Introduction
Mitochondrial acetoacetyl-CoA thiolase (T2) (EC
2.3.1.9, gene symbol ACAT1) deficiency (Online Mendelian
Inheritance in Man [OMIM] #203750, *607809), well known as
β-ketothiolase deficiency, is an autosomal recessive disorder
affecting the isoleucine catabolic pathway and ketone body
metabolism (1,2). T2 plays important role in ketone body
utilization in extrahepatic tissues through thiolysis of
acetoacetyl-CoA to acetyl-CoA. In hepatic ketogenesis, T2 mediates
interconversion between acetoacetyl-CoA and acetyl-CoA. Another
thiolase, mitochondrial 3-ketoacyl-CoA thiolase, can compensate for
T2 deficiency in ketogenesis but to a lesser extent in ketolysis.
Accordingly, T2 deficiency results in ketoacidosis (1,2).
Since 1971 (3), more than 100
T2-deficient patients have been identified worldwide (1,
unpublished observation). This disorder is clinically characterized
by intermittent ketoacidotic episodes, often triggered by
infections. Patients usually have no symptoms between episodes
unless a prior episode caused a lasting neurological impairment
(2). The distinctive laboratory
feature is an increased urinary excretion of
2-methyl-3-hydroxybutyrate (2M3HB), 2-methylacetoacetate, and
tiglylglycine, derived from intermediates of isoleucine catabolism.
Blood acylcarnitine analysis commonly show elevated C5:1 carnitine
derived from tiglyl-CoA and C5-OH carnitine from
2-methyl-3-hydroxybutyryl-CoA (2).
However, some patients with atypical clinical and/or laboratory
findings have been reported (4).
The severity of the clinical features varies from patient to
patient, but follow-up studies revealed that T2 deficiency, in
general, has a favorable outcome (1,5).
The human ACAT1 gene is located at chromosome
11q22.3-23.1. It spans approximately 27 kb and contains 12 exons
and 11 introns (6). T2 cDNA is
approximately 1.5 kb long and encodes a precursor protein of 427
amino acids (7). So far, more than
70 ACAT1 mutations have been identified, 20% of which cause
aberrant splicing (8).
Herein, we report the molecular basis of T2
deficiency in a patient, in whom a minigene experiment demonstrated
that a single nucleotide substitution of T to A in the
polypyrimidine stretch at the −13 position of the splice acceptor
site of intron 2 (c.121-13T>A) causes exon 3 skipping in
ACAT1 gene. We also discuss why it was difficult to predict
the effect of this mutation on splicing using in silico
tools.
Materials and methods
Clinical summary of patient
The female patient was born to second-degree
consanguineous Indian parents. She experienced age-appropriate
development with no significant illnesses until 10 months of age,
when she was admitted to a hospital with vomiting, loose stools,
poor feeding, fast breathing, and lethargy for one day. Her
laboratory investigations showed normal hemogram levels, in
addition to levels of blood glucose of 5.4 mmol/l, serum sodium of
138 mEq/l, serum potassium of 4.5 mEq/l, lactate of 0.6 mmol/l,
marginally elevated ammonia of 146 µmol/l, and urinary ketone of
3+. An arterial blood gas analysis showed a severe metabolic
acidosis with pH 6.88, pO2 127 mmHg, pCO2 10
mmHg, and HCO3− 4.5 mmol/l. Blood cultures
and C-reactive protein were negative.
The patient was successfully treated with
intravenous fluids (10% dextrose with electrolytes), bicarbonate
infusion, and intravenous carnitine supplementation (100
mg/kg/day); blood glucose level was kept at a high normal range to
induce insulin secretion which suppressed ketogenesis. The acidosis
gradually resolved, and the patient's condition improved. Urinary
gas chromatography-mass spectrometry revealed an increased
excretion of lactic acid, 3-hydroxybutyrate, and 2M3HB. Plasma
acylcarnitine profiling by tandem mass spectrometry showed an
elevated C5-OH carnitine and C5-OH/C2 ratio. She is now 3-years
old, and has attained age-appropriate development without any
further ketoacidotic episodes thus far.
Ethical consideration
This study was approved by the Ethics Committee of
the Graduate School of Medicine, Gifu University (Gifu, Japan) and
was carried out in accordance with the principles contained within
the Declaration of Helsinki. The participant provided informed
consent to participate in the study.
Enzyme assay and immunoblot
analysis
The patient's and controls' fibroblasts were
cultured in an Eagle's minimal essential medium containing 10%
fetal calf serum. Protein concentration was determined by the
method of Lowry using bovine serum albumin as a standard. Enzyme
assay for T2 activity was performed as described (9). In brief, using supernatants from cell
extracts, we spectrophotometrically monitored the decrease of
acetoacetyl-CoA absorbance at 303 nm, which is the result of
thiolysis of acetoacetyl-CoA to acetyl-CoA. We measured the
difference of thiolase activity in the absence and the presence of
potassium ions, which specifically stimulate T2; such a difference
represents T2 activity (9).
Immunoblot analysis was performed using rabbit polyclonal
antibodies and Proto Blot® Western Blot AP system
(Promega Corp., Madison, WI, USA) as described by Fukao et
al (10); a mixture of an
anti-human T2 antibody and anti-human succinyl-CoA: 3-oxoacid CoA
transferase antibody was used as the first antibody. We applied 30
µg proteins extracted from each of patient's and two controls'
fibroblasts. Kaleidoscope Prestained Standards (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) were used as molecular size
markers.
Mutation analysis at the genomic DNA
level
Genomic DNA was purified from the patient's
fibroblasts using a Sepa Gene® (EIDIA Co., Ltd., Tokyo,
Japan). A mutation screening was performed at the genomic level by
PCR and direct sequencing using a set of primer pairs that amplify
exons with their intron boundaries as previously described
(11). The genomic ACAT1
sequence was obtained from NCBI Reference Sequence no.
NG_009888.1.
cDNA analysis
The patient's and controls' fibroblasts were
cultured in two flasks, one of which was treated with 200 µg/ml of
cycloheximide (CHX) (Sigma, St. Louis, MO, USA) for 5 h before RNA
extraction, as previously described (12). Total RNA was isolated from
fibroblasts using ISOGEN kit (Nippon Gene Co., Ltd., Tokyo, Japan).
Thereafter, the isolated RNA (5 µg) was reverse-transcribed in 20
µl of 50 mM Tris-HCl pH 7.5, 75 mM KCl, 3 mM MgCl2, 10
mM dithiothretiol, 0.5 mM dNTPs, and 200 U of M-MLV reverse
transcriptase (Life Technologies, Rockville, MD, USA) with a primer
mixture including 5 pmol of both a ACAT1-specific antisense
primer, T2-135 (5′-c.*76 TGACCCACAGTAGTCACAC-3′), and
oligo dT primers.
The above preparation was incubated at 37°C for 1 h.
A total of 1 µl of this cDNA solution served as a PCR template. The
positions of the PCR primers were numbered in relation to the
adenine of the initiation methionine codon, which was assigned
position +1. The ACAT1 cDNA fragment was amplified
(c.-40-646) using Primer T2-4 (sense) (5′-c.-40AGT CTA
CGC CTG TGG AGC-3′) and Primer T2-64 (antisense)
(5′-c.646TAG CAT AAG CGT CCT GTT CA-3′) (13). The ACAT1 cDNA sequence was
obtained from Gen Bank accession number NM_000019.3. After 30 PCR
cycles, amplified fragments were separated by electrophoresis on a
5% (w/v) polyacrylamide gel. The amplified fragments were also
separated in 1% (w/v) agarose gel, extracted using a
Geneclean® II kit (BIO 101, Vista, CA, USA), and
analyzed by direct sequencing.
Minigene splicing experiment
An ACAT1 segment, from exon 2 to 4 (exon
2-intron 2-exon 3-intron 3-exon 4; approximately 2.4 kb long), was
amplified from control genomic DNA using an Ex taq®
(Takara Bio, Inc., Otsu, Japan). The used primers included the
EcoRI linker sequence: a sense primer (Exon2-EcoRI)
(5′-cgttcgaattcc.77TAA GAT ATGTGG AAC GGA GTT ATG-3′)
and an antisense primer (Exon4-EcoRI)
(5′-cgttcgaattcc.321TGC CTG CCT TGT AGG AGC-3′). After
sequence confirmation in a pGEM®-T Easy Vector (Promega
Corp.), the EcoRI fragment was subcloned into a eukaryote
expression vector pCAGGS (14).
This was defined as a wild-type minigene splicing construct. We
used a KOD-Plus-Mutagenesis Kit® (Toyobo Co., Ltd.,
Osaka, Japan) to make three mutant constructs: c.121-13T>A,
T>C, and T>G. A total of 2 µg of each minigene splicing
vector was transfected into 5×105 cells of
SV40-transformed T2-deficient fibroblasts using
Lipofectamine® 2000 (Invitrogen, San Diego, CA, USA). At
48 h after transfection, RNA was extracted from the cells. Our
minigene constructs should produce human T2-rabbit β-globin fusion
mRNAs. The first strand cDNA was transcribed with the rabbit
β-globin-specific antisense primer (β-glo2) (5′-461AGC
CAC CAC CTT CTG ATA-3′) and then amplified with the
Exon2-EcoRI primer on ACAT1 exon 2, and another
rabbit-specific antisense primer (β-glo3) (5′-443GGC AGC
CTG CAC CTG AGG AGT-3′) to amplify the chimeric cDNA of human
ACAT1 and rabbit β-globin. A study of normal and aberrant
splicing in the four constructs was performed by sequencing and 5%
polyacrylamide gel electrophoresis of the PCR-amplified cDNA
fragments.
Results
Enzyme assay and immunoblot
analysis
The potassium ion-activated acetoacetyl-CoA thiolase
activity was markedly reduced in the patient's fibroblasts; in the
absence of potassium (−K+) it was 11.5, and in the
presence of potassium (+K+) it was 12.6 nmol/min/mg of
protein, leading to a +K+/-K+ ratio of 1.1
(the control fibroblasts -K+ 5.1, +K+ 8.0
nmol/min/mg of protein, +K+/-K+ ratio of
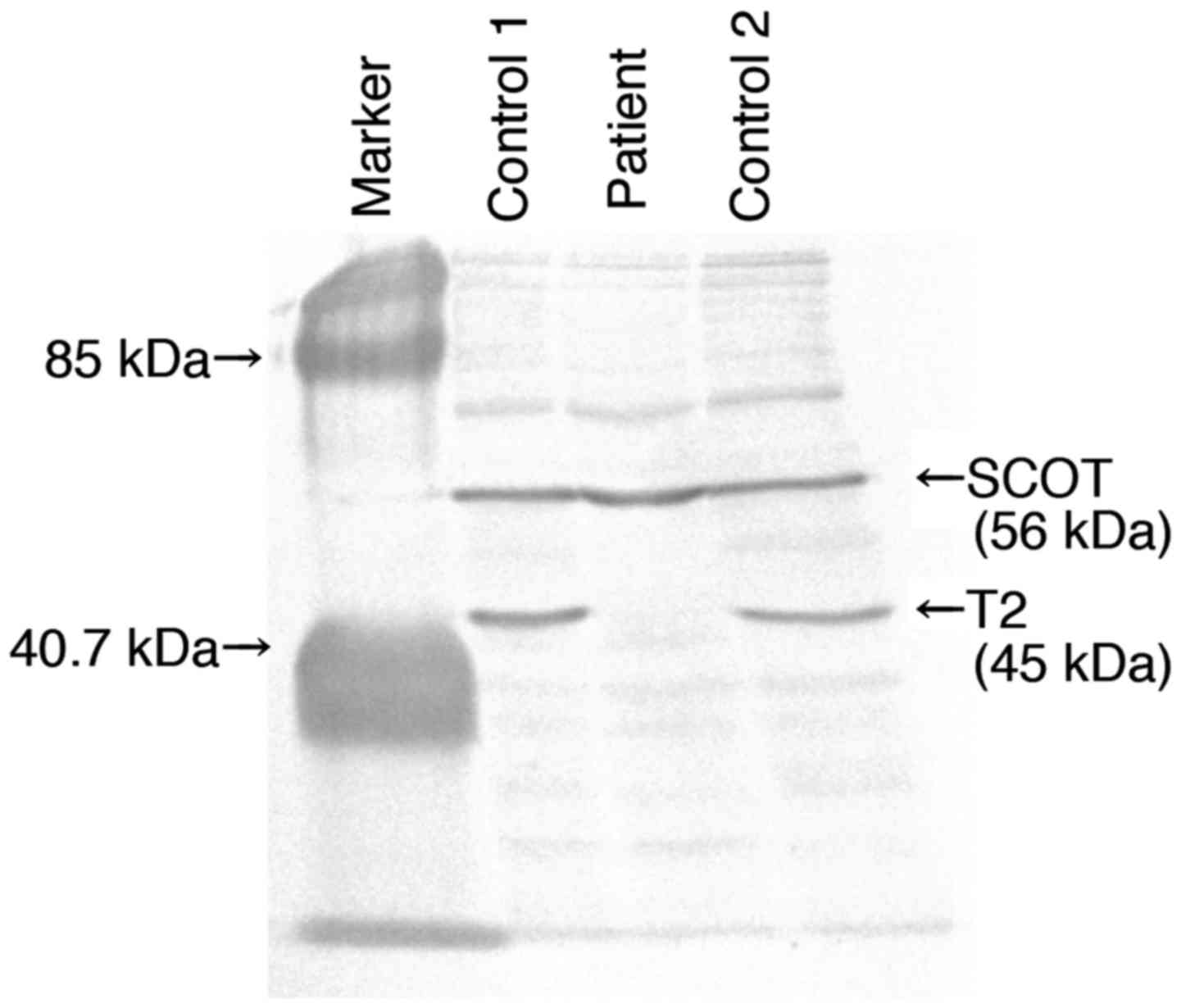
1.6). In immunoblot analysis, no T2 protein was detected in the
patient's fibroblasts (Fig. 1).
These data confirm the diagnosis of T2 deficiency in the
patient.
Mutation analysis at the genomic DNA
level
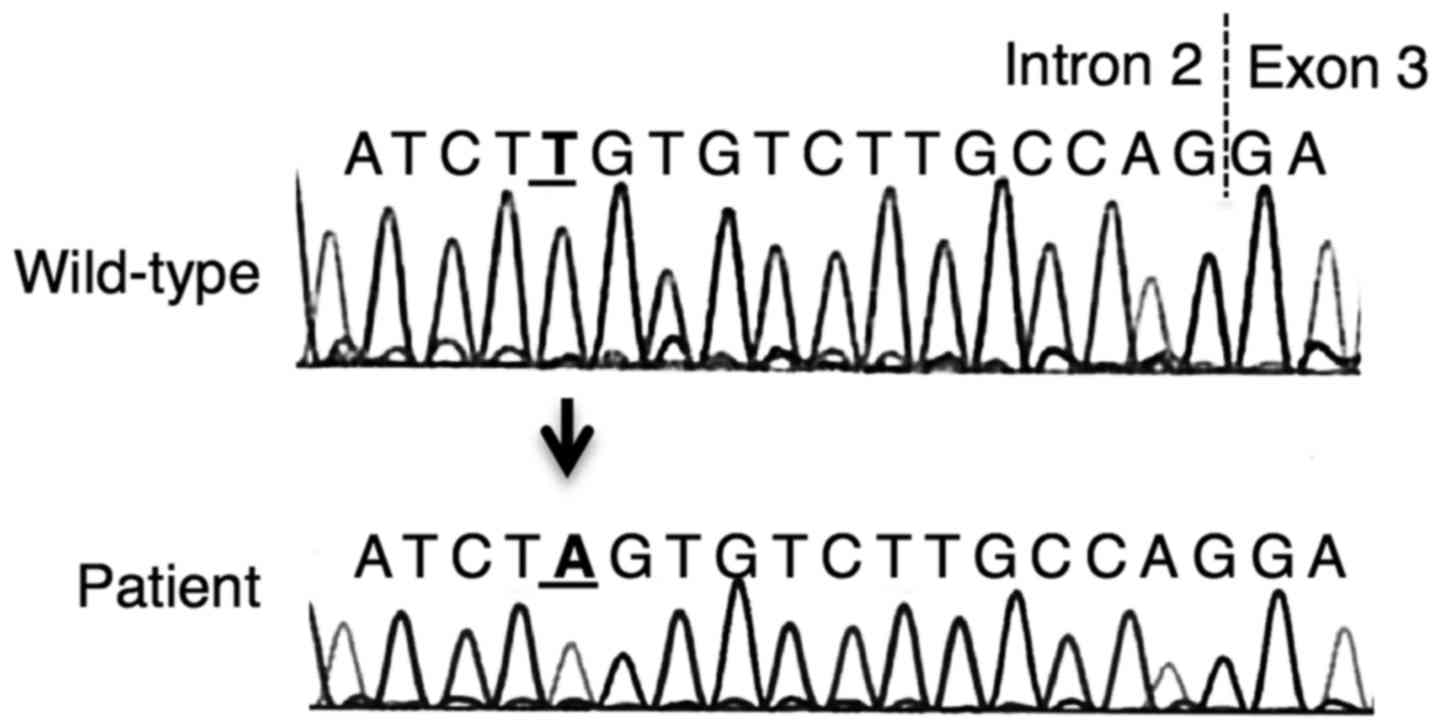
As shown in Fig. 2,
a homozygous substitution, c.121-13T>A, was revealed in the DNA
fragment around exon 3 of ACAT1. No other mutations were
identified.
cDNA analysis
The amplification of the ACAT1 cDNA
(c.-40-646) from control's fibroblasts produced a single fragment
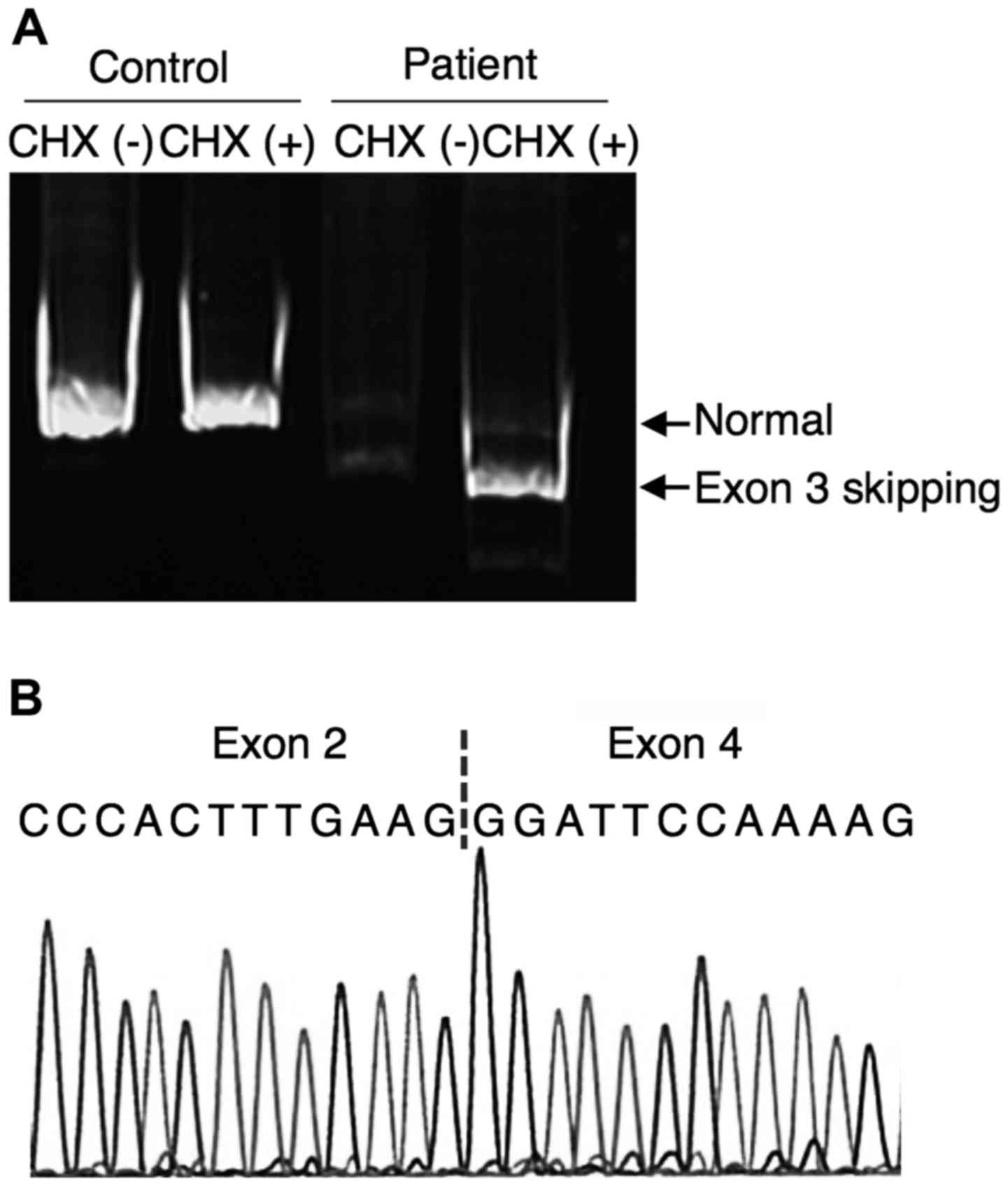
with the expected size of 686 bp. However, two faint fragments were
amplified from CHX-untreated patient's fibroblasts; one had the
same mobility as that of control's fibroblasts and the other was
smaller (568 bp). The smaller fragment was predominantly amplified
from CHX-treated patient's fibroblasts (Fig. 3A). The sequencing of this smaller
fragment revealed an exon 3 skipping (Fig. 3B). Since exon 3 skipping causes
frame-shift, ACAT1 mRNAs with exon 3 skipping was subjected
to nonsense-mediated mRNA decay (NMD).
Minigene splicing experiment
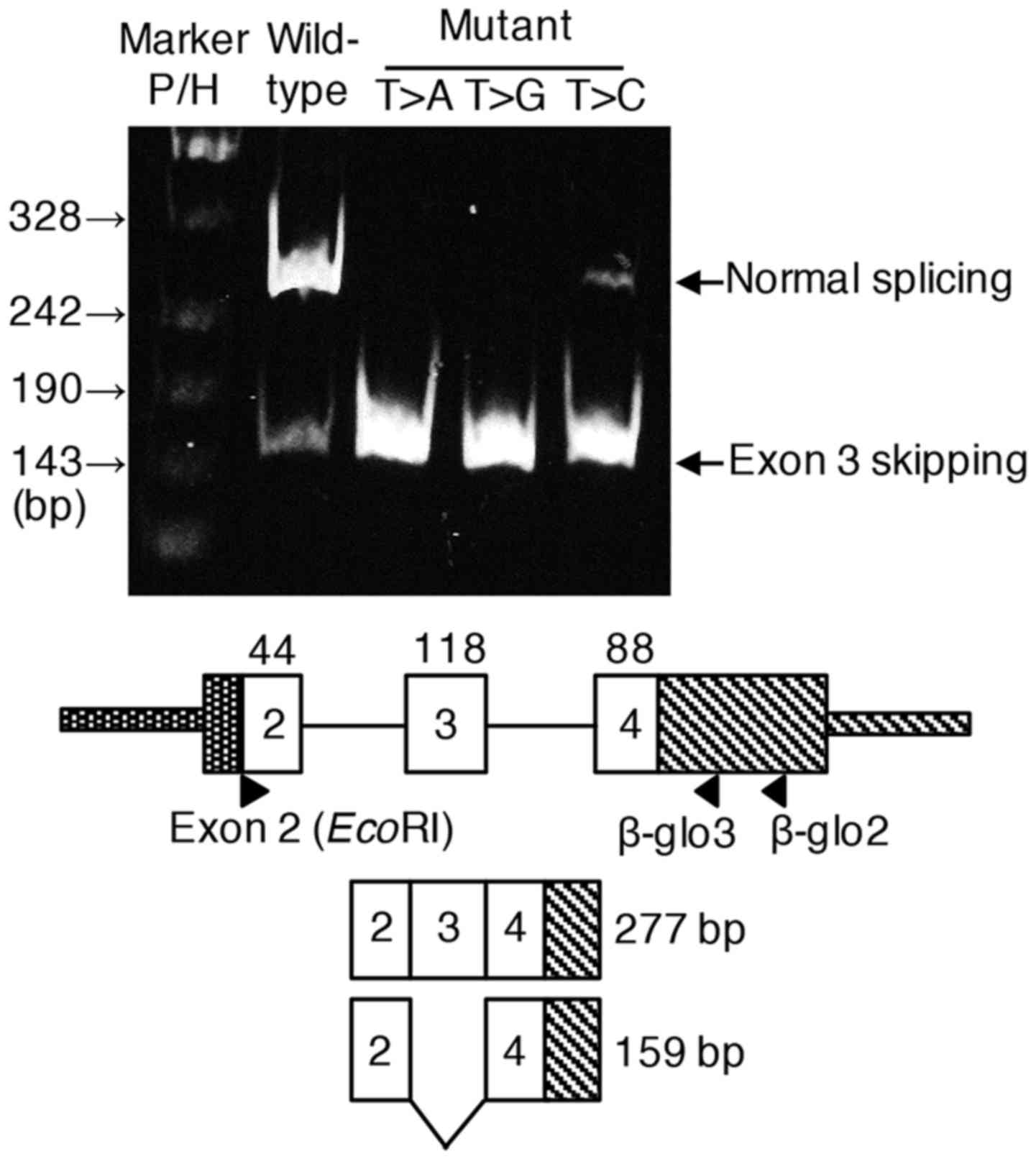
We hypothesized that the patient's mutation,
c.121-13T>A, in intron 2 caused exon 3 skipping, hence minigene
splicing experiment was performed. In case of wild-type minigene
splicing construct, normal splicing was predominantly observed
although small amount of transcript with exon 3 skipping was also
detected. In case of c.121-13T>A construct, almost all
transcripts skipped exon 3, confirming this mutation identified in
the patient was responsible for the exon skipping. Furthermore,
c.121-13T>G and unexpectedly c.121-13T>C also resulted in
predominate exon 3 skipping (Fig.4).
Discussion
Aberrant splicing-causing mutations are increasingly
recognized in several human diseases. These mutations result in the
complete skipping of an exon, holding of an intron, or an
introduction of a new splice site within an exon or intron
(15). Most of these mutations,
including that of ACAT1, are located at the highly conserved
sequences at splice sites: the −1/-2 position (ag) at the splice
acceptor site (13,16) and the +1/+2/+5 position at the
splice donor site (13,17,18).
However, some exonic mutations have been also demonstrated to
result in aberrant splicing by activating cryptic splice sites
within their exons (19,20) or by altering the consensus
sequences of exonic splice enhancer sites (8).
We identified herein a novel homozygous ACAT1
mutation, c.121-13T>A, that caused aberrant splicing. Using
Human Gene Mutation Database and Single Nucleotide Polymorphism
Database, this mutation has not yet been reported in ACAT1
gene. This c.121-13T>A mutation is located at the polypyrimidine
tract of intron 2. Splice acceptor site mutations in the
polypyrimidine tract have not been so commonly reported. However,
we previously reported a homozygous SLC25A20 mutation,
c.199-10T>G, in two patients with carnitine-acylcarnitine
translocase deficiency (21). The
c.199-10T>G mutation has been shown to reside at a consensus
branch point sequence, resulting in skipping of exons 3 and 4 or
exon 3 alone, which leads to a truncated protein (22).
T2 deficiency was confirmed in our patient by
demonstrating the lack of T2 activity and protein in the patient's
fibroblasts. cDNA analysis using RNA extracted from patient's
fibroblasts revealed two faint fragments on electrophoresis,
compared to a clearly-visible single fragment of control's
fibroblasts. To investigate whether the observed decline of
transcripts was due to NMD (23),
we treated fibroblasts with CHX, a general protein translation
inhibitor, before RNA extraction. cDNA analysis using CHX-treated
patient's fibroblasts showed a predominance of the
smaller-than-normal fragment, in which exon 3 skipping was
confirmed by sequencing (r.121_238del); unless treated by CHX, such
fragment was mostly subjected to NMD.
We hypothesized that the patient's mutation,
c.121-13T>A, weakens the recognition of the splice acceptor site
in intron 2. For elucidating the causal relationship between this
mutation and exon 3 skipping by a splicing experiment, we made a
wild-type minigene construct, including an ACAT1 segment
from exon 2 to 4, and the mutant constructs, c.121-13T>A, and
another two constructs, c.121-13T>C, and c.121-13T>G
substitutions for comparison. Minigene splicing experiment clearly
showed that not only c.121-13T>A but also c.121-13T>C, and
c.121-13T>G substitutions resulted in exon 3 skipping.
We examined whether in silico tools can
predict exon 3 skipping caused by these polypyrimidine-tract
mutations (Table I). It is
noteworthy that the c.121-13T>A, T>C, and T>G mutations
resulted in only a modest decrease in the score calculated using
both the Shapiro and Senapathy method (24) and Analyzer Splice Tool (http://ibis.tau.ac.il/ssat/SpliceSiteFrame.htm). The
Human Splicing Finder (HSF) (http://www.umd.be/HSF3/HSF.html), which combines 12
different algorithms to identify and predict the effects of
mutations on splicing signals (25), also failed to predict the effects
of c.121-13T>C and T>G on splicing. Based on these results,
it was difficult to predict exon 3 skipping caused by
c.121-13T>A, T>C, and T>G mutations that was clearly
demonstrated by our minigene experiment (26).
 | Table I.Analysis of in silico tools
for splicing defect prediction. |
Table I.
Analysis of in silico tools
for splicing defect prediction.
|
| Human splicing
finder (HSF) |
|
|---|
|
|
|
|
|---|
| Minigene
construct | Shapiro and
senapathy score | Analyzer splice
tool | HSF matrices
(acceptor site) | MaxEnt (3′
Motif) | Exon 3 skipping
identified by minigene splicing experiment |
|---|
| Wild-type | 92.50
(TTGTGTCTTGCCAG/G) | 89.93 |
|
| A small amount of
transcripts |
| c.121-13T>A | 86.88
(TAGTGTCTTGCCAG/G) | 84.22 | Activates a cryptic
intronic acceptor site | A new acceptor site
or broken wild-type acceptor site | Almost all
transcripts |
| c.121-13T>G | 86.88
(TGGTGTCTTGCCAG/G) | 84.38 | No impact on
splicing | No impact on
splicing | Almost all
transcripts |
| c.121-13T>C | 90.63
(TCGTGTCTTGCCAG/G) | 86.72 | No impact on
splicing | No impact on
splicing | Most
transcripts |
In fact, bioinformatics predictions with in
silico tools have been widely used to evaluate the potentially
pathogenic effects of genetic mutations on splicing. However, the
predictions of these algorithms mostly are not consistent with each
other, or with the results of minigene splice experiments (27–29).
Most in silico tools generate a numerical score as a measure
of the strength of splicing motifs, but the score itself is
meaningless without a clear threshold. Several criteria were
developed to set a cutoff value to define aberrant-splicing causing
mutations; however, these values are commonly arbitrary across
various tools and studies, which may explain the inconsistency
among different tools. Although most prediction tools perform
successfully for mutations at the highly conserved splice sites
(ag/gt), mutations at more distant sites, like c.121-13T>A in
our study, are still representing a challenge. It seems that our
knowledge about the splicing process has yet to be optimized
(26,30).
We demonstrated by a minigene splicing experiment
that a novel mutation (c.121-13T>A) results in aberrant splicing
with exon 3 skipping in ACAT1 gene. The c.121-13 position of
ACAT1 gene appears to be an originally low-recognized site.
In the routine diagnostic practice, in silico tools can
predict the potential consequences of mutations on splicing, but
their results are not so reliable. The minigene splicing experiment
remains the most reliable method to unravel splicing
abnormalities.
The present study has some limitations. We only
identified this mutation (c.121-13T>A) in one patient. However,
in other disorders, similar splice site variants, especially in the
polypyrimidine tract of the splice acceptor site should be also
considered as disease-causing mutations. In addition, the technique
of minigene splicing experiment, which is the most reliable method
to detect aberrant splicing, is not widely available. Accordingly,
bioinformatics predictions with in silico tools remains
useful, but we should consider their current restrictions.
Acknowledgements
The authors thank Ms. Naomi Sakaguchi for her
technical assistance. The present study was supported in part by a
Grant-in-Aid for Scientific Research from the Ministry of
Education, Culture, Sports, Science and Technology of Japan (nos.
26114708, 24591505, 16K09962 and 15K01693), Health and Labour
Science Research Grants for Research on Intractable Diseases from
the Ministry of Health, Labour and Welfare of Japan, and the
Practical Research Project for Rare/Intractable Diseases from Japan
Agency for Medical Research and Development (AMED).
Glossary
Abbreviations
Abbreviations:
|
2M3HB
|
2-methyl-3-hydroxybutyrate
|
|
CHX
|
cycloheximide
|
|
NMD
|
nonsense-mediated mRNA decay
|
|
T2
|
mitochondrial acetoacetyl-CoA
thiolase
|
References
|
1
|
Hori T, Yamaguchi S, Shinkaku H, Horikawa
R, Shigematsu Y, Takayanagi M and Fukao T: Inborn errors of ketone
body utilization. Pediatr Int. 57:41–48. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fukao T, Mitchell G, Sass JO, Hori T, Orii
K and Aoyama Y: Ketone body metabolism and its defects. J Inherit
Metab Dis. 37:541–551. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Daum RS, Lamm PH, Mamer OA and Scriver CR:
A ‘new’ disorder of isoleucine catabolism. Lancet. 2:1289–1290.
1971. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Abdelkreem E, Otsuka H, Sasai H, Aoyama Y,
Hori T, El Aal MA, Mahmoud S and Fukao T: Beta-ketothiolase
deficiency: Resolving challenges in diagnosis. J Inborn Errors
Metab Screen. 4:1–9. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fukao T, Scriver CR and Kondo N: t2
Collaborative Working Group: The clinical phenotype and outcome of
mitochondrial acetoacetyl-CoA thiolase deficiency
(beta-ketothiolase or T2 deficiency) in 26 enzymatically proved and
mutation-defined patients. Mol Genet Metab. 72:109–114. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kano M, Fukao T, Yamaguchi S, Orii T,
Osumi T and Hashimoto T: Structure and expression of the human
mitochondrial acetoacetyl-CoA thiolase-encoding gene. Gene.
109:285–290. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fukao T, Yamaguchi S, Kano M, Orii T,
Fujiki Y, Osumi T and Hashimoto T: Molecular cloning and sequence
of the complementary DNA encoding human mitochondrial
acetoacetyl-coenzyme A thiolase and study of the variant enzymes in
cultured fibroblasts from patients with 3-ketothiolase deficiency.
J Clin Invest. 86:2086–2092. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fukao T, Horikawa R, Naiki Y, Tanaka T,
Takayanagi M, Yamaguchi S and Kondo N: A novel mutation
(c.951C>T) in an exonic splicing enhancer results in exon 10
skipping in the human mitochondrial acetoacetyl-CoA thiolase gene.
Mol Genet Metab. 100:339–344. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Williamson DH, Bates MW, Page MA and Krebs
HA: Activities of enzymes involved in acetoacetate utilization in
adult mammalian tissues. Biochem J. 121:41–47. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fukao T, Song XQ, Mitchell GA, Yamaguchi
S, Sukegawa K, Orii T and Kondo N: Enzymes of ketone body
utilization in human tissues: Protein and messenger RNA levels of
succinyl-coenzyme A (CoA): 3-ketoacid CoA transferase and
mitochondrial and cytosolic acetoacetyl-CoA thiolases. Pediatr Res.
42:498–502. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fukao T, Nakamura H, Song XQ, Nakamura K,
Orii KE, Kohno Y, Kano M, Yamaguchi S, Hashimoto T, Orii T and
Kondo N: Characterization of N93S, I312T, and A333P missense
mutations in two Japanese families with mitochondrial
acetoacetyl-CoA thiolase deficiency. Hum Mutat. 12:245–254. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hori T, Fukao T, Murase K, Sakaguchi N,
Harding CO and Kondo N: Molecular basis of two-exon skipping (exons
12 and 13) by c.1248+5g>a in OXCT1 gene: Study on intermediates
of OXCT1 transcripts in fibroblasts. Hum Mutat. 34:473–480. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fukao T, Yamaguchi S, Orii T, Schutgens
RB, Osumi T and Hashimoto T: Identification of three mutant alleles
of the gene for mitochondrial acetoacetyl-coenzyme A thiolase. A
complete analysis of two generations of a family with
3-ketothiolase deficiency. J Clin Invest. 89:474–479. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Niwa H, Yamamura K and Miyazaki J:
Efficient selection for high-expression transfectants with a novel
eukaryotic vector. Gene. 108:193–199. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Baralle D and Baralle M: Splicing in
action: Assessing disease causing sequence changes. J Med Genet.
42:737–748. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fukao T, Yamaguchi S, Orii T, Osumi T and
Hashimoto T: Molecular basis of 3-ketothiolase deficiency:
Identification of an AG to AC substitution at the splice acceptor
site of intron 10 causing exon 11 skipping. Biochim Biophys Acta.
1139:184–188. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fukao T, Song XQ, Yamaguchi S, Kondo N,
Orii T, Matthieu JM, Bachmann C and Hashimoto T: Identification of
three novel frameshift mutations (83delAT, 754insCT, and 435+1G to
A) of mitochondrial acetoacetyl-coenzyme A thiolase gene in two
Swiss patients with CRM-negative beta-ketothiolase deficiency. Hum
Mutat. 9:277–279. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Thümmler S, Dupont D, Acquaviva C, Fukao T
and de Ricaud D: Different clinical presentation in siblings with
mitochondrial acetoacetyl-CoA thiolase deficiency and
identification of two novel mutations. Tohoku J Exp Med. 220:27–31.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nakamura K, Fukao T, Perez-Cerda C, Luque
C, Song XQ, Naiki Y, Kohno Y, Ugarte M and Kondo N: A novel
single-base substitution (380C>T) that activates a 5-base
downstream cryptic splice-acceptor site within exon 5 in almost all
transcripts in the human mitochondrial acetoacetyl-CoA thiolase
gene. Mol Genet Metab. 72:115–121. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fukao T, Boneh A, Aoki Y and Kondo N: A
novel single-base substitution (c.1124A>G) that activates a
5-base upstream cryptic splice donor site within exon 11 in the
human mitochondrial acetoacetyl-CoA thiolase gene. Mol Genet Metab.
94:417–421. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vatanavicharn N, Yamada K, Aoyama Y, Fukao
T, Densupsoontorn N, Jirapinyo P, Sathienkijkanchai A, Yamaguchi S
and Wasant P: Carnitine-acylcarnitine translocase deficiency: Two
neonatal cases with common splicing mutation and in vitro
bezafibrate response. Brain Dev. 37:698–703. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ogawa A, Yamamoto S, Kanazawa M,
Takayanagi M, Hasegawa S and Kohno Y: Identification of two novel
mutations of the carnitine/acylcarnitine translocase (CACT) gene in
a patient with CACT deficiency. J Hum Genet. 45:52–55. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maquat LE: Nonsense-mediated mRNA decay in
mammals. J Cell Sci. 118:1773–1776. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shapiro MB and Senapathy P: RNA splice
junctions of different classes of eukaryotes: Sequence statistics
and functional implications in gene expression. Nucleic Acids Res.
15:7155–7174. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Desmet FO, Hamroun D, Lalande M,
Collod-Béroud G, Claustres M and Béroud C: Human splicing finder:
An online bioinformatics tool to predict splicing signals. Nucleic
Acids Res. 37:e672009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jian X, Boerwinkle E and Liu X: In silico
tools for splicing defect prediction: A survey from the viewpoint
of end users. Genet Med. 16:497–503. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Théry JC, Krieger S, Gaildrat P, Révillion
F, Buisine MP, Killian A, Duponchel C, Rousselin A, Vaur D, Peyrat
JP, et al: Contribution of bioinformatics predictions and
functional splicing assays to the interpretation of unclassified
variants of the BRCA genes. Eur J Hum Genet. 19:1052–1058. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ben Rhouma F, Azzouz H, Petit FM, Khelifa
MB, Chehida AB, Nasrallah F, Parisot F, Lasram K, Kefi R, Bouyacoub
Y, et al: Molecular and biochemical characterization of a novel
intronic single point mutation in a Tunisian family with glycogen
storage disease type III. Mol Biol Rep. 40:4197–4202. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nouri N, Fazel-Najafabadi E, Behnam M,
Nouri N, Aryani O, Ghasemi M, Nasiri J and Sedghi M: Use of in
silico tools for classification of novel missense mutations
identified in dystrophin gene in developing countries. Gene.
535:250–254. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Houdayer C, Caux-Moncoutier V, Krieger S,
Barrois M, Bonnet F, Bourdon V, Bronner M, Buisson M, Coulet F,
Gaildrat P, et al: Guidelines for splicing analysis in molecular
diagnosis derived from a set of 327 combined in silico/in vitro
studies on BRCA1 and BRCA2 variants. Hum Mutat. 33:1228–1238. 2012.
View Article : Google Scholar : PubMed/NCBI
|