Introduction
Cerebral ischemic injury is one of the leading
causes of human mortality and disability worldwide (1). Restoration of blood flow to the
ischemic brain is often used to treat patients in clinical
experiments. However, reperfusion itself also has the potential to
produce additional injuries in the ischemic brain due to
overproduction of reactive oxygen species (ROS). The potential
pathological mechanisms of ischemia/reperfusion (I/R) injury
include glutamate excitotoxicity, calcium overload, nitric oxide
(NO) production, oxidative stress, inflammation and apoptosis,
which eventually lead to cell death (2). Oxidative stress and apoptosis
following cerebral I/R are the two major processes that induce
neuronal injury (3,4). For this reason, multifunctional
molecules with anti-oxidative and anti-apoptotic properties are
ideal neuroprotective agents.
α-lipoic acid (LA), an endogenous short-chain fatty
acid, is a cofactor for multiple mitochondrial dehydrogenase
enzymes, including pyruvate dehydrogenase (5). It is an ROS scavenger, which is able
to stimulate the insulin signaling pathway, chelate metal and
regenerate endogenous natural antioxidants (6). In previous decades, using several
different experimental models of I/R, studies have demonstrated the
protective effects of LA against I/R-induced injury, including
myocardial injury (7), peripheral
nerve injury (8), testicular
injury (9) and retinal injury
(10). In addition, it has been
reported that LA can activate phosphatidylinositol-4,5-bisphosphate
3-kinase (PI3K)/Akt and extracellular signal-regulated kinase 1/2
(ERK1/2) pathways to induce protection against I/R injury in other
organs (11–13). However, to the best of our
knowledge, few studies were reported to address whether LA has
neuroprotective effects against cerebral I/R-induced injury and its
potential mechanisms.
In the present study, the neuroprotective effects of
LA in rats were investigated with 90 min middle cerebral artery
occlusion (MCAO)/24 h reperfusion-induced injury. Furthermore, the
hypothesis that the neuroprotective effect of LA is associated with
a reduction in oxidative stress and inhibition of apoptosis though
activation of brain-derived neurotrophic factor (BDNF), PI3K/Akt
and ERK1/2 in rats was assessed. Therefore, the effect of LA on
infarct size, neurological deficit score (NDS) and brain water
content were investigated. In addition, to examine the mechanisms
activated by LA, oxidative parameters, including malondialdehyde
(MDA), NO and total antioxidant capacity (T-AOC), superoxide
dismutase (SOD) and the expression of cleaved caspase-3, BDNF,
PI3K, p-Akt and p-ERK1/2 proteins were measured in rat brains.
Materials and methods
Animals and drug administration
Adult male Sprague-Dawley rats, 10–12 weeks old,
weighing 250–280 g, were purchased from the Animal Center of
Southern Medical University (Guangzhou, China). All protocols were
approved by the animal care committee of Southern Medical
University and undertaken according to the guidelines for the Care
and Use of Laboratory Animals of the National Institutes of Health
(Bethesda, MD, USA). The rats were kept under constant laboratory
conditions (20–25°C, 60±5% humidity) and a 12 h light/dark cycle.
They were allowed free access to food and water up until 12 h prior
to surgery at which point only water was available.
LA powder (Shanghai Modern Pharmaceutical Co., Ltd.,
Shanghai, China) was mixed with sterile saline. Subsequently, 1 M
NaOH was added until the suspension dissolved. The pH was lowered
to 7.4 using hydrochloric acid (14). Rats were administered with LA or
saline via subcutaneous injection 2 h prior to MCAO (15).
Experimental groups
In preliminary experiments, different dosages of LA
were administered to determine the optimal dosage. Rats were
randomly assigned to the following groups (n=6): i) Sham group; ii)
I/R group, wherein the animals received I/R and a vehicle
treatment; iii) LA group, in which the rats received I/R and LA (50
mg/kg); iv) LA group, in which the rats received I/R and LA (70
mg/kg); v) LA group, in which the rats received I/R and LA (100
mg/kg). Following determining 100 mg/kg as the optimal dosage, rats
were randomly divided into the following groups (n=9): i) Sham
group; ii) I/R group, in which the rats received I/R and a vehicle
treatment; iii) LA group, wherein the rats received I/R and LA (100
mg/kg).
Establishment of the MCAO model
Rats fasted overnight, but were allowed free access
to water prior to the MCAO procedure. MCAO was induced as described
previously (16). Briefly, rats
were anesthetized with 10% chloral hydrate (3.5 ml/kg) by
intraperitoneal injection. The skin and surrounding fur were
disinfected with 75% ethyl alcohol. Following incision to the skin,
the left common carotid artery (CCA) was exposed and carefully
separated from the vagal nerves. The left external carotid artery
(ECA) and the left internal carotid artery (ICA) were carefully
isolated and the ECA was ligated. In addition, the CCA was
temporally occluded with 3-0 silk thread. The left MCA was occluded
for 90 min with a paraffin-coated nylon filament (17), which was introduced into the ICA
for 18–20 mm until resistance was detected. After 90 min of
occlusion, the ECA was permanently occluded. The silk thread
occluding the CCA was removed for reperfusion for 24 h. A heating
lamp was used to maintain body temperature at 37°C during
surgery.
Determination of infarct size and
neurological function
After 90 min of ischemia followed by 24 h of
reperfusion animals were anesthetized with 10% chloral hydrate (3.5
ml/kg) and decapitated. The brains were quickly removed and frozen
at −20°C for 20 min, dissected into 2 mm coronal slices and
immediately incubated in 2% 2,3,5-triphenyltetrazolium chloride
(TTC; Mbchem, Shanghai, China) at 37°C for 10 min as described
previously (16). Following TTC
staining, the normal brain tissue was dark red, whereas infarcted
tissue was unstained. Following TTC staining, the tissues were then
fixed in 4% paraformaldehyde (Mbchem) for 24 h and scanned with a
digital camera (Canon, Inc., Tokyo, Japan). Infarct size was
calculated using Image J software (National Institutes of Health,
Bethesda, MD, USA) by an individual blinded to the identity of the
experimental groups and the result was expressed as a percentage of
infarct area to total brain area.
Neurological deficit evaluations were performed at
the end of the experiment by an observer masked to the identity of
experimental groups using the following criteria as previously
described (16): 0, no neurologic
deficit or normal function; 1, failure to extend right forepaw
fully; 2, circling to right; 3, leaning to right; 4, absence of
spontaneous motor activity. Therefore, a higher score was
associated with poorer neurological function.
Measurement of brain water content
Brain water content was measured as described
previously (18). Briefly, after
24 h of reperfusion animals were anesthetized with 10% chloral
hydrate (3.5 ml/kg) and decapitated. The brains were rinsed with
saline and separated into ischemic and non-ischemic hemispheres,
then immediately weighed to gain the wet weight (WW). The brains
were placed in an oven at 100°C for 24 h and weighed to obtain the
dry weight (DW). The brain water content (%) was measured using the
following formula: (WW − DW)/WW × 100%.
Measurement of the oxidative parameters
MDA, NO, T-AOC and SOD
After 24 h of reperfusion, animals were anesthetized
with 10% chloral hydrate (3.5 ml/kg) and decapitated. The rat
brains were quickly removed and detached, then rinsed with cold
saline. The brains were then homogenized in cold saline (1:10,
wt/vol) and centrifuged at 3,000 × g for 10 min to prepare tissue
homogenate. The levels of MDA, NO and the activities of T-AOC and
SOD were determined using commercially available assay kits
(Nanjing Jiancheng Bioengineering Institute, Nanjing, China)
according to the manufacturer’s instructions.
Western blotting
Animals were anesthetized with 10% chloral hydrate
(3.5 ml/kg) and decapitated 24 h after reperfusion. The rat brains
were quickly removed and detached, then rinsed with cold saline.
Tissue samples were lysed in ice-cold RIPA buffer (Nanjing Keygen
Biotech, Co., Ltd., Nanjing, China) containing 1%
phenylmethylsulfonyl fluoride, 1% phosphatase inhibitors and 0.1%
protease inhibitor with a glass homogenizer on ice, repeated for 5
min and incubated on ice for 10 min, then centrifuged at 13,000 × g
for 20 min at 4°C. The supernatant was aliquoted and stored at
−80°C. Protein concentration was measured using the BCA kit (Pierce
Biotechnology, Inc., Rockford, IL, USA). The protein (30 μg) was
then separated on 8–15% polyacrylamide SDS gels and transferred
onto a polyvinylidene difluoride membrane. The membranes were
treated with blocking solution (5% non-fat dry milk or 5% bovine
serum albumin in Tris-buffered saline with Tween® 20 and
incubated at room temperature for 2 h. Following this, the
membranes were incubated with a rabbit anti-rat polyclonal cleaved
caspase-3 antibody (1:1,000; Cell Signaling Technology, Inc.,
Beverly, MA, USA), a rabbit anti-rat polyclonal BDNF antibody
(1:1,000; Chemicon, Temecula, CA, USA), a rabbit anti-rat
polyclonal PI3K antibody (1:1,000; Cell Signaling Technology,
Inc.), a rabbit anti-rat polyclonal p-Akt antibody (1:2,000; Cell
Signaling Technology, Inc.) and a rabbit anti-rat polyclonal
p-ERK1/2 antibody (1:2,000; Cell Signaling Technology, Inc.) at 4°C
overnight, followed by incubation with the corresponding goat
anti-rabbit secondary antibodies (Wuhan Boster Bio-Engineering Co.,
Ltd., Wuhan, China) at room temperature for 1 h. GADPH was used as
a control to ensure equal protein loading. The blots were
visualized with ECL-Plus reagent (Pierce Biotechnology, Inc.) and
the exposures were transferred to radiographic films. The
radiographic films were scanned by a cannon scanner (Canon, Inc.)
and analyzed using Image J software (National Institutes of
Health).
Statistical analysis
All data were analyzed using SPSS 13.0 software
(SPSS, Inc., Chicago, IL, USA) and are expressed as the mean ±
standard deviation. Statistical analysis was performed by one-way
analysis of variance and P<0.05 was considered to indicate a
statistically significant difference.
Results
LA attenuates infarct size
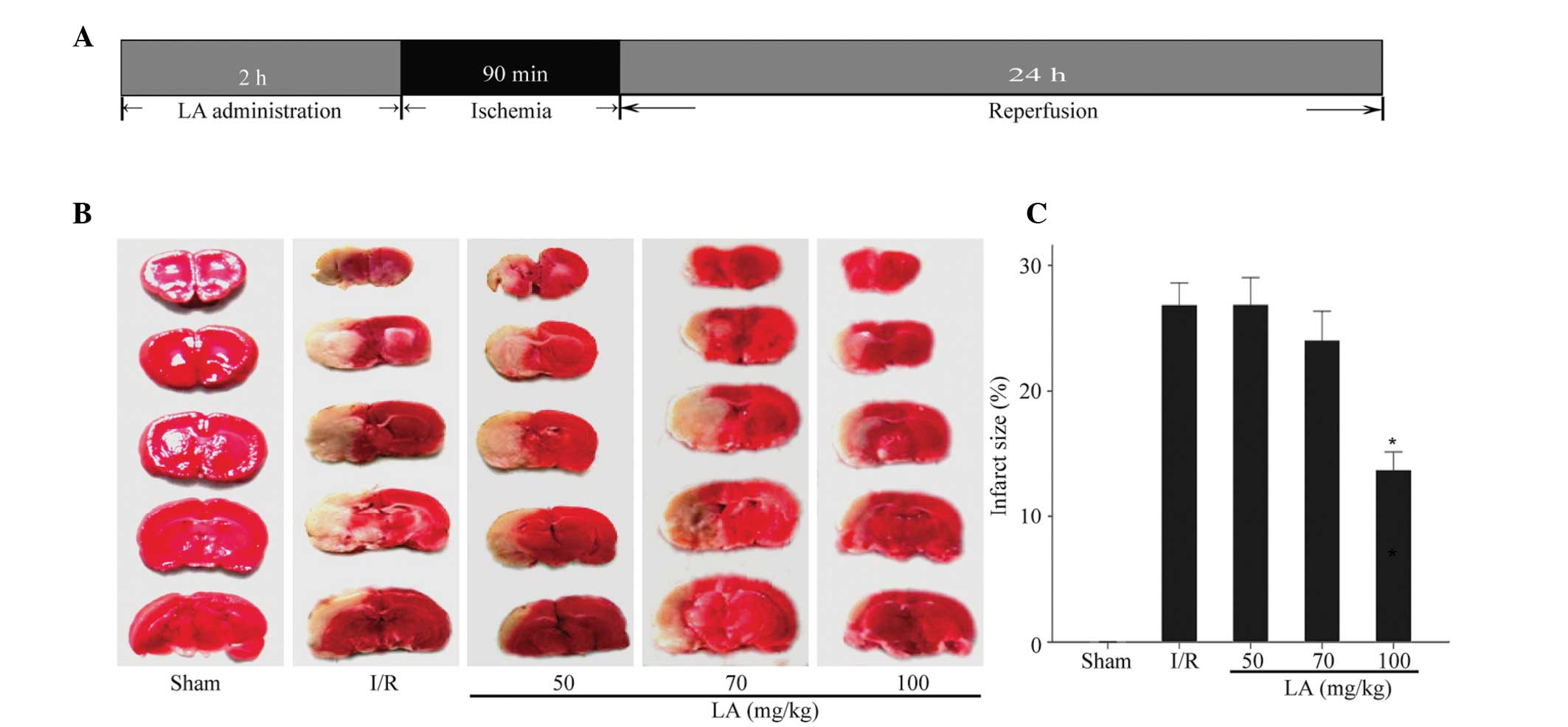
The experimental procedure is shown in Fig. 1A. In order to investigate the
effect of LA on infarct size, the brain infarct area was analyzed
by TCC staining after 24 h of reperfusion. There was no infarct
area in the sham rat brain (Fig.
1B). Pretreatment with 100 mg/kg LA significantly reduced total
infarct size by 51.3% compared with ischemic rats with vehicle
treatment from 26.9±1.7% to 13.8±1.4% (P<0.05), while 50 mg/kg
and 70 mg/kg did not reveal a significant effect (26.9±2.1% and
24.1±2.3%, respectively; Fig. 1C).
Therefore, 100 mg/kg LA was selected as the optimum dosage for
further investigation.
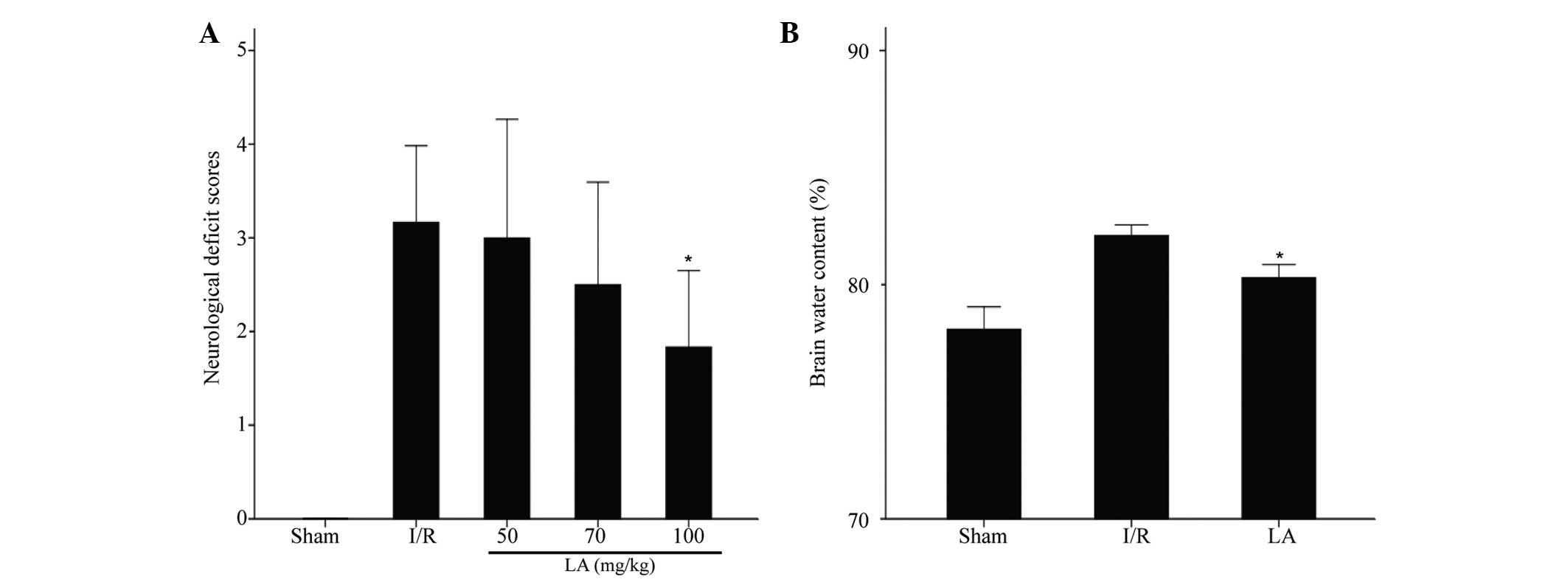
LA improves neurological function
As shown in Fig.
2A, 24 h after reperfusion, the neurological score was
3.17±0.41 in the I/R group, while this significantly decreased
following treatment with LA (100 mg/kg) to 1.83±0.41 (P<0.05),
however, 50 and 70 mg/kg LA did not have a significant effect
(3.0±0.63 and 2.5±0.55, respectively).
LA ameliorates brain edema
Brain water content was quantified using the dry-wet
weight method and the result is shown in Fig. 2B. The brain water content in the
ischemic area of the I/R group was significantly higher than that
of the sham group. Pretreatment of rats with 100 mg/kg LA
significantly reduced brain edema in the rats that underwent I/R
(82.1±0.5% vs. 80.3±0.6%; P<0.05).
LA improves the oxidative parameters MDA,
NO, T-AOC and SOD
As shown in Fig. 3,
the content of MDA and NO significantly increased in the I/R group
(P<0.05) compared with that in the sham group and decreased in
the LA-pretreated group (P<0.05). The activities of T-AOC and
SOD were significantly higher in the LA-treated group (P<0.05)
than in the I/R group.
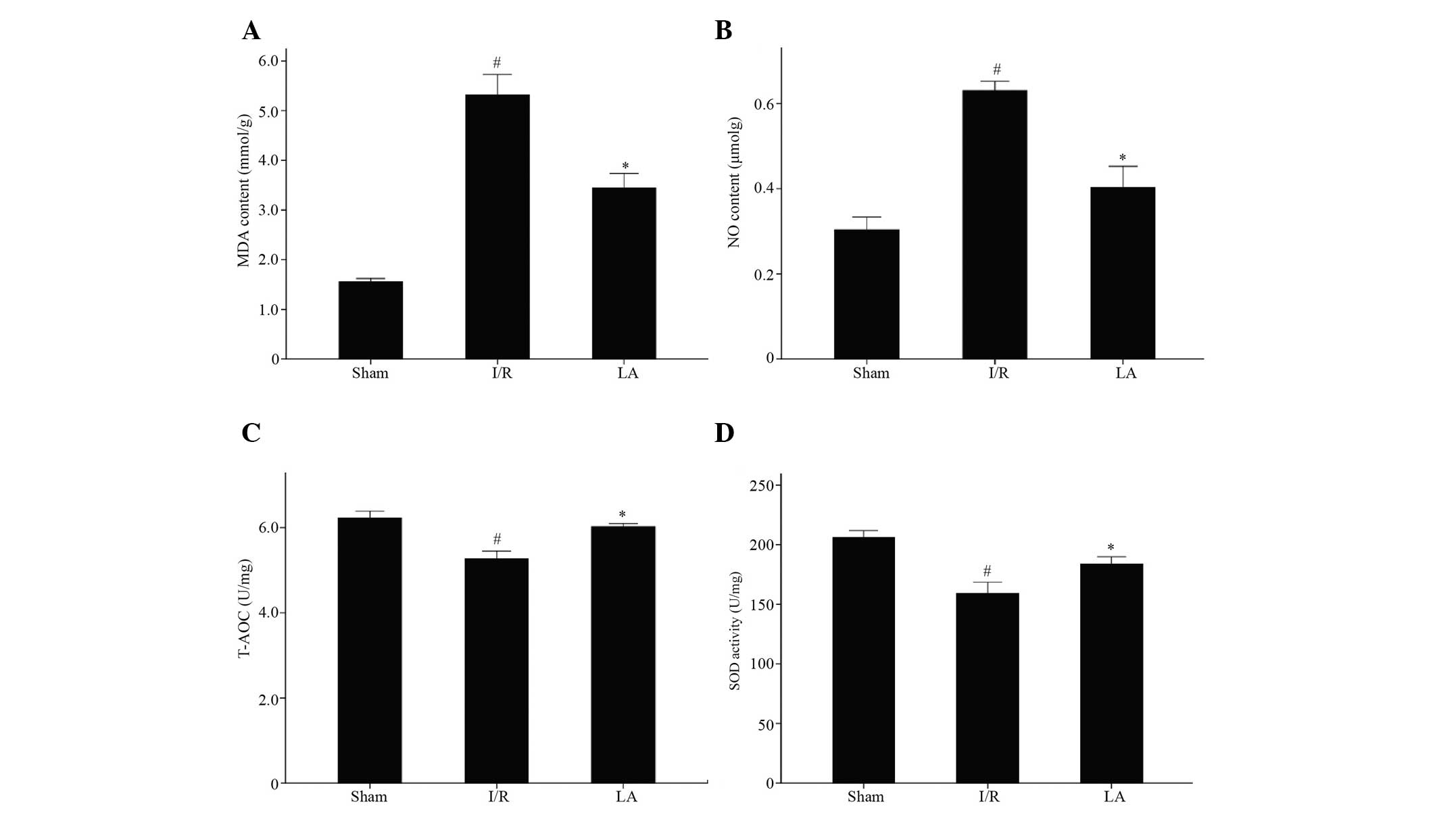 | Figure 3Effects of LA therapy on the
oxidative parameters (A) MDA, (B) NO, (C) T-AOC and (D) SOD in
ischemic rat brains after 90 min middle cerebral artery occlusion
and 24 h reperfusion. Data are presented as the mean ± standard
deviation (n=3; #P<0.05, I/R group vs. sham group;
*P<0.05, LA group vs. I/R group) LA, α-lipoic acid;
I/R, ischemia/reperfusion; MDA, malondialdehyde; NO, nitric oxide;
T-AOC, total antioxidant capacity; SOD, superoxide dismutase. |
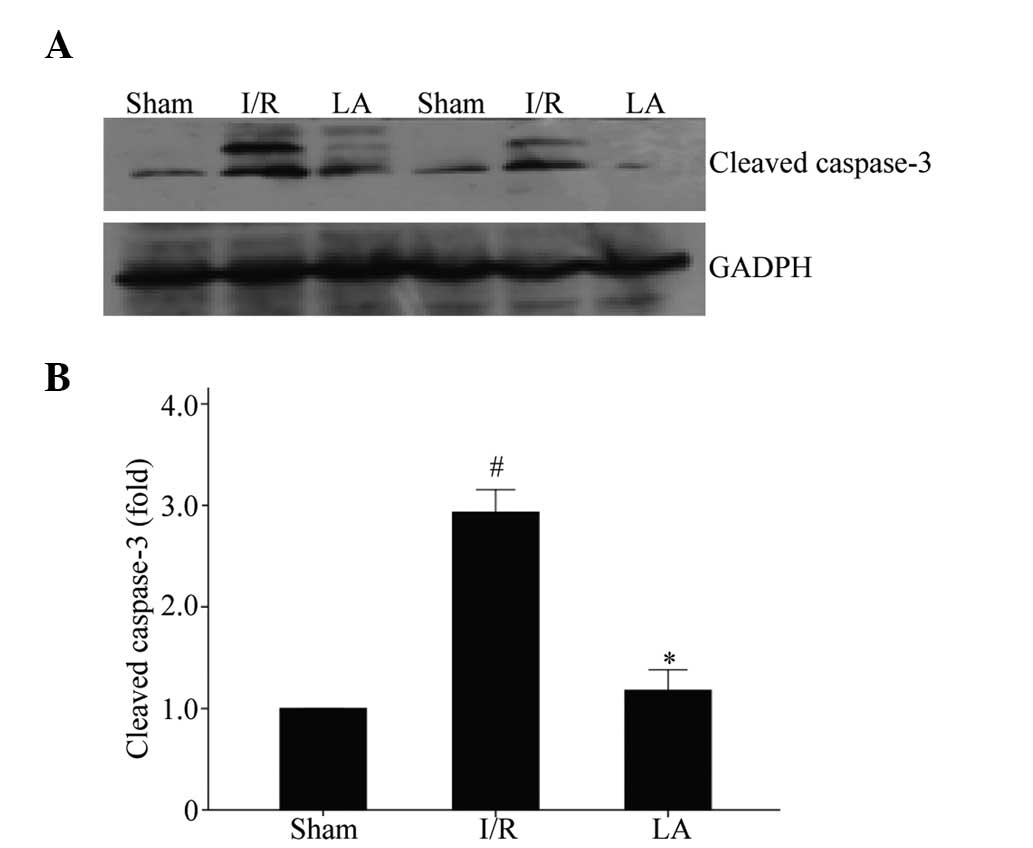
LA suppresses caspase-independent
apoptosis
Cleaved caspase-3, activated from caspase-3, has
been identified as a key mediator of apoptosis and is considered to
be one of the final steps in cell apoptosis (19,20).
As shown in Fig. 4, LA
significantly suppressed the level of cleaved caspase-3 compared
with the I/R group (P<0.05).
LA upregulates the expression of BDNF,
PI3K, p-Akt and p-ERK1/2
The expression of BDNF, PI3K, p-Akt and p-ERK1/2
were examined using western blotting (Fig. 5A). As shown in Fig. 5B, compared with the I/R group, LA
significantly increased the level of BDNF and p-Akt (P<0.05).
The results demonstrated that the levels of PI3K and p-ERK1/2 were
significantly downregulated following cerebral I/R, however,
pretreatment with LA was able to reverse this effect and increase
the expression of these proteins (Fig.
5C–E; P<0.05).
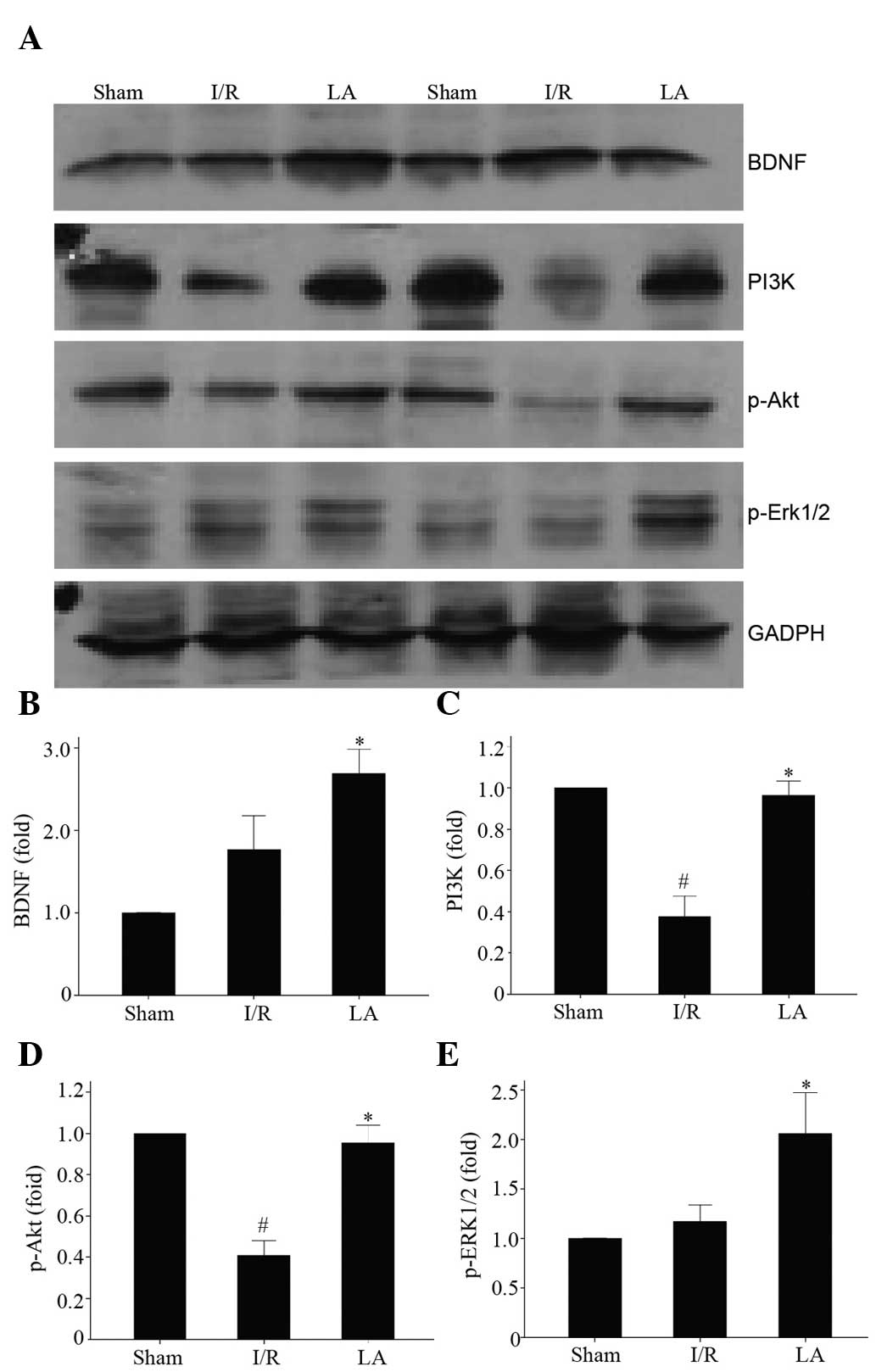 | Figure 5Effects of LA therapy on the protein
expression of BDNF, PI3K, p-Akt and p-ERK1/2 in the ischemic brain
of rats after 90 min middle cerebral artery occlusion and 24 h
reperfusion. (A) Representative images of western blotting of BDNF,
PI3K, p-Akt and p-ERK1/2. (B–E) Quantitative data are expressed as
the intensity ratio of target proteins to GADPH. GADPH was used as
a control to ensure equal protein loading. Data are presented as
the mean ± standard deviation (n=3; #P<0.05, I/R
group vs. sham group; *P<0.05, LA group vs. I/R
group). LA, α-lipoic acid; BDNF, brain-derived neurotrophic factor;
I/R, ischemia/reperfusion; PI3K,
phosphatidylinositol-4,5-bisphosphate 3-kinase; ERK1/2,
extracellular signal-regulated kinase 1/2. p-ERK1/2, phosphoylated
extracellular signal-regulated kinase 1/2. |
Discussion
The present study demonstrated that pretreatment
with LA has protective effects against neuronal injury caused by
cerebral I/R in rats. Therefore, as a preventive agent, LA is
beneficial to cerebral I/R-induced injury. This finding is
consistent with previous studies on the beneficial effects of LA
against cerebral ischemia (21,22).
The results demonstrated that the optimal dose of LA
was 100 mg/kg. This was not in accordance with a previous study in
which only 5 mg/kg LA had the capacity to protect against cerebral
I/R-induced injury (22). This
difference may be due to the fact that the time period of cerebral
I/R is longer in the present study (30 min of ischemia followed by
5.5 h of reperfusion vs. 90 min of ischemia followed by 24 h of
reperfusion), leading to a more severe neuronal injury. Therefore,
a higher dose of LA (100 mg/kg) is required to protect against
cerebral I/R-induced injury.
Oxidative stress, is the main pathological process
of cerebral I/R injury due to the balance between the formation and
elimination of ROS, including superoxide, hydrogen peroxide and
peroxynitrite (23). The increased
concentration of ROS can cause cellular damage and subsequent cell
death since ROS may oxidize crucial cellular components, including
lipids, proteins and DNA (24).
The levels of MDA and NO increased indicating that severe oxidative
damage was caused by I/R (25). It
has been reported that LA has the capability to scavenge MDA and NO
within brain tissue (26). In the
present study, pretreatment with LA decreased the levels of MDA and
NO in rat brains. The endogenous antioxidant enzymes, including SOD
and glutathione (GSH) peroxidase and low-molecular weight ROS
scavengers, including GSH, are critical in attenuating the injury
induced by I/R (25,27). In the present study, LA enhanced
the activities of T-AOC and SOD in rat brains. These data provided
further evidence that LA could inhibit I/R-induced oxidative
stress, which may contribute to the attenuation of I/R-induced
injury.
Apoptosis has a critical pathogenic role in I/R
injury. Cleaved caspase-3, which is activated from caspase-3, is an
important molecule involved in apoptosis in I/R injury (28). It is reported that activation of
caspase-3 at the final execution phase of apoptosis leads to DNA
fragmentation and cell death (29). It was observed that the level of
cleaved caspase-3 markedly increased in the brain following
cerebral I/R indicating extensive apoptosis caused by I/R. The
present data indicated that pretreatment with LA reversed the
increased level of cleaved caspase-3. The result suggested that LA
had anti-apoptotic capacity, which was beneficial to cerebral
I/R-induced injury.
BDNF is a member of the neurotrophin family and is
important in neuronal survival, differentiation, axon growth,
dendritic spine development and synaptic plasticity. When a neuron
is damaged, BDNF performs a variety of biological effects,
including preventing cell death in damaged neurons, improving the
pathological state of neurons and promoting the regeneration of
damaged neurons (30). In the case
of cerebral I/R, BDNF may protect neurons (31–33).
BDNF can activate the TrkB receptor, leading to the activation of
several intracellular signaling pathways, including PI3K/Akt and
RAS/ERK pathways (34). The
PI3K/Akt and ERK1/2 signaling pathways are important in regulating
cell growth, proliferation and apoptosis (35–37).
Numerous studies have reported that the activation of the PI3K/Akt
and ERK1/2 pathways are markedly associated with protection from
cerebral I/R injury (38,39). Akt, which is a direct downstream
target of PI3K when phosphorylated can suppress apoptosis by
activation of anti-apoptotic substrates, including B-cell lymphoma
2 family members and inhibition of pro-apoptotic substrates,
including cytochrome c (40). When
cells suffer ischemic insults, the phosphorylation of Akt may have
a protective effect (41). ERK1/2
is a member of the mitogen-activated protein kinase superfamily
that can mediate cell proliferation and apoptosis (42). The activation of ERK1/2 and
subsequent downstream signaling targets, is important in
ROS-mediated cell death (43). In
the present study, it was demonstrated that LA significantly
increased the level of BDNF. The results revealed that expression
of PI3K and p-Akt was significantly downregulated in brains
following cerebral I/R, while LA could reverse this situation and
increase the levels of PI3K and p-Akt. In addition, the
phosphorylation of ERK1/2 increased with administration of LA. The
results suggested that LA pretreatment provided protective effects
against cerebral I/R-induced injury possibly by promoting the
BDNF-PI3K/Akt-ERK1/2 signaling pathway.
In conclusion, the present study demonstrated that
LA could afford protection against cerebral I/R-induced injury by
attenuation of oxidative stress and caspase-dependent apoptosis.
Furthermore, the results suggest that administration of LA induces
a neuroprotective effect in association with the activation of the
BDNF-PI3K/Akt-ERK1/2 signaling pathway.
Acknowledgements
This study was supported by the National Science
Foundation of China (grant no. 81370449), the Industry, Education
and Research of Guangdong Province (grant no. 2011B090400015) and
the Science and Technology Development project of Guangzhou (grant
no. 2010UI-E00531-7).
References
|
1
|
Tu Q, Wang R, Ding B, Zhong W and Cao H:
Protective and antioxidant effect of Danshen polysaccharides on
cerebral ischemia/reperfusion injury in rats. Int J Biol Macromol.
60:268–271. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sahota P and Savitz SI: Investigational
therapies for ischemic stroke: neuroprotection and neurorecovery.
Neurotherapeutics. 8:434–451. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mattson MP, Duan W, Pedersen WA and
Culmsee C: Neurodegenerative disorders and ischemic brain diseases.
Apoptosis. 6:69–81. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhao ZQ: Oxidative stress-elicited
myocardial apoptosis during reperfusion. Curr Opin Pharmacol.
4:159–165. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Perham RN: Swinging arms and swinging
domains in multifunctional enzymes: catalytic machines for
multistep reactions. Annu Rev Biochem. 69:961–1004. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Smith AR, Shenvi SV, Widlansky M, Suh JH
and Hagen TM: Lipoic acid as a potential therapy for chronic
diseases associated with oxidative stress. Curr Med Chem.
11:1135–1146. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang X, Yu Y, Ji L, Liang X, Zhang T and
Hai CX: Alpha-lipoic acid protects against myocardial
ischemia/reperfusion injury via multiple target effects. Food Chem
Toxicol. 49:2750–2757. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mitsui Y, Schmelzer JD, Zollman PJ, Mitsui
M, Tritschler HJ and Low PA: Alpha-lipoic acid provides
neuroprotection from ischemia-reperfusion injury of peripheral
nerve. J Neurol Sci. 163:11–16. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ozbal S, Ergur BU, Erbil G, Tekmen I,
Bagrıyanık A and Cavdar Z: The effects of α-lipoic acid against
testicular ischemia-reperfusion injury in rats.
ScientificWorldJournal. 2012:4892482012. View Article : Google Scholar
|
|
10
|
Chidlow G, Schmidt KG, Wood JP, Melena J
and Osborne NN: Alpha-lipoic acid protects the retina against
ischemia-reperfusion. Neuropharmacology. 43:1015–1025. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Deng C, Sun Z, Tong G, et al: α-Lipoic
acid reduces infarct size and preserves cardiac function in rat
myocardial ischemia/reperfusion injury through activation of
PI3K/Akt/Nrf2 pathway. PLoS One. 8:e583712013. View Article : Google Scholar
|
|
12
|
Oh SK, Yun KH, Yoo NJ, et al:
Cardioprotective effects of alpha-lipoic acid on myocardial
reperfusion injury: suppression of reactive oxygen species
generation and activation of mitogen-activated protein kinase.
Korean Circ J. 39:359–366. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xie R, Li X, Ling Y, et al: Alpha-lipoic
acid pre- and post-treatments provide protection against in vitro
ischemia-reperfusion injury in cerebral endothelial cells via
Akt/mTOR signaling. Brain Res. 1482:81–90. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cameron NE, Cotter MA, Horrobin DH and
Tritschler HJ: Effects of alpha-lipoic acid on neurovascular
function in diabetic rats: interaction with essential fatty acids.
Diabetologia. 41:390–399. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wolz P and Krieglstein J: Neuroprotective
effects of alpha-lipoic acid and its enantiomers demonstrated in
rodent models of focal cerebral ischemia. Neuropharmacology.
35:369–375. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zuo XL, Wu P and Ji AM: Nylon filament
coated with paraffin for intraluminal permanent middle cerebral
artery occlusion in rats. Neurosci Lett. 519:42–46. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lan R, Xiang J, Wang GH, et al:
Xiao-Xu-Ming decoction protects against blood-brain barrier
disruption and neurological injury induced by cerebral ischemia and
reperfusion in rats. Evid Based Complement Alternat Med.
2013:6297822013.PubMed/NCBI
|
|
19
|
Namura S, Zhu J, Fink K, et al: Activation
and cleavage of caspase-3 in apoptosis induced by experimental
cerebral ischemia. J Neurosci. 18:3659–3668. 1998.PubMed/NCBI
|
|
20
|
D’Amelio M, Cavallucci V and Cecconi F:
Neuronal caspase-3 signaling: not only cell death. Cell Death
Differ. 17:1104–1114. 2010. View Article : Google Scholar
|
|
21
|
Clark WM, Rinker LG, Lessov NS, Lowery SL
and Cipolla MJ: Efficacy of antioxidant therapies in transient
focal ischemia in mice. Stroke. 32:1000–1004. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Connell BJ, Saleh M, Khan BV and Saleh TM:
Lipoic acid protects against reperfusion injury in the early stages
of cerebral ischemia. Brain Res. 1375:128–136. 2011. View Article : Google Scholar
|
|
23
|
Yabuki Y and Fukunaga K: Oral
administration of glutathione improves memory deficits following
transient brain ischemia by reducing brain oxidative stress.
Neuroscience. 250:394–407. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou XQ, Zeng XN, Kong H and Sun XL:
Neuroprotective effects of berberine on stroke models in vitro and
in vivo. Neurosci Lett. 447:31–36. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang PR, Wang JS, Zhang C, Song XF, Tian N
and Kong LY: Huang-Lian-Jie-Du-Decotion induced protective
autophagy against the injury of cerebral ischemia/reperfusion via
MAPK-mTOR signaling pathway. J Ethnopharmacol. 149:270–280. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Packer L, Tritschler HJ and Wessel K:
Neuroprotection by the metabolic antioxidant alpha-lipoic acid.
Free Radic Biol Med. 22:359–378. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang G, Chan PH, Chen J, et al: Human
copper-zinc superoxide dismutase transgenic mice are highly
resistant to reperfusion injury after focal cerebral ischemia.
Stroke. 25:165–170. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Haylor JL, Harris KP, Nicholson ML, Waller
HL, Huang Q and Yang B: Atorvastatin improving renal ischemia
reperfusion injury via direct inhibition of active caspase-3 in
rats. Exp Biol Med (Maywood). 236:755–763. 2011. View Article : Google Scholar
|
|
29
|
Thornberry NA and Lazebnik Y: Caspases:
enemies within. Science. 281:1312–1316. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Binder DK and Scharfman HE: Brain-derived
neurotrophic factor. Growth Factors. 22:123–131. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ferrer I, Krupinski J, Goutan E, Marti E,
Ambrosio S and Arenas E: Brain-derived neurotrophic factor reduces
cortical cell death by ischemia after middle cerebral artery
occlusion in the rat. Acta Neuropathol. 101:229–238.
2001.PubMed/NCBI
|
|
32
|
Muller HD, Hanumanthiah KM, Diederich K,
Schwab S, Schabitz WR and Sommer C: Brain-derived neurotrophic
factor but not forced arm use improves long-term outcome after
photothrombotic stroke and transiently upregulates binding
densities of excitatory glutamate receptors in the rat brain.
Stroke. 39:1012–1021. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ploughman M, Windle V, MacLellan CL, White
N, Doré JJ and Corbett D: Brain-derived neurotrophic factor
contributes to recovery of skilled reaching after focal ischemia in
rats. Stroke. 40:1490–1495. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Patapoutian A and Reichardt LF: Trk
receptors: mediators of neurotrophin action. Curr Opin Neurobiol.
11:272–280. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Burgering BM and Coffer PJ: Protein kinase
B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction.
Nature. 376:599–602. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Franke TF, Yang SI, Chan TO, et al: The
protein kinase encoded by the Akt proto-oncogene is a target of the
PDGF-activated phosphatidylinositol 3-kinase. Cell. 81:727–736.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Arslan F, Lai RC, Smeets MB, et al:
Mesenchymal stem cell-derived exosomes increase ATP levels,
decrease oxidative stress and activate PI3K/Akt pathway to enhance
myocardial viability and prevent adverse remodeling after
myocardial ischemia/reperfusion injury. Stem Cell Res. 10:301–312.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhou L and Miller CA: Mitogen-activated
protein kinase signaling, oxygen sensors and hypoxic induction of
neurogenesis. Neurodegener Dis. 3:50–55. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Franke TF, Hornik CP, Segev L, Shostak GA
and Sugimoto C: PI3K/Akt and apoptosis: size matters. Oncogene.
22:8983–8998. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu H, Liu X, Wei X, et al: Losartan, an
angiotensin II type 1 receptor blocker, ameliorates cerebral
ischemia-reperfusion injury via PI3K/Akt-mediated eNOS
phosphorylation. Brain Res Bull. 89:65–70. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mebratu Y and Tesfaigzi Y: How ERK1/2
activation controls cell proliferation and cell death: Is
subcellular localization the answer? Cell Cycle. 8:1168–1175. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dong J, Ramachandiran S, Tikoo K, Jia Z,
Lau SS and Monks TJ: EGFR-independent activation of p38 MAPK and
EGFR-dependent activation of ERK1/2 are required for ROS-induced
renal cell death. Am J Physiol Renal Physiol. 287:F1049–F1058.
2004. View Article : Google Scholar : PubMed/NCBI
|