblastoma. BMC Genomics. 11:2982010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guicheney P, Vignier N, Zhang X, He Y,
Cruaud C, Frey V, Helbling-Leclerc A, Richard P, Estournet B,
Merlini L, et al: PCR based mutation screening of the laminin
alpha2 chain gene (LAMA2): application to prenatal diagnosis and
search for founder effects in congenital muscular dystrophy. J Med
Genet. 35:211–217. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hellemans J, Mortier G, De Paepe A,
Speleman F and Vandesompele J: qBase relative quantification
framework and software for management and automated analysis of
real-time quantitative PCR data. Genome Biol. 8:R192007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Katsanis SH and Katsanis N: Molecular
genetic testing and the future of clinical genomics. Nat Rev Genet.
14:415–426. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vasli N and Laporte J: Impacts of
massively parallel sequencing for genetic diagnosis of
neuromuscular disorders. Acta Neuropathol. 125:173–185. 2013.
View Article : Google Scholar
|
|
33
|
Bell CJ, Dinwiddie DL, Miller NA, et al:
Carrier testing for severe childhood recessive diseases by
next-generation sequencing. Sci Transl Med. 3:65ra42011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Umbarger MA, Kennedy CJ, Kennedy CJ,
Saunders P, et al: Next-generation carrier screening. Genet Med.
16:132–140. 2014. View Article : Google Scholar :
|
|
35
|
Oliveira J, Santos R, Soares-Silva I,
Jorge P, et al: LAMA2 gene analysis in a cohort of 26 congenital
muscular dystrophy patients. Clin Genet. 74:502–512. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Di Blasi C, Piga D, Brioschi P, et al:
LAMA2 gene analysis in congenital muscular dystrophy: new
mutations, prenatal diagnosis, and founder effect. Arch Neurol.
62:1582–1586. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yamamoto LU, Gollop TR, Naccache NF, et
al: Protein and DNA analysis for the prenatal diagnosis of
alpha2-laminin-deficient congenital muscular dystrophy. Diagn Mol
Pathol. 13:167–171. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Engvall E and Wewer UM: Domains of
laminin. J Cell Biochem. 61:493–501. 1996. View Article : Google Scholar : PubMed/NCBI
|