Introduction
Atherosclerosis is the primary cause of heart
disease and stroke, and it is also one of the main causes of death
and disability around the world (1,2). The
phenotype transformation of vascular smooth muscle cell (VSMC)
plays an important role in the pathology process of
atherosclerosis. In the atherosclerotic lesion, there is a large
amount of the synthetic phenotype of VSMC in the walls of blood
vessel. These synthetic phenotype VSMC tends to form foam-like
cells and accelerate lesion progression (3,4).
First, these cells secrete a variety of proteins (MMP-2, MMP-9,
collagen type I and OPN), which increase lipid content and enhance
the accumulation of monocytes and macrophages, second, they are
believed to be activated because of enhancing ability of
proliferation, immigration, and phagocytosis (3).
Galectin-3 (gal-3), a galactoside-binding protein,
can be widely expressed by different kind of cells. Oxidized
low-density lipoprotein (oxLDL) can also promote the expression of
gal-3 in macrophages (5). In human
umbilical vein endothelial cells (HUVEC), gal-3 plays an important
role in vascular endothelial growth factor (VEGF)-and basic
fibroblast growth factor (bFGF)-mediated angiogenesis (6). More importantly, Gal-3 is an
important player in Aldo-induced vascular inflammation and mediates
aldo-induced vascular fibrosis, silencing gal-3 blocks aldo-induced
collagen type I deposition both in vivo and in vitro
(7).
Gal-3 can regulate Wnt signaling (8). Gal-3 is a key regulator in the
Wnt/β-catenin signaling pathway and interact with axin-2, GSK-3β
and β-catenin immediately (9–11).
Furthermore, gal-3 can in direct contact with β-catenin to
stimulate cyclin D1 and c-myc expression (9). Tatsuo even find that human gal-3
sequence has a structural similarity to β-catenin in the breast
cancer cells (10).
Recently, the role of gal-3 in cardiovascular
disease has also been widely studied by some researchers (12–17).
In the setting of atherosclerotic disease, gal-3 seems to promote
atherogenesis. Gal-3 is an useful biomarker which can predict the
subsequent infarction after first myocardial infarction (MI)
(18). Furthermore, in the absence
of gal-3, the development of atherosclerotic pathological change in
the ApoE-deficient mouse is reduced (19). Patients with type 2 diabetes and
arterial hypertension have higher levels of gal-3 in plasma
(20). Gal-3 has been proved to be
a promising biomarker for detecting prediabetes and diabetes
(21). Until now the mechanism of
gal-3 in atherogenesis is still not clear yet, we presumed that
gal-3 was a high risk factors of atherosclerosis and could promote
pathological process of atherosclerosis. Considering the importance
of phenotype transformation of SMC, does gal-3 induce
atherosclerosis development by modulate cellular phenotype? If it
does, what is the mechanism involved?
Thus, in this study, we tried to investigate the
effects of exogenous gal-3 inHUSMCs. Furthermore, we examined
whether β-catenin, a reported signaling associated with phenotype
transformation of vascular smooth muscle cells, is involved in this
gal-3 activity.
Materials and methods
Reagents
The Dulbecco's modified Eagle's medium (DMEM), fetal
bovine serum (FBS), and penicillin/streptomycin (pen/strep, 10,000
U/ml each) were purchased from Gibco (Carlsbad, CA, USA). oxLDL was
obtained from Peking Union-Biology Co., Ltd. (Beijing China).
TRIzol reagent for RNA isolation was purchased from Invitrogen
(Carlsbad, CA, USA). XAV939 was purchased from Sigma-Aldrich (St.
Louis, MO, USA). The reverse transcriptase kit (RR037A) and SYBR
Premix Ex Taq (DRR420S) were purchased from Takara (Dalian China).
The Cell Counting Kit-8 (CCK-8) assay kit was purchased from
Dojindo (Kumamoto, Japan). Recombinant human gal-3 was purchased
from PeproTech (Rocky Hill, NJ, USA). The primary antibody against
gal-3 (no. sc-20157), SMA (no. sc-32251), cyclin D1 (no. sc-8396)
and GAPDH (no. sc-48166) were obtained from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). Anti-phospho GSK3β (Ser9)
(no. CST-9322), anti-GSK3β (no. CST-9315), anti-nonphospho (active)
β-catenin (no. CST-4270), and anti-β-catenin (no. CST-9582) were
acquired from Cell Signalling Technology, Inc. (Danvers, MA, USA).
The antibody against osteopontin (OPN; no. ab91655), calponin (no.
ab46794) and axin2 (no. ab109307) were obtained from Abcam
(Cambridge, UK). The goat anti-rabbit secondary antibody (no.
A-21109) and the goat anti-mouse secondary antibody (no. A-21058)
were provided by Invitrogen. All other chemicals were from
commercial sources.
Cells culture
A primary culture of human SMCs was established by
explant outgrowth of a segment of human umbilical cord retrieved at
the time of caesarean section (22). Endothelial cells were removed by
scraping the luminal surface of the vessel with a cotton swab, and
the adventitia was mechanically stripped away. Primary cultures
were maintained in DMEM supplemented with 20% FBS and 1%
antibiotics (penicillin/streptomycin) (all from Gibco). Cells
between 4th and 10th passages were used in these experiments. The
trail confirmed with the principle of the Declaration of Helsinki
and was approved by the Ethics Committee of the Shanghai Ninth
Hospital.
siRNA interference
β-catenin expression was inhibited by transfection
with a siRNA specific to β-catenin. β-catenin siRNA was transiently
transfected into the cells using Lipofectamine® 2000
(Invitrogen Life Technologies), according to the manufacturer's
instructions. Briefly, 5×105 HUSMCs per well were
cultured in 6-well plates to 75% confluence. The cells were then
transfected with 100 pmol siRNA duplexes using 5 µl
Lipofectamine® 2000 and 500 µl DMEM (reduced serum
medium). The process of transfected was without antibiotics.
Following a 72-h incubation at 37°C, the cells were harvested for
analysis. The human β-catenin siRNA sequence was 5-CAT GUG UTG GUA
AGC UCUA-3 and the scrambled siRNA sequence was 5-GCA ACA GTT GCA
GAG AGGU-3. They were synthesized by Biotend (Shanghai, China).
Cell proliferation assay
Cell proliferation was measured with the CCK-8 assay
kit. In brief, 5,000 cells were plated in each well of a 96-well
plate and allowed to attach for 24 h. Then, HUSMCs were
subsequently incubated for 0, 12, 24 or 48 h in the presence of
gal-3. Subsequently, the plate was incubated with CCK-8 for 4 h at
37°C. At last, the absorbance of wave length was taken at 450
nm.
Migration assay
The assay was performed as previously described
(23). Migration assays were
performed in a chamber, HUSMCs were resuspended in 200 µl
serum-free DMEM medium and 5×104 HUSMCs were loaded into
the upper chambers. The lower chamber was filled with 400 µl of
DMEM in the presence or absence of 10 µg/ml recombinant gal-3. The
chamber was incubated at 37°C for 24 h. Then, the lower side of the
filter was washed with PBS and fixed with 4% paraformaldeyde.
Nuclei were stained with 4′,6-diamino-2-phenylindole (DAPI,
1:1,000; Sigma) for 5 min at room temperature. The cells were
counted in three random high-power fields (x100) in each well.
Real-time RT-PCR analysis
Total RNA was extracted using TRIzol reagent
(Invitrogen) according to the manufacturer's protocol. Total RNA
was reverse-transcribed into cDNA using reverse transcriptase, and
quantitative real-time PCR was performed using SYBR-Green master
mix, on an Applied Biosystems 7500 real-time PCR system, according
to the manufacturer's instructions. Specific primers for human
gal-3, β-catenin, OPN, axin2, calponin, SMA and GAPDH were as
follows: Gal-3 forward, 5′-GGCCACTGATTGTGCCTTAT-3′ and reverse,
5′-TGCAACCTTGAAGTGGTCAG-3′; β-catenin forward,
5′-GCCGGCTATTGTAGAAGCTG-3′ and reverse, 5′-GAGTCCCAAGGAGACCTTCC-3′;
OPN forward, 5′-TGAGTCTGGAAATAACTAATGTGTTTGA-3′ and reverse,
5′-GAACATAGACATAACCCTGAAGCTTTT-3′; axin2 forward,
5′-CTCTCTACCTCATTTCCCGAGAAC-3′ and reverse,
5′-CGAGATCAGCTCAGCTGCAA-3′; calponin forward,
5′-ATGTGAGGAGGGAAGAGTGTG-3′ and reverse, 5′-CGGTTGAAGTGAGCAGAGG-3′;
SMA forward, 5′-AGCGTGGCTACTCCTTCGTGAC-3′ and reverse,
5′-GCTCGTTGCCGATGGTGATGAC-3′; cyclin D1 forward,
5′-AATGACCCCGCACGATTTC-3′ and reverse, 5′-TCAGGTTCAGGCCTTGCAC-3′;
GAPDH forward, 5′-TGATGACATCAAGAAGGTGGTGAAG-3′ and reverse,
5′-TCCTTGGAGGCCATGTGGGCCAT-3′.
Western blot analysis
Cells were lysed in a lysis buffer containing 150 mM
of NaCl, 10 mM of Tris (pH 7.5), 5 mM of EDTA, 1% Triton X-100, 1
mM of PMSF, 10 mg/ml of leupeptin, 10 mg/ml of pepstatin, and 10
mg/ml of aprotinin for 30 min on ice. The nuclear and cytoplastic
protein were separately extracted by using the kit (P 0028;
Beyotime Institute of Biotechnology (Shanghai, China). Protein
concentrations were measured with the BCA Protein Assay (Pierce
Biotechnology Inc., Rockford, IL, USA). The lysates (20 µg) were
electrophoresed on 10% SDS-PAGE and transferred to nitrocellulose
membranes (Merck Millipore, Danvers, MA, USA). The membrane was
blocked with 5% nonfat dry milk in TBST buffer (100 mM NaCl, 10 mM
Tris-HCl, pH 7.4, and 0.1% Tween-20) for 1 h at room temperature.
The blots were then incubated with various 1000-fold diluted
primary antibodies at a dilution of 1:1,000 in TBST at 4°C
overnight, and then washed twice with TBST buffer at room
temperature and incubated for 1 h with the appropriate
peroxidase-conjugated secondary antibody (1:5,000 dilution). All
signals were detected by Odyssey (LI-COR Biosciences, Lincoln, NE,
USA). To quantity the protein, band intensity was assessed by
Quantity One 4.6.2 software.
Statistical analysis
All data are expressed as the mean ± SD. Statistics
were performed using the SPSS 13.0 software (SPSS, Inc., Chicago,
IL, USA). One-way ANOVA followed by the Student-Newman-Keuls post
hoc analyses were used. For data that not passed the normality
test; non-paramatric ANOVA (Kruskal-Wallis test) was used. A value
of P<0.05 was considered statistically significant. All
experiments were performed at least three times.
Results
Exogenous galectin-3 promotes HUSMCs
proliferation and migration
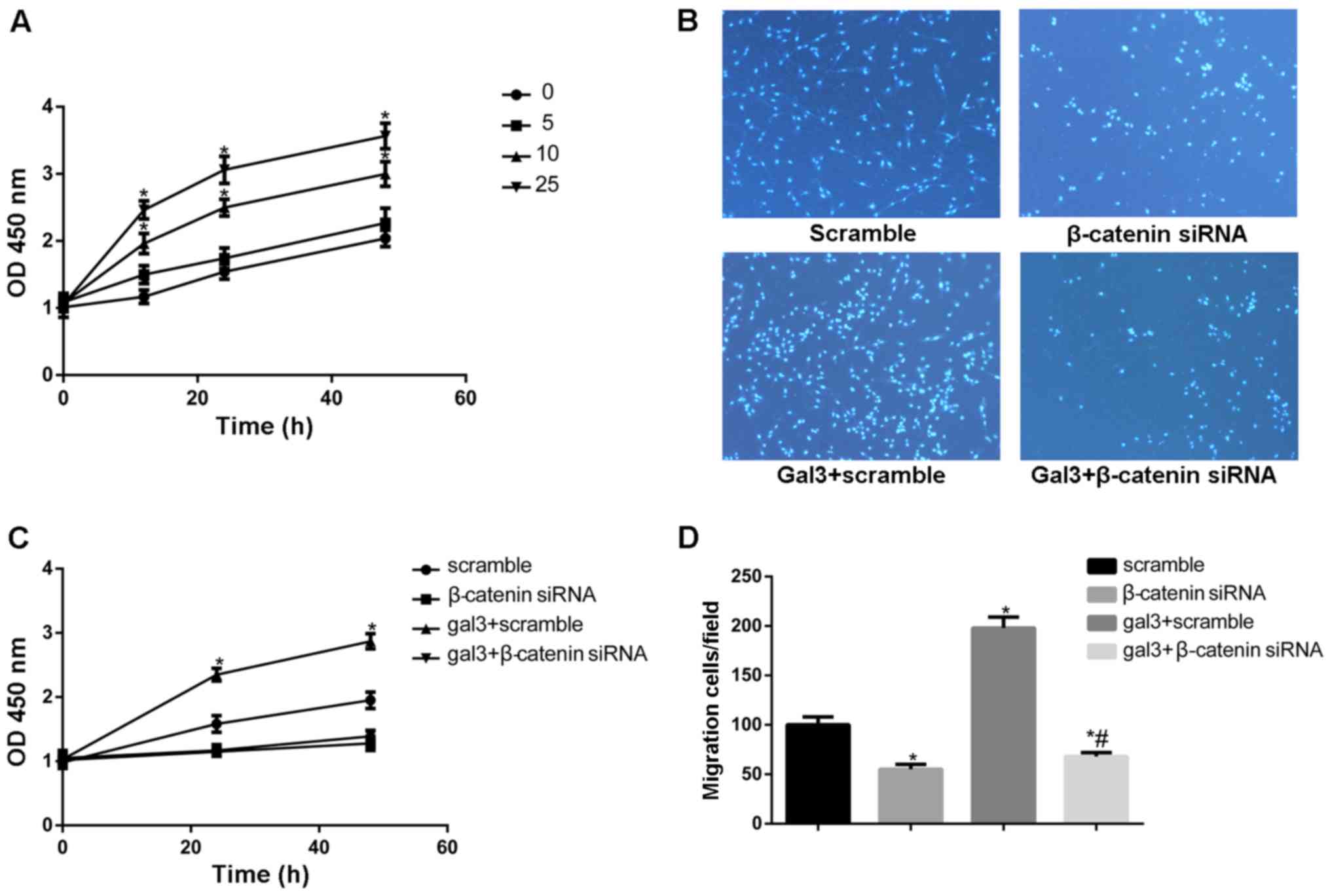
First, we used increasing concentrations of gal-3
(up to 25 µg/ml) to stimulate cells. Cell proliferation was
determined at 12, 24, and 48 h using an CCK-8 assay. As shown in
Fig. 1A, the proliferation of
HUSMCs was significantly increased in a concentration dependent
manner. The Transwell migration assay was used to investigate the
effect of recombined gal-3 on cell migration. As shown in Fig. 1B and C, exogenous gal-3 enhanced
HUSMCs migration significantly.
Galectin-3 promotes the phenotype
transformation of vascular smooth muscle cells
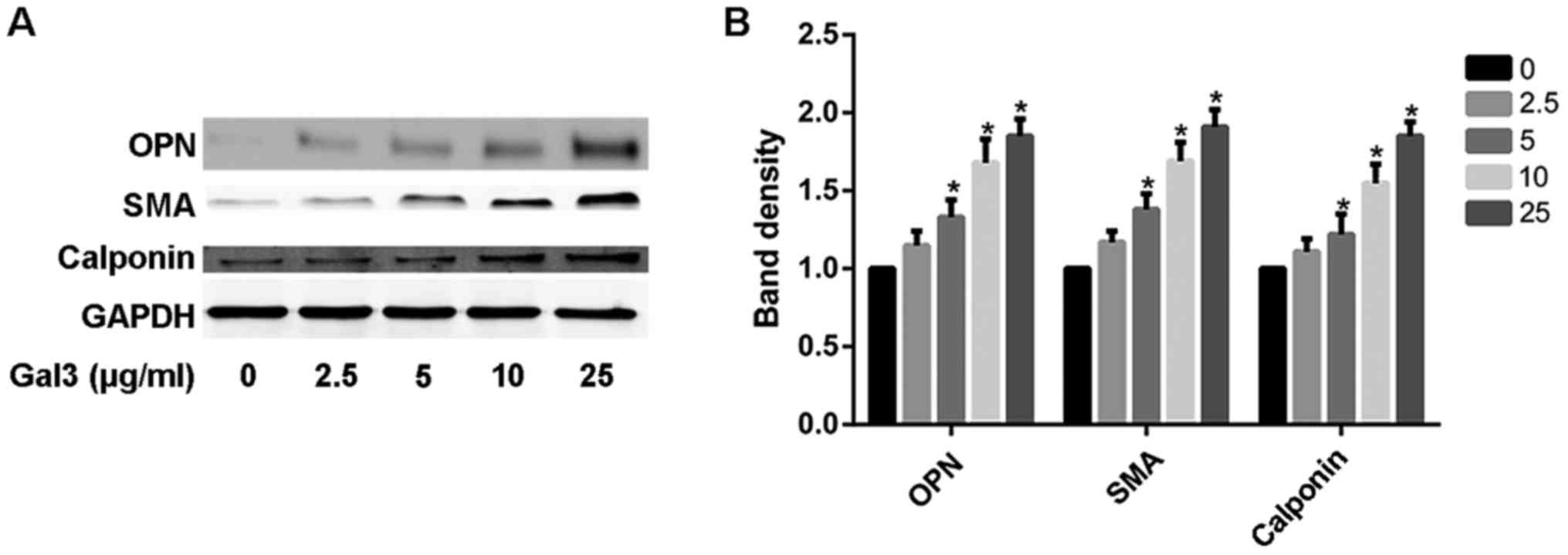
We also investigated the role of exogenous gal-3 in
the phenotype transformation of HUSMC. First of all, the HUSMCs
were cultured in the DMEM without serum for 12 h. After that, the
serum-starved HUSMCs were treated with gal-3 for 48 h. As shown in
the Fig. 2, the expression of the
smooth muscle synthetic proteins, OPN rose significantly over a
range of gal-3 concentration (0–25 µg/ml). Concomitant with the
increased OPN expression, the protein markers of contractile
phenotype were also increased in gal-3-treated cells, as measured
by SMA and calponin expression.
Galectin-3 activates canonical Wnt
signaling in human umbilical vascular smooth muscle cells
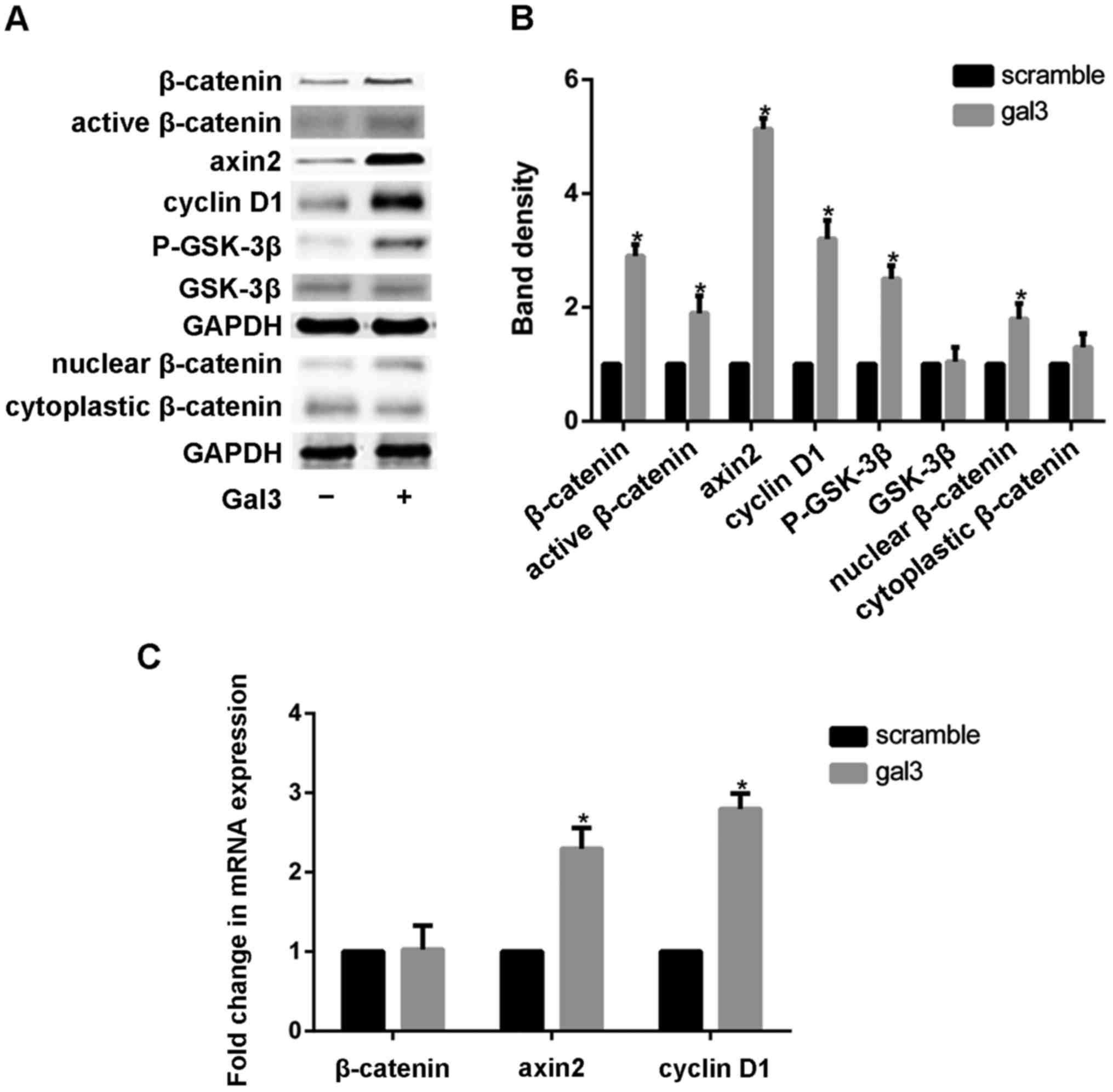
Canonical Wnt signal activation could be evaluated
by recording β-catenin protein levels (24). The expression of β-catenin was
tested by western blotting after cells were treated with gal-3. The
distribution of β-catenin in cytoplasm and nucleu were examined by
western blot analysis seperately. Gal3 obviousely increased the
expression of β-catenin in nucleu, but it has relatively little
effect on the cytoplasmic β-catenin. To further confirm the
activation of canonical Wnt signaling by gal-3, we also detected
the expression of axin2 (regulating β-catenin stability) (25) and cyclin D1 (β-catenin target gene)
by using realtime-PCR and western blotting. Our results showed that
recombinant gal-3 significantly increased expression of axin2 and
cyclin D1 (Fig. 3). Gal-3 induced
a marked increase in the protein expression of nonphosphorylated
(active) and total β-catenin However, the mRNA level of β-catenin
was almost unchanged in the gal-3-treated cells (Fig. 3C). In order to confirm that
β-catenin expression was modulated by gal-3 at the
post-translational level, we also detected the activity of GSK-3β
which promotes the degradation of β-catenin. Phosphorylation GSK-3β
was 2.5-fold increased by gal-3, while total GSK-3β were almostly
unchanged.
Canonical Wnt/β-catenin signaling
mediates the effects of galectin-3 in HUSMCs
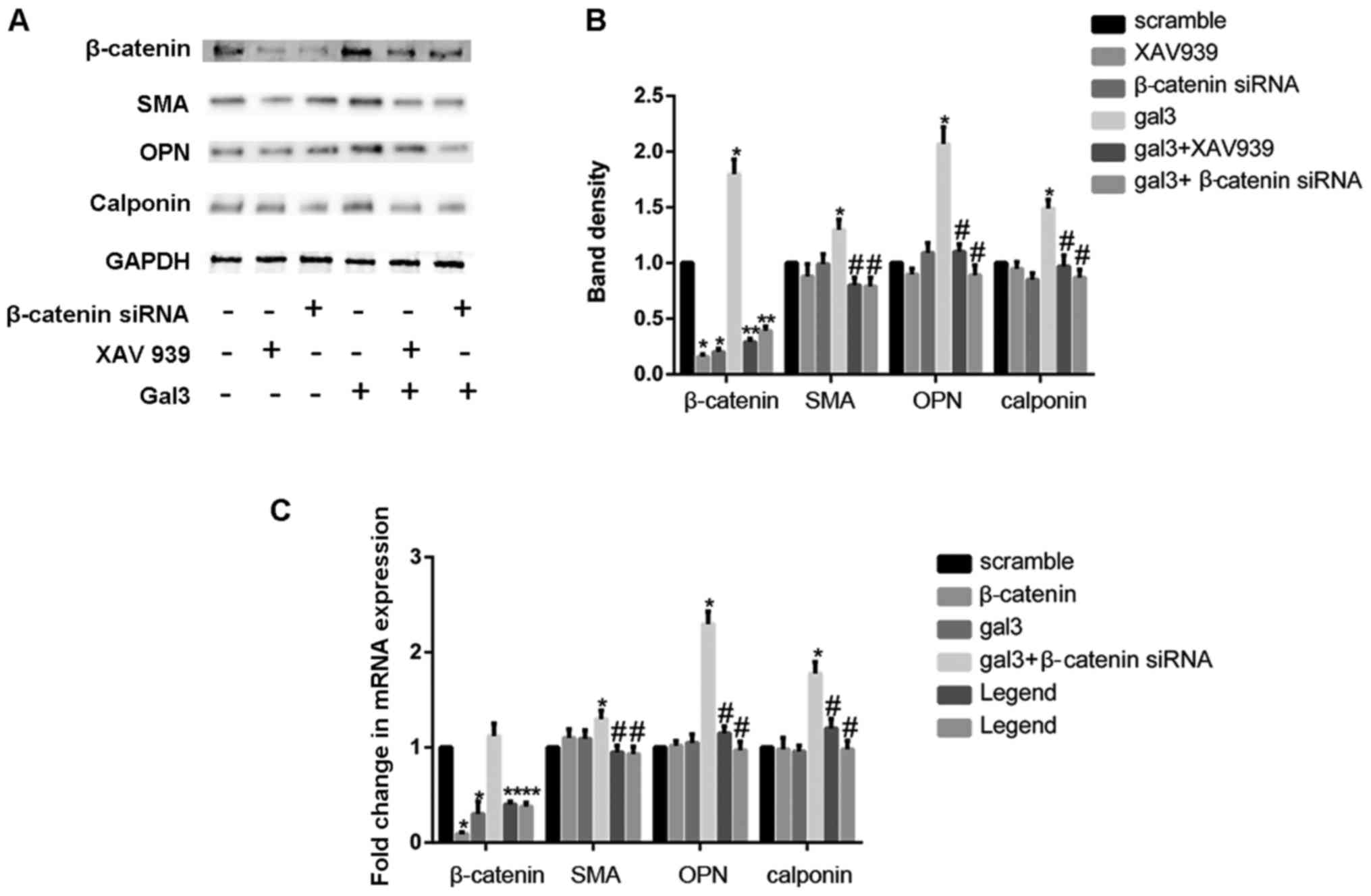
To further evaluate the role of β-catenin in
gal-3-mediated change of HUSMC, the expression of β-catenin was
inhibited by siRNA strategy in HUSMCs. The siRNA of β-catenin
reduced the expression of β-catenin mRNA and protein by 91 and 83%,
respectively. These results indicated that it effectively
antagonized the activation of Wnt/β-catenin signaling pathway. We
then preformed cell proliferation and migration assays on these
cells, the silencing of β-catenin decreased the proliferation and
migration of HUSMCs (Fig. 1B and
D). Furthermore, the increased ability of migration and
proliferation in gal-3-treated cells were also eliminated after
silencing of β-catenin. We then detected the role of β-catenin in
the phenotypic switch induced by gal-3. β-catenin knockdown
efficiently blocked the increase expression of OPN, SMA and
calponin induced by gal-3 in both mRNA and protein levels, as
demonstrated by western blot and realtime PCR. In order to further
confirm the role the Wnt signaling pathway in gal-3 induced
phenotype transformation of VSMC, XAV939, a inhibitor of Wnt
signaling pathway, was also used. XAV939 similarly attenuated the
effect of gal-3 in HUSMCs (Fig.
4A-C).
Discussion
In this study, we demonstrate, for the first time,
that gal-3 can activate the canonical Wnt signaling pathway in
HUSMCs. More importantly, we find that gal-3 promote the phenotype
transformation of HUSMCs, including increasing proliferation and
migration and upregulating a series of phenotype related proteins.
While using the strategy of β-catenin siRNA can obviousely block
the gal-3 induces phenotype transformation of HUSMCs. Taken all
these data together, we conclude that gal-3 promotes the activation
and phenotype transformation of HUSMCs through canonical
Wnt/β-catenin signaling.
Gal-3 has been reported to affect the function of
almost all the cells in the wall of blood vessel. In human
umbilical vein endothelial cells (HUVEC), gal-3 is a mediator of
vascular endothelial growth factor (VEGF)- and basic fibroblast
growth factor (bFGF)-mediated angiogenesis (6,26).
Gal-3 can also induce the migration of macrophage in mouse
(27). Atherosclerosis is
characterized by the activation and accumulation of smooth muscle
cells in the intimal layer of blood vessels where they internalized
a lot of lipids (28). The
enhanced proliferation and migration of smooth muscle cells are the
symbol of the early pathology of atherosclerosis. In this study, we
also showed that exogenous gal-3 can obviously promote the
proliferation and migration of HUSMCs. In order to further explore
the role of gal-3 in the HUSMCs, we also detected the expression of
some widely used SMC marker proteins.
Traditionally, smooth muscle cells are thought of
being exist in two totally different phenotypes: Contractile and
synthetic phenotype. It is now being recognized that there are a
diversity of SMC phenotypes, ranging from contractile to synthetic
(4,29,30).
In fact, these so-called contractile and synthetic phenotypes
should be envisaged as ‘idealized’ phenotypes. Higher growth rates
and stronger migratory activity are usually considered typical of
the ‘synthetic’ phenotype, and OPN is also identified as a
synthetic-related protein (3).
Actually, recent studies proved that different synthetic and
contractile markers could be upregulated at the same time (4,26).
In some instances, contractile differentiation can be up-regulated
in the ‘synthetic’ phenotype and contractile differentiation
markers may express with matrix synthesis (28,31).
In our study, we found that gal-3 stimulated the expression of OPN.
Interesting, at the same time, gal-3 also increased the
contractile-related protein, calponin and SMA. Therefore, we
believe that galectin-3 can promote expression of both smooth
muscle synthetic and contractile proteins.
There are two different subclasses of Wnt
signalling: The non-canonical pathway (which is β-catenin
dependent) and the canonical pathway (β-catenin independent). The
activation of non-canonical Wnt pathway can involve the
intra-cytoplasmic release of Ca2+ and the activation of
Jun N-terminal kinase (JNK). The canonical Wnt signalling pathway
involves the inhibition of β-catenin degradation complex, therefore
resulting in the translocation of β-catenin from the cytoplasm to
the nucleus, and binding to downstream target genes. Earlier
reports have shown that β-catenin can regulate the expression of
OPN, SMA and calponin expression, induce cell proliferation and
migration in many types of cells (28,32).
Recently, the role of gal-3 in vascular smooth muscle cells
osteogenic differentiation has also been confirmed (32). Thus, we explored the role of
β-catenin in gal-3-treated HUSMCs. In our study, gal-3 activated
the canonical Wnt signaling pathway by upregulating the total and
nonphosphorylated β-catenin. We also found that the expression of
β-catenin downstream gene axin2 and cyclin D1 was upregulated by
gal-3 (28,32). It further indicated that
Wnt/β-catenin signaling pathway was activated by gal-3 in the
HUSMCs. Our results showed that gal-3 induced the expression of
total β-catenin at protein level but not mRNA level. It is
consensus with previous findings. Gal-3, a binding of β-catenin,
increases phosphorylation of GSK-3β (12,33,34).
Inactivation of GSK-3β can reduce the degradation of β-catenin, and
increase cellular β-catenin levels besides, we found that gal-3
indeed increased the phosphorylated level of GSK-3β. In order to
prove the role of β-catenin in gal-3-induced phenotype
transformation of HUSMCs, we used the siRNA strategy and XAV939 to
decrease the expression of β-catenin. Our results demonstrated that
silencing of β-catenin inhibited gal-3-induced protein expression
and blocked gal-3-mediated cell proliferation and migration of
vascular smooth muscle cells. Thus, we conclude that recombinant
gal-3 induces phenotype transformation of HUSMCs via canonical
Wnt/β-catenin pathway.
In conclusion, our results show that gal-3 promotes
the activation and the phenotype transformation in HUSMCs. This
activity is through the canonical Wnt signaling pathway. Recently,
gal-3 has been proved to be related to the atherosclerosis and
coronary heart disease. As smooth muscle cells plays an important
role in a variety of plausible mechanisms of atherosclerosis and
chronic heart diseases. Our research could help to clarify the role
of gal-3 in the pathological process of atherosclerosis.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (81270376); Shanghai Hospital
Development Center (SHDC12012312); Ministry of Education Science
and Technology Development Center (20130073110016); Shanghai
Science and Technology Commission (Chinese medicine) (12401905200)
and the Fund of Ninth People's Hospital (grant no. 2013A02).
References
|
1
|
Lusis AJ: Atherosclerosis. Nature.
407:233–241. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Karagiannis GS, Weile J, Bader GD and
Minta J: Integrative pathway dissection of molecular mechanisms of
moxLDL-induced vascular smooth muscle phenotype transformation. BMC
Cardiovasc Disord. 13:42013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Doran AC, Meller N and McNamara CA: Role
of smooth muscle cells in the initiation and early progression of
atherosclerosis. Arterioscler Thromb Vasc Biol. 28:812–819. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hao H, Gabbiani G and Bochaton-Piallat ML:
Arterial smooth muscle cell heterogeneity: Implications for
atherosclerosis and restenosis development. Arterioscler Thromb
Vasc Biol. 23:1510–1520. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim K, Mayer EP and Nachtigal M:
Galectin-3 expression in macrophages is signaled by Ras/MAP kinase
pathway and up-regulated by modified lipoproteins. Biochim Biophys
Acta. 1641:13–23. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Markowska AI, Liu FT and Panjwani N:
Galectin-3 is an important mediator of VEGF- and bFGF-mediated
angiogenic response. J Exp Med. 207:1981–1993. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Calvier L, Miana M, Reboul P, Cachofeiro
V, Martinez-Martinez E, de Boer RA, Poirier F, Lacolley P, Zannad
F, Rossignol P and López-Andrés N: Galectin-3 mediates
aldosterone-induced vascular fibrosis. Arterioscler Thromb Vasc
Biol. 33:67–75. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dumic J, Dabelic S and Flögel M:
Galectin-3: An open-ended story. Biochim Biophys Acta.
1760:616–635. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shimura T, Takenaka Y, Tsutsumi S, Hogan
V, Kikuchi A and Raz A: Galectin-3, a novel binding partner of
beta-catenin. Cancer Res. 64:6363–6367. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shimura T, Takenaka Y, Fukumori T,
Tsutsumi S, Okada K, Hogan V, Kikuchi A, Kuwano H and Raz A:
Implication of galectin-3 in Wnt signaling. Cancer Res.
65:3535–3537. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song S, Mazurek N, Liu C, Sun Y, Ding QQ,
Liu K, Hung MC and Bresalier RS: Galectin-3 mediates nuclear
beta-catenin accumulation and Wnt signaling in human colon cancer
cells by regulation of glycogen synthase kinase-3beta activity.
Cancer Res. 69:1343–1349. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Francia P, Adduci C, Semprini L, Borro M,
Ricotta A, Sensini I, Santini D, Caprinozzi M, Balla C, Simmaco M
and Volpe M: Osteopontin and galectin-3 predict the risk of
ventricular tachycardia and fibrillation in heart failure patients
with implantable defibrillators. J Cardiovasc Electrophysiol.
25:609–616. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
de Boer RA, Edelmann F, Cohen-Solal A,
Mamas MA, Maisel A and Pieske B: Galectin-3 in heart failure with
preserved ejection fraction. Eur J Heart Fail. 15:1095–1101. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Felker GM, Fiuzat M, Shaw LK, Clare R,
Whellan DJ, Bettari L, Shirolkar SC, Donahue M, Kitzman DW, Zannad
F, et al: Galectin-3 in ambulatory patients with heart failure:
Results from the HF-ACTION study. Circ Heart Fail. 5:72–78. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mayr A, Klug G, Mair J, Streil K,
Harrasser B, Feistritzer HJ, Jaschke W, Schocke M, Pachinger O and
Metzler B: Galectin-3: Relation to infarct scar and left
ventricular function after myocardial infarction. Int J Cardiol.
163:335–337. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sanchez-Mas J, Lax A, Asensio-Lopez MC,
Fernandez-Del Palacio MJ, Caballero L, Garrido IP, Pastor F,
Januzzi JL and Pascual-Figal DA: Galectin-3 expression in cardiac
remodeling after myocardial infarction. Int J Cardiol.
172:e98–e101. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van Kimmenade RR, Januzzi JL Jr, Ellinor
PT, Sharma UC, Bakker JA, Low AF, Martinez A, Crijns HJ, MacRae CA,
Menheere PP and Pinto YM: Utility of amino-terminal pro-brain
natriuretic peptide, galectin-3, and apelin for the evaluation of
patients with acute heart failure. J Am Coll Cardiol. 48:1217–1224.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Szadkowska I, Wlazel RN, Migala M,
Bajon-Laskowska K, Szadkowski K, Zielińska M, Paradowski M and
Pawlicki L: The association between galectin-3 and occurrence of
reinfarction early after first myocardial infarction treated
invasively. Biomarkers. 18:655–659. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nachtigal M, Ghaffar A and Mayer EP:
Galectin-3 gene inactivation reduces atherosclerotic lesions and
adventitial inflammation in ApoE-deficient mice. Am J Pathol.
172:247–255. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Seferovic JP, Lalic NM, Floridi F, Tesic
M, Seferovic PM, Giga V, Lalic K, Jotic A, Jovicic S, Colak E, et
al: Structural myocardial alterations in diabetes and hypertension:
The role of galectin-3. Clin Chem Lab Med. 52:1499–1505.
2014.PubMed/NCBI
|
|
21
|
Yilmaz H, Cakmak M, Inan O, Darcin T and
Akcay A: Increased levels of galectin-3 were associated with
prediabetes and diabetes: New risk factor? J Endocrinol Invest.
38:527–533. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guyton JR, Lenz ML, Mathews B, Hughes H,
Karsan D, Selinger E and Smith CV: Toxicity of oxidized low density
lipoproteins for vascular smooth muscle cells and partial
protection by antioxidants. Atherosclerosis. 118:237–249. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Leavesley DI, Schwartz MA, Rosenfeld M and
Cheresh DA: Integrin beta 1- and beta 3-mediated endothelial cell
migration is triggered through distinct signaling mechanisms. J
Cell Biol. 121:163–170. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tickenbrock L, Schwäble J, Strey A, Sargin
B, Hehn S, Baas M, Choudhary C, Gerke V, Berdel WE, Müller-Tidow C
and Serve H: Wnt signaling regulates transendothelial migration of
monocytes. J Leukoc Biol. 79:1306–1313. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schaale K, Neumann J, Schneider D, Ehlers
S and Reiling N: Wnt signaling in macrophages: Augmenting and
inhibiting mycobacteria-induced inflammatory responses. Eur J Cell
Biol. 90:553–559. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nangia-Makker P, Honjo Y, Sarvis R,
Akahani S, Hogan V, Pienta KJ and Raz A: Galectin-3 induces
endothelial cell morphogenesis and angiogenesis. Am J Pathol.
156:899–909. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jia W, Kidoya H, Yamakawa D, Naito H and
Takakura N: Galectin-3 accelerates M2 macrophage infiltration and
angiogenesis in tumors. Am J Pathol. 182:1821–1831. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Carthy JM, Luo Z and McManus BM: WNT3A
induces a contractile and secretory phenotype in cultured vascular
smooth muscle cells that is associated with increased gap junction
communication. Lab Invest. 92:246–255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rensen SS, Doevendans PA and van Eys GJ:
Regulation and characteristics of vascular smooth muscle cell
phenotypic diversity. Neth Heart J. 15:100–108. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Matsushita T, Rama A, Charolidi N, Dupont
E and Severs NJ: Relationship of connexin43 expression to
phenotypic modulation in cultured human aortic smooth muscle cells.
Eur J Cell Biol. 86:617–628. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rama A, Matsushita T, Charolidi N, Rothery
S, Dupont E and Severs NJ: Up-regulation of connexin43 correlates
with increased synthetic activity and enhanced contractile
differentiation in TGF-beta-treated human aortic smooth muscle
cells. Eur J Cell Biol. 85:375–386. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Menini S, Iacobini C, Ricci C, Fantauzzi C
Blasetti, Salvi L, Pesce CM, Relucenti M, Familiari G, Taurino M
and Pugliese G: The galectin-3/RAGE dyad modulates vascular
osteogenesis in atherosclerosis. Cardiovasc Res. 100:472–480. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kobayashi T, Shimura T, Yajima T, Kubo N,
Araki K, Tsutsumi S, Suzuki H, Kuwano H and Raz A: Transient gene
silencing of galectin-3 suppresses pancreatic cancer cell migration
and invasion through degradation of β-catenin. Int J Cancer.
129:2775–2786. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang D, Chen ZG, Liu SH, Dong ZQ, Dalin
M, Bao SS, Hu YW and Wei FC: Galectin-3 gene silencing inhibits
migration and invasion of human tongue cancer cells in vitro via
downregulating β-catenin. Acta Pharmacol Sin. 34:176–184. 2013.
View Article : Google Scholar : PubMed/NCBI
|