Introduction
Coronary heart disease, particularly acute
myocardial infarction, is one of the leading causes of human
morbidity and mortality. Myocardial ischemia/reperfusion (I/R) has
been identified as a major pathological process that contributes to
further damage to myocardial tissues in patients with coronary
heart disease, including patients who require extracorporeal
circulation surgery, off-pump coronary artery bypass surgery or
percutaneous coronary intervention (1). The underlying mechanisms of
myocardial I/R injury have been demonstrated to involve
intracellular overload of calcium, excessive production of free
radicals, impaired function of mitochondria and the endoplasmic
reticulum, and exaggerated inflammation (2,3). All
of these mechanisms induce cardiomyocyte apoptosis and may
eventually lead to the development of heart failure (4).
MicroRNAs (miRNAs/miRs) are a family of
single-stranded, small non-protein-coding RNAs of ~22 nt that
regulate gene expression post-transcriptionally by degrading or
inhibiting target miRNAs (5,6).
Many previous studies indicated that miRNAs serve significant roles
in cardiovascular diseases, including myocardial I/R injury
(7), myocardial hypertrophy
(8), arrhythmia (9) and heart failure (10). For example, miR-499 inhibits
cardiomyocyte apoptosis by suppressing calcineurin-mediated
dephosphorylation of dynamin-related protein 1 via targeting of
α-and β-isoforms of the calcineurin catalytic subunit (11), whereas overexpression of miR-320
increases the infarction size in mouse hearts and enhances
cardiomyocyte apoptosis by targeting heat shock protein 20
(12). Additionally, miR-214 is
upregulated during I/R injury, and knockdown of miR-214 decreases
cardiac contractility and increases cardiomyocyte apoptosis in
response to I/R injury (13). The
cardioprotective effect of miR-214 is attributed to the suppression
of sodium/calcium exchanger 1 and inhibition of the downstream
Ca2+ signaling pathway (13). Furthermore, transduction of an
miR-125b-expressing lentivirus into mouse hearts attenuates
I/R-induced apoptosis by decreasing the levels of p53, B-cell
lymphoma 2 (Bcl2)-antagonist/killer-1, Bcl2-associated X protein,
Fas and tumor necrosis factor receptor-associated factor 6
(14).
In addition, miRNAs have well-established roles in
cancer. miR-15a, which is known to suppress proliferation and
increase the apoptosis of tumor cells, is involved in the
pathogenesis of many types of cancer, including pituitary tumors,
colorectal cancers and non-small cell lung cancer (15–17).
In human liver cells, miR-15a has been demonstrated to target
mothers against decapentaplegic homolog 7 (SMAD7) (18). A microarray assay additionally
indicated that miR-15a is upregulated in an animal model of I/R
injury (19). However, the
detailed mechanisms of how miR-15a may be involved in cardiac I/R
injury are not well established.
SMAD proteins were initially identified as key
downstream effectors in the transforming growth factor-β1 (TGF-β1)
signaling pathway (20). For
example, upon phosphorylation by TGF-β type I receptor, SMAD2/SMAD4
translocates to the nucleus to promote downstream gene
transcription (21). However,
SMAD7 serves as an inhibitory molecule by blocking TGF-β1-induced
SMAD activation and interfering with the interactions between other
SMAD proteins and receptors (21).
Furthermore, SMAD7 blocks nuclear factor-κB (NF-κB) activation by
increasing inhibitor of NF-κB (IκB) production (22). A recent study indicated that
upregulated levels of SMAD7 decreases NF-κB protein levels and
attenuates hypoxia/reoxygenation (H/R)-induced cardiomyocyte
apoptosis (23).
In the present study, the expression of miR-15a and
its role in myocardial injury and apoptosis induced by H/R in
cultured H9c2 rat cardiomyocytes were examined. The results
suggested that miR-15a induces H/R-induced apoptosis of
cardiomyocytes by targeting SMAD7; therefore, inhibition of miR-15a
may have therapeutic benefits.
Materials and methods
Cell culture
H9c2 rat cardiomyoblasts (Cell Bank of the Chinese
Academy of Sciences, Shanghai, China) were cultured at 37°C in 5%
CO2 in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% fetal bovine serum (FBS; Biowest Europe,
Nuaillé, France). The cells were plated at a density of
1.5×105 cells/cm2 in 6-well plates.
H/R model
Cells were subjected to hypoxia for 24 h at 37°C in
1% O2, 94% N2 and 5% CO2 using a
modular incubator (Model 3131; Forma; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). Subsequently, reoxygenation (5%
CO2; 37°C) was performed for 24 h. Cells under normoxia
served as a control. Ischemia and reperfusion were simulated using
DMEM containing 1 or 10% FBS, respectively.
miRNA transfection
Transfection was performed using a lentivirus
(Novobio Scientific, Inc., Shanghai, China). The sense, antisense
and negative control duplex oligonucleotides of miR-15a were
recombined using a BLOCK-iTTM Pol II miR RNAi Expression Vector kit
(Invitrogen; Thermo Fisher Scientific, Inc.). The lentivirus
expression vector additionally expressed green fluorescent protein
for assessment of infection efficiency. Recombinant lentiviruses
were packaged and produced in 293T cells (Novobio Scientific,
Inc.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA, including miRNA, was extracted using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. Reverse
transcriptase and oligo (dT) primers were employed to synthesize
cDNA from 5 µg total RNA in a volume of 12 µl, and then the
reaction mixture was finally adjusted to 20 µl for RT-qPCR. RT-qPCR
analysis was performed on a Rotor-Gene 3000 real-time cDNA
detection system (Qiagen, Inc., Valencia, CA, USA) to examine the
expression of miR-15a with SYBR®−Green (Invitrogen;
Thermo Fisher Scientific, Inc.). qPCR was performed under the
following conditions: Initiation at 95°C for 2 min, followed by 40
cycles of denaturation at 95°C for 10 sec; annealing at 60°C for 30
sec; and extension at 70°C for 45 sec. The quantitation cycle (Cq)
value was set within the exponential phase of PCR and normalized
against a U6 housekeeping gene (24). The following primer sequences were
used: rno-miR-15a-5p-RT,
CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAAACCATT (reverse primer);
rno-miR-15a-5p-F, AGCTGGGTAGCAGCACATAATGGTTT (forward primer).
Western blot analysis
Cell lysates were extracted by centrifugation at
12,000 × g for 15 min at 4°C. Whole lysates isolated from cultured
H9c2 cells were prepared in ice cold lysis buffer (BioTeke
Corporation, Beijing, China) with protease inhibitors (Pierce;
Thermo Fisher Scientific, Inc.) Protein concentration was measured
by performing a bicinchoninic assay (Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. Subsequently,
20 mg/well protein was separated by 10% SDS-PAGE and transferred to
polyvinylidene fluoride membranes. The membranes were blocked in 5%
non-fat milk and Tris-buffered saline with 0.05% Tween-20 at room
temperature for 1 h prior to incubation with primary antibodies
against SMAD7 (cat. no. bs-0566R; 1:1,000; BIOSS, Beijing, China),
NF-κB (cat. no. AN365-1; 1:1,000; Beyotime Institute of
Biotechnology, Haimen, China), GAPDH (cat. no. ab-9485; 1:2,500;
Abcam, Cambridge, UK) or lamin B1 (cat. no. ab-16048; 1:1,000;
Abcam) at 4°C overnight, followed by incubation with the
horseradish peroxidase-conjugated goat anti-rabbit secondary
antibody (catalog no. ab-6721; 1:1,000; Abcam) for 1 h at room
temperature. Bands were visualized using an Enhanced
Chemiluminescence kit (EMD Millipore, Billerica, MA, USA) according
to the manufacturer's instructions, with GAPDH as the control.
Nuclear NF-κB was normalized to lamin B1 as a loading control.
Experiments were repeated three times and the band intensity was
quantified using Image-Pro Plus 6.0 (Media Cybernetics, Inc.,
Rockville, MD, USA).
Measurement of cell injury and
apoptosis
Cell injury was assessed by measuring the
concentrations of lactate dehydrogenase (LDH) and malonaldehyde
(MDA) in the culture medium using detection kits LDH Cytotoxicity
Assay and Lipid Peroxidation MDA Assay kits (Beyotime Institute of
Biotechnology). Cell apoptosis was detected by Annexin
V-phycoerythrin (PE)/7-aminoactinomycin D (AAD) dual staining.
Briefly, following washing twice with PBS, the cells were
resuspended in binding buffer. The cells were stained using an
Annexin V-PE/7-AAD apoptosis kit (Nanjing KeyGen Biotech Co., Ltd.,
Nanjing, China) according to the manufacturer's instructions, and
then examined using a fluorescence microscope (IX51; Olympus
Corporation, Tokyo, Japan). Undyed cells, cells under H/R
conditions and cells transfected with lentivirus PDS019 under H/R
conditions were used as a blank control, positive control and
negative control, respectively. Subsequently, the cells were
assayed with a flow cytometer (BD Biosciences, Franklin Lakes, NJ,
USA).
Luciferase assays
Putative miR-15a target was predicted by
bioinformatics analysis using MiRanda (http://www.microrna.org), miRDB (http://www.mirdb.org/miRDB/), miRwalk (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/)
and TargetScan (http://www.targetscan.org). Following transfection of
miR-15a or anti-miR-15a into H9c2 cells, both the
CL358-SMAD7-3′UTR-WT (wild-type) and CL440-SMAD7-3′UTR-MU (mutant
type) vector expressing firefly luciferase, and the pRL-cmv vector
expressing Renilla luciferase (GeneChem, Co., Ltd., Shanghai,
China) were co-transfected using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.). Luciferase activity
was measured with the Dual Luciferase Reporter Assay system
(Promega Corporation, Madison, WI, USA) and Renilla luciferase
activity served as an internal control.
Statistics and data analysis
Data are presented as the mean ± standard deviation.
Student's t-test and one-way analysis of variance followed by Tukey
post hoc tests were used for statistical analysis. P<0.05 was
considered to indicate a statistically significant difference. SPSS
software (version 19.0; IBM SPSS, Armonk, MY, USA) was employed in
statistical analyses. All experiments were performed at least three
times.
Results
Upregulation of miR-15a expression in
response to H/R in H9c2 cells
To identify the potential role of miR-15a in H/R,
myocardial I/R injury was simulated by exposing H9c2 rat
cardiomyoblasts to 24 h hypoxia, followed by 24 h reoxygenation.
The expression of miR-15a was subsequently measured by RT-qPCR. The
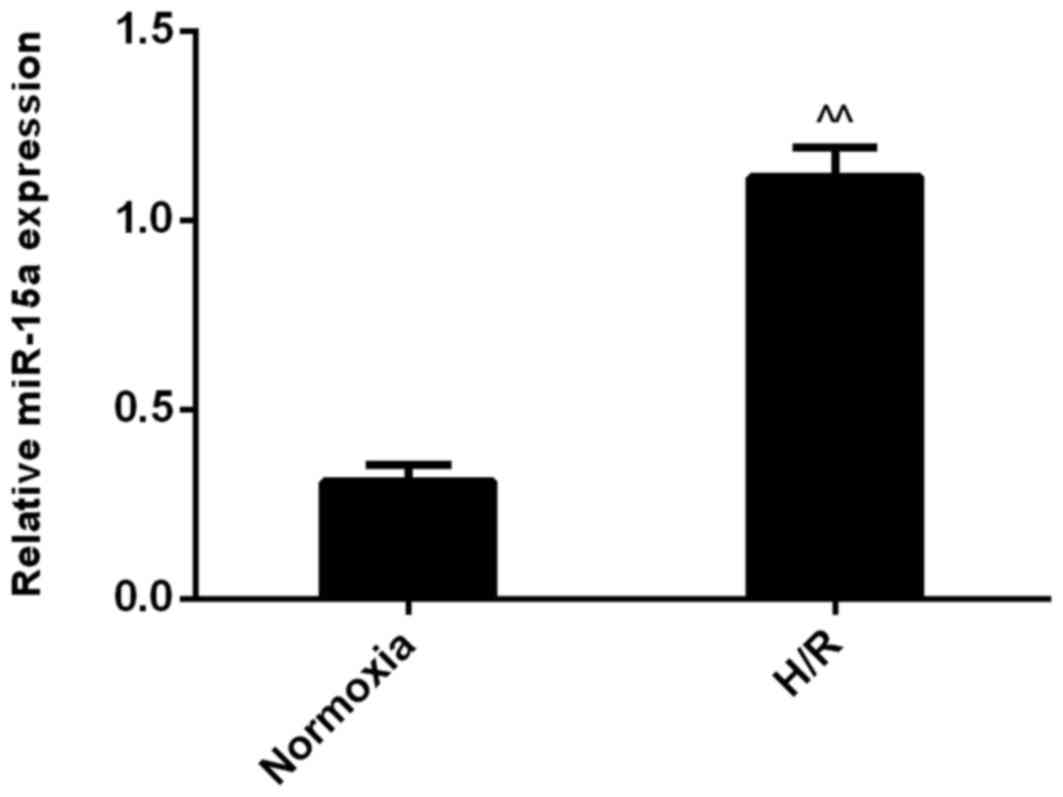
results demonstrated that miR-15a expression was significantly
increased compared with the normoxia group (Fig. 1). These results are consistent with
microarray assays indicating that miR-15a is upregulated in an
animal model of I/R injury (17).
On the basis of these findings, it was hypothesized that miR-15a
may serve a role in the H/R response in H9c2 cells.
Inhibition of miR-15a protects H9c2
cells and reduces H/R-induced cell apoptosis
To directly determine whether miR-15a is involved in
the H/R response in H9c2 cells, a lentivirus expressing miR-15a and
anti-miR-15a was prepared. Transduction with the lentivirus
expressing miR-15a significantly increased miR-15a levels in H9c2
cells under H/R conditions. Conversely, anti-miR-15a markedly
decreased the levels of miR-15a (Fig.
2A).
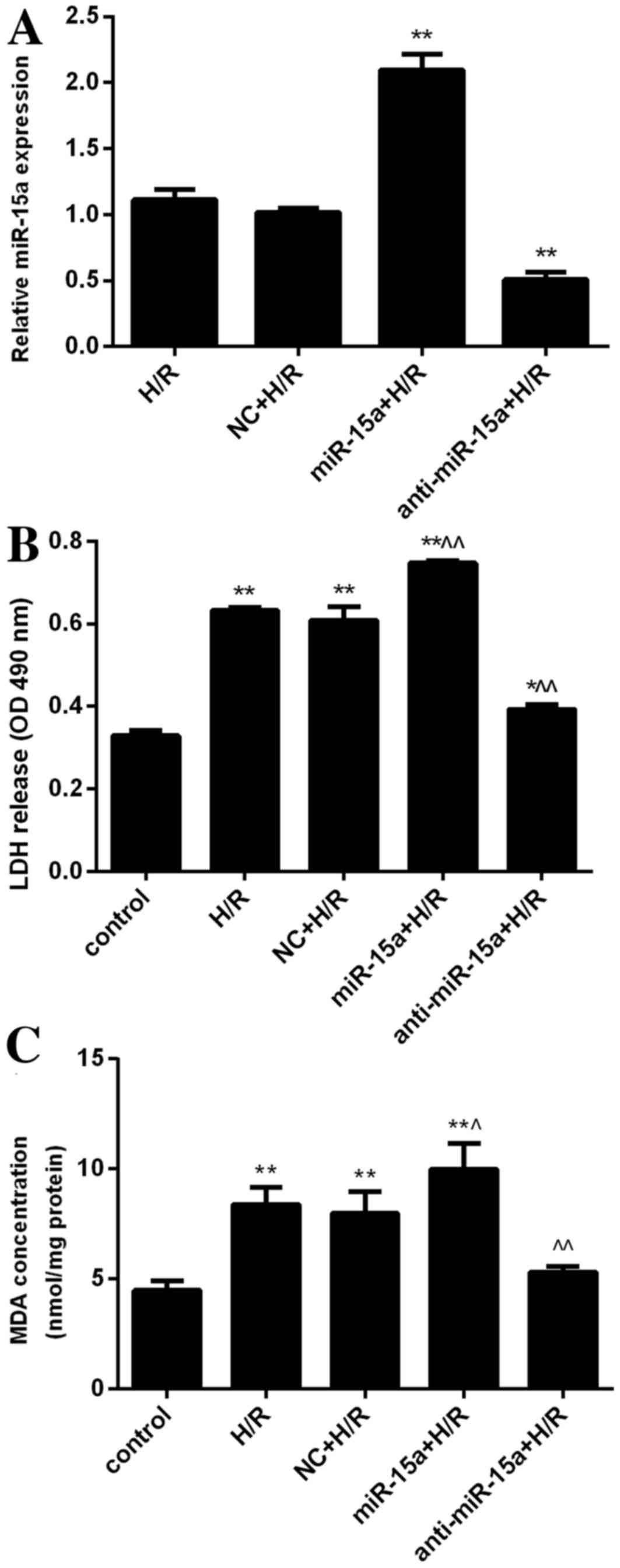 | Figure 2.Inhibition of miR-15a is
cytoprotective. (A) H9c2 cells were transfected with lentivirus
expressing miR-15a or anti-miR-15a. The effect of overexpression
and inhibition of miR-15a under H/R was examined. Results were
normalized to U6 and expressed as the fold change relative to the
control (non-transfected H/R-treated cells). **P<0.01 vs. H/R.
(B) LDH release of control H9c2 cells, cells exposed to H/R, and
cells exposed to H/R following transfection with a control
lentivirus (NC+H/R), a miR-15a-expressing lentivirus (miR-15a+H/R)
or an anti-miR-15a-expressing virus (anti-miR-15a+H/R). *P<0.05
and **P<0.01 vs. control; ^P<0.05 and
^^P<0.01 vs. H/R. (C) MDA release of the cells from
panel B. *P<0.05 and **P<0.01 vs. control;
^P<0.05 and ^^P<0.01 vs. H/R. Data are
presented as the mean ± standard deviation from three independent
experiments. H/R, hypoxia/reoxygenation. H/R,
hypoxia/reoxygenation; miR-15a, microRNA-15a; LDH, lactate
dehydrogenase; OD, optical density; NC, negative control; MDA,
malondialdehyde. |
To determine the effects of modulating miR-15 levels
on H9c2 cell growth, the effects of the lentiviruses on LDH and MDA
release into the culture media were assessed. H/R significantly
increased LDH release (OD, 0.632±0.007 vs. 0.329±0.012 in normoxia
condition; P<0.01; Fig. 2B).
Furthermore, overexpression of miR-15a further increased LDH levels
induced by H/R (OD, 0.747±0.007; P<0.01 vs. the H/R group),
whereas anti-miR-15a significantly decreased LDH levels induced by
H/R (OD, 0.393±0.012; P<0.01 vs. the H/R group). In addition,
H/R treatment raised MDA levels (8.381±0.771 vs. 4.486±0.437
nmol/mg protein under normoxic conditions; P<0.01). MDA release
was further increased by miR-15a overexpression (9.979±1.190
nmol/mg protein; P<0.05 vs. the H/R group), but was
significantly suppressed by anti-miR-15a expression (5.324±0.263
nmol/mg protein, P<0.01 vs. the H/R group; Fig. 2C). These results suggested that
miR-15a expression inversely correlates with H9c2 cell growth under
conditions of H/R, and that inhibition of miR-15a has protective
effects against H/R-induced cellular damage.
To further determine whether the regulation of cell
growth by miR-15a occurs at the level of cell apoptosis, Annexin
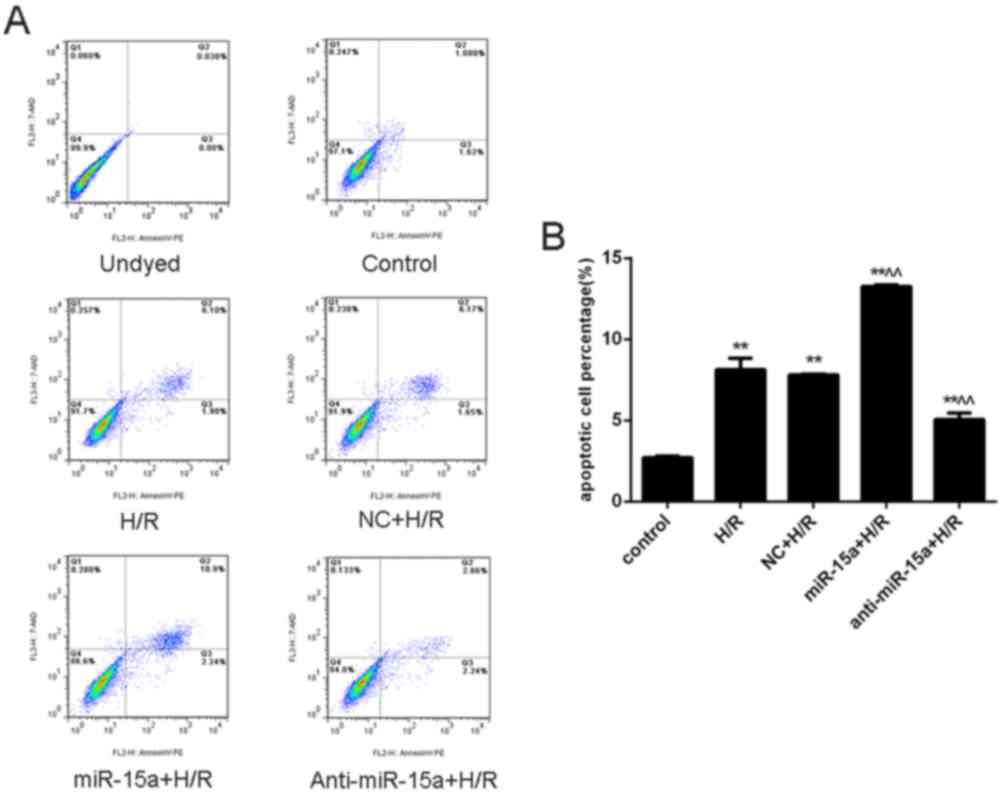
V-PE/7-AAD dual staining was performed. As expected, apoptosis
levels were greater in H/R-treated cells than in control normoxia
H9c2 cells (8.150±0.070% vs. 2.72±0.093%; P<0.01; Fig. 3A and B). Furthermore, miR-15a
expression increased the apoptosis rate (13.250±0.105%; P<0.01
vs. the H/R group), whereas anti-miR-15a expression decreased the
apoptosis rate (5.050±0.407%; P<0.01 vs. the H/R group). Taken
together, these results indicated that inhibition of miR-15a
protects H9c2 cells against H/R-induced apoptosis.
SMAD7 is a target of miR-15a whose
regulation correlates inversely with NF-κB translocation
To further elucidate the mechanism of miR-15a in
myocardial I/R injury, bioinformatics analysis was performed using
MiRanda (http://www.microrna.org), miRDB
(http://www.mirbd.org/miRDB/), miRwalk
(http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/)
and TargetScan (http://www.targetscan.org). The results demonstrated
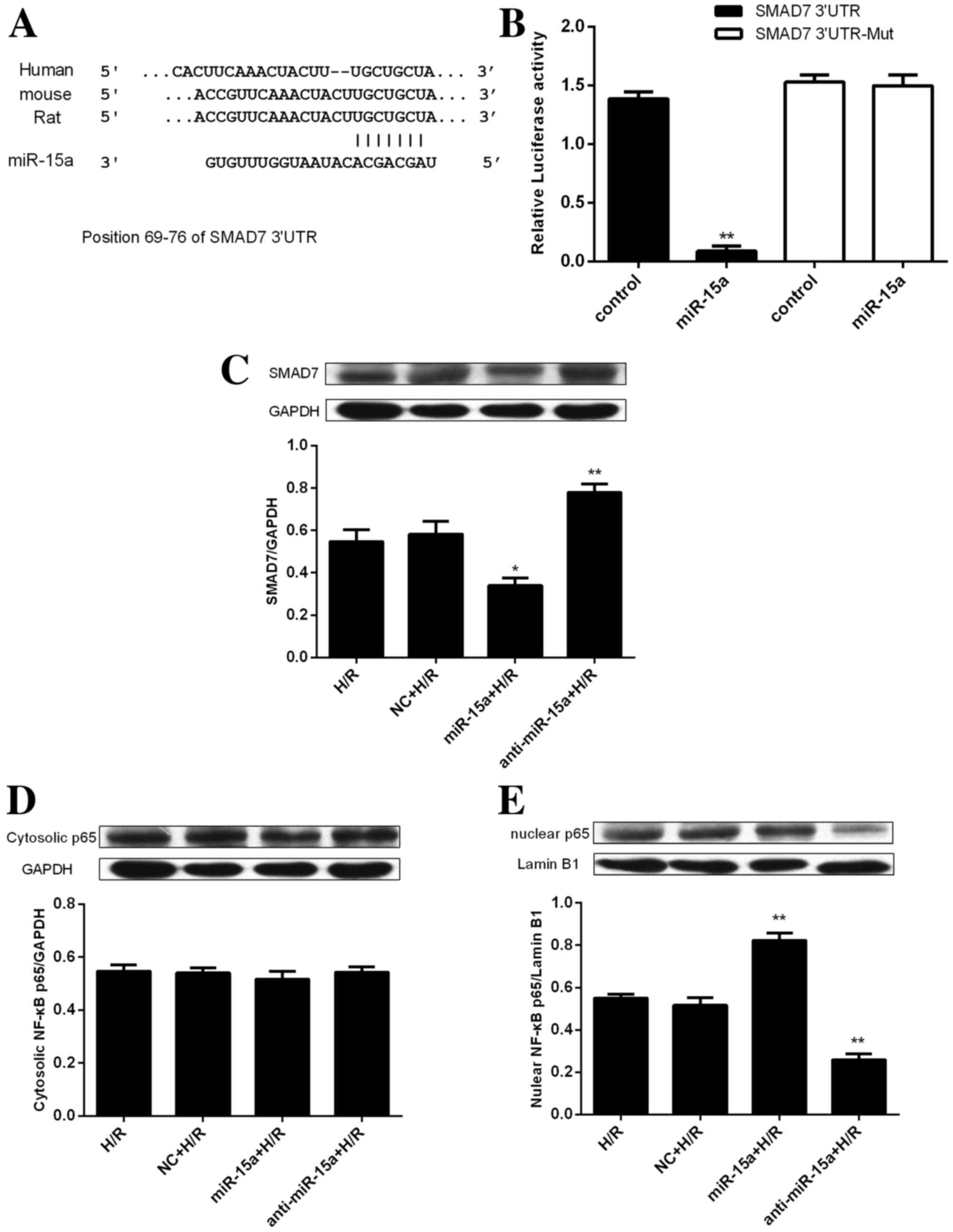
that the SMAD7 3′-untranslated region (UTR) has a conserved binding
site for miR-15a (Fig. 4A). To
confirm that SMAD7 is a target of miR-15a in H9c2 cells, a firefly
luciferase reporter plasmid was constructed by cloning the 3′-UTR
SMAD7 segment harboring the miR-15a binding sequence (SMAD7 3′-UTR)
or a mutated miR-15a binding site (SMAD7 3′-UTR-Mut). The current
results indicated that miR-15a overexpression significantly reduced
the activity of the wild-type luciferase reporter, but not the
mutant luciferase reporter (Fig.
4B).
To verify the ability of miR-15a to regulate the
expression of SMAD7, the levels of endogenous SMAD7 protein in
H/R-treated H9c2 cells were assessed following expression of
miR-15a or anti-miR-15a. Overexpression of miR-15a significantly
downregulated SMAD7, whereas anti-miR-15a expression upregulated
SMAD7 (Fig. 4C). These results
demonstrated the miR-15a regulates SMAD7 and that inhibition of
miR-15a may release its repression of SMAD7 under conditions of
H/R.
Based on a previous study demonstrating that SMAD7
protects against apoptosis by inhibiting NF-κB activation (23), the effects of miR-15a on the
SMAD7/NF-κB p65 signaling pathway were investigated by western
blot. Protein expression levels of cytosolic NF-κB p65 remained
unaltered following miR-15a or anti-miR-15a transfection (Fig. 4D). However, nuclear NF-κB p65
protein expression levels were significantly increased by miR-15a
ectopic expression and inhibited by anti-miR-15a expression
(Fig. 4E). Therefore, the present
results suggested that miR-15a targeting of SMAD7 in H/R-induced
H9c2 cells correlates with activation of NF-κBp65, whereas
inhibition of miR-15a releases SMAD7 and ameliorates cell injury,
potentially via restoration of SMAD7-dependent NF-κB p65
inhibition.
Discussion
Myocardial apoptosis serves a critical role in
myocardial I/R injury (25);
however, the mechanisms underlying the associated pathological
alterations remain unclear. At the cellular level, I/R injury often
results in myocardial apoptosis, and multiple miRNAs are known to
be involved in the pathological process. In the current study, the
potential role of miR-15a in myocardial I/R injury was investigated
in vitro, and a miR-15a-controlled apoptotic signaling
pathway involving SMAD7 and NF-κB p65 was identified.
The miRNA-15 family members, which include miR-15a,
miRNA-15b, miRNA-16, miRNA-195, miRNA-424 and miRNA-497, were
initially suggested to contribute to the inhibition of
cardiomyocyte proliferation by repressing several cell cycle
regulators (26). Hullinger et
al (19) demonstrated that the
levels of miR-15 family members were increased following
ischemia-induced cardiomyocyte cell death, and that inhibition of
miR-15 family members reduced the infarct size following I/R
injury. In addition, miR-15a and miR-15b were demonstrated to be
upregulated in an animal model of I/R injury (27). Furthermore, miRNA-15b was indicated
to impair mitochondrial function by repressing ADP-ribosylation
factor-like protein 2 (28). Liu
et al (29) revealed that
miRNA-15b may enhance H/R-induced apoptosis of cardiomyocytes by
targeting the anti-apoptotic factor Bcl2. The present study
demonstrated that the expression of miR-15a is sensitive to H/R in
H9c2 cells. Following 24 h hypoxia and 24 h reoxygenation, the
expression of miR-15a increased significantly compared with the
expression in the normoxia control group, which is consistent with
previous observations suggesting that the miR-15 family is involved
in cardiac I/R injury.
Although previous studies have confirmed the
pro-apoptotic activity of miR-15a in certain cancers, prior to the
present study, it was unclear how miR-15a is involved in I/R
injury. To elucidate the underlying mechanism of miR-15a in
H/R-induced cardiomyocyte apoptosis, miR15a expression was
regulated in H9c2 cells using a lentivirus. The presented results
demonstrated that miR-15a inhibition decreased LDH and MDA release
and reduced the cell apoptosis rate, whereas overexpression of
miR-15a had the opposite effect. Therefore, inhibition of miR-15a
may attenuate cell injuries and protect against H/R-induced cell
apoptosis.
Using bioinformatics analysis, SMAD7 was identified
as a potential target of miR-15a. Additionally, using luciferase
assays, SMAD7 was confirmed to serve as a target of miR-15a.
However, because miRNAs typically have multiple targets, other
targets may contribute to the detrimental effects of miR-15a on I/R
injury. As an inhibitory SMAD, SMAD7 is known to block NF-κB
activation by inducing IκB expression and simultaneously inhibiting
the TGF-β-induced SMAD signaling pathway. Conversely, the NF-κB
subunit p65 may suppress TGF-β-SMAD signaling via upregulation of
SMAD7 (30). NF-κB, which
comprises five subunits including p65, RelB, c-Rel, p50 and p52,
regulates genes that are associated with immune response,
inflammation, cell survival and proliferation (31,32).
NF-κB is intimately involved in apoptosis and possesses pro- and
anti-apoptotic effects in different types of cells and with
different stimuli (33–35). Among the five subunits, multiple
studies demonstrated that NF-kB p65 activation in cardiomyocytes is
pro-apoptotic (23,34). In resting conditions, NF-κB is
localized in the cytoplasm and is bound to IκB. However, upon
stimulation, IκB ubiquitination and degradation are triggered by
IκB kinase-mediated IκB phosphorylation. This results in nuclear
translocation of NF-κB, which activates the transcription of target
genes. The current study demonstrated that the expression of
miR-15a in H/R-induced H9C2 cells inhibits the expression of
endogenous SMAD7 and activates the nuclear localization of NF-κB
p65, whereas anti-miR-15a has the opposite effect. These results
indicated that the upregulation of SMAD7 expression by miR-15a
inhibition may provide a significant approach for the amelioration
of H/R induced myocardial injury.
In conclusion, the present findings indicated that
miR-15a inhibition protects cardiomyocytes against H/R-induced
apoptosis by upregulating the expression of its target, SMAD7, and
downregulating the NF-κB p65 subunit. Future studies should further
elucidate the interaction between SMAD7 and NF-κB p65 in this
process, how the pathway is integrated into apoptosis, and the
potential interaction with the TGF-β signaling pathway. This may
provide evidence for miR-15a as a potential therapeutic target for
the treatment of cardiac I/R injury.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81572248).
References
|
1
|
Cannon RO III: Mechanisms, management and
future directions for reperfusion injury after acute myocardial
infarction. Nat Clin Pract Cardiovasc Med. 2:88–94. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wu N, Zhang X, Guan Y, Shu W, Jia P and
Jia D: Hyper-cholesterolemia abrogates the cardioprotection of
ischemic postconditioning in isolated rat heart: Roles of glycogen
synthase kinase-3β and the mitochondrial permeability transition
pore. Cell Biochem Biophys. 69:123–130. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hu X, Zhou X, He B, Xu C, Wu L, Cui B, Wen
H, Lu Z and Jiang H: Minocycline protects against myocardial
ischemia and reperfusion injury by inhibiting high mobility group
box 1 protein in rats. Eur J Pharmacol. 638:84–89. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Farh KK, Grimson A, Jan C, Lewis BP,
Johnston WK, Lim LP, Burge CB and Bartel DP: The widespread impact
of mammalian MicroRNAs on mRNA repression and evolution. Science.
310:1817–1821. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kataoka M and Wang DZ: Non-coding RNAs
including miRNAs and lncRNAs in cardiovascular biology and disease.
Cells. 3:883–898. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Boon RA and Dimmeler S: MicroRNAs in
myocardial infarction. Nat Rev Cardiol. 12:135–142. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Heymans S, Corsten MF, Verhesen W, Carai
P, van Leeuwen RE, Custers K, Peters T, Hazebroek M, Stöger L,
Wijnands E, et al: Macrophage microRNA-155 promotes cardiac
hypertrophy and failure. Circulation. 128:1420–1432. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Danielson LS, Park DS, Rotllan N,
Chamorro-Jorganes A, Guijarro MV, Fernandez-Hernando C, Fishman GI,
Phoon CK and Hernando E: Cardiovascular dysregulation of miR-17-92
causes a lethal hypertrophic cardiomyopathy and arrhythmogenesis.
FASEB J. 27:1460–1467. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thome JG, Mendoza MR, Cheuiche AV, La
Porta VL, Silvello D, Dos Santos KG, Andrades ME, Clausell N, Rohde
LE and Biolo A: Circulating microRNAs in obese and lean heart
failure patients: A case-control study with computational target
prediction analysis. Gene. 574:1–10. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang JX, Jiao JQ, Li Q, Long B, Wang K,
Liu JP, Li YR and Li PF: miR-499 regulates mitochondrial dynamics
by targeting calcineurin and dynamin-related protein-1. Nat Med.
17:71–78. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ren XP, Wu J, Wang X, Sartor MA, Qian J,
Jones K, Nicolaou P, Pritchard TJ and Fan GC: MicroRNA-320 is
involved in the regulation of cardiac ischemia/reperfusion injury
by targeting heat-shock protein 20. Circulation. 119:2357–2366.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Aurora AB, Mahmoud AI, Luo X, Johnson BA,
van Rooij E, Matsuzaki S, Humphries KM, Hill JA, Bassel-Duby R,
Sadek HA and Olson EN: MicroRNA-214 protects the mouse heart from
ischemic injury by controlling Ca2+ overload and cell death. J Clin
Invest. 122:1222–1232. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang X, Ha T, Zou J, Ren D, Liu L, Zhang
X, Kalbfleisch J, Gao X, Williams D and Li C: MicroRNA-125b
protects against myocardial ischaemia/reperfusion injury via
targeting p53-mediated apoptotic signalling and TRAF6. Cardiovasc
Res. 102:385–395. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bozok Çetintaş V, Tetik Vardarlı A, Düzgün
Z, Tezcanlı Kaymaz B, Açıkgöz E, Aktuğ H, Kosova Can B, Gündüz C
and Eroğlu Z: miR-15a enhances the anticancer effects of cisplatin
in the resistant non-small cell lung cancer cells. Tumour Biol.
37:1739–1751. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Renjie W and Haiqian L: MiR-132, miR-15a
and miR-16 synergistically inhibit pituitary tumor cell
proliferation, invasion and migration by targeting Sox5. Cancer
Lett. 356:568–578. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
de Groen FL, Timmer LM, Menezes RX,
Diosdado B, Hooijberg E, Meijer GA, Steenbergen RD and Carvalho B:
Oncogenic role of miR-15a-3p in 13q amplicon-driven colorectal
adenoma-to-carcinoma progression. PLoS One. 10:e01324952015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu N, Jiao T, Huang Y, Liu W, Li Z and Ye
X: Hepatitis B virus regulates apoptosis and tumorigenesis through
the microRNA-15a-Smad7-transforming growth factor beta pathway. J
Virol. 89:2739–2749. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hullinger TG, Montgomery RL, Seto AG,
Dickinson BA, Semus HM, Lynch JM, Dalby CM, Robinson K, Stack C,
Latimer PA, et al: Inhibition of miR-15 protects against cardiac
ischemic injury. Circ Res. 110:71–81. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yamamura Y, Hua X, Bergelson S and Lodish
HF: Critical role of Smads and AP-1 complex in transforming growth
factor-beta-dependent apoptosis. J Biol Chem. 275:36295–36302.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ng YY, Hou CC, Wang W, Huang XR and Lan
HY: Blockade of NFkappaB activation and renal inflammation by
ultrasound-mediated gene transfer of Smad7 in rat remnant kidney.
Kidney Int Suppl. S83–S91. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang B, Zhou M, Li C, Zhou J, Li H, Zhu
D, Wang Z, Chen A and Zhao Q: MicroRNA-92a inhibition attenuates
hypoxia/reoxygenation-induced myocardiocyte apoptosis by targeting
Smad7. PLoS One. 9:e1002982014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Olivetti G, Abbi R, Quaini F, Kajstura J,
Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski
S, et al: Apoptosis in the failing human heart. N Engl J Med.
336:1131–1141. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Porrello ER, Johnson BA, Aurora AB,
Simpson E, Nam YJ, Matkovich SJ, Dorn GW II, van Rooij E and Olson
EN: MiR-15 family regulates postnatal mitotic arrest of
cardiomyocytes. Circ Res. 109:670–679. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu LF, Liang Z, Lv ZR, Liu XH, Bai J,
Chen J, Chen C and Wang Y: MicroRNA-15a/b are up-regulated in
response to myocardial ischemia/reperfusion injury. J Geriatr
Cardiol. 9:28–32. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nishi H, Ono K, Iwanaga Y, Horie T, Nagao
K, Takemura G, Kinoshita M, Kuwabara Y, Mori RT, Hasegawa K, et al:
MicroRNA-15b modulates cellular ATP levels and degenerates
mitochondria via Arl2 in neonatal rat cardiac myocytes. J Biol
Chem. 285:4920–4930. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu L, Zhang G, Liang Z, Liu X, Li T, Fan
J, Bai J and Wang Y: MicroRNA-15b enhances
hypoxia/reoxygenation-induced apoptosis of cardiomyocytes via a
mitochondrial apoptotic pathway. Apoptosis. 19:19–29. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Freudlsperger C, Bian Y, Contag Wise S,
Burnett J, Coupar J, Yang X, Chen Z and Van Waes C: TGF-β and NF-κB
signal pathway cross-talk is mediated through TAK1 and SMAD7 in a
subset of head and neck cancers. Oncogene. 32:1549–1559. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Valen G: Signal transduction through
nuclear factor kappa B in ischemia-reperfusion and heart failure.
Basic Res Cardiol. 99:1–7. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen LF and Greene WC: Shaping the nuclear
action of NF-kappaB. Nat Rev Mol Cell Biol. 5:392–401. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hong S, Lee C and Kim SJ: Smad7 sensitizes
tumor necrosis factor induced apoptosis through the inhibition of
antiapoptotic gene expression by suppressing activation of the
nuclear factor-kappaB pathway. Cancer Res. 67:9577–9583. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hamid T, Guo SZ, Kingery JR, Xiang X, Dawn
B and Prabhu SD: Cardiomyocyte NF-κB p65 promotes adverse
remodelling, apoptosis, and endoplasmic reticulum stress in heart
failure. Cardiovasc Res. 89:129–138. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ling H, Gray CB, Zambon AC, Grimm M, Gu Y,
Dalton N, Purcell NH, Peterson K and Brown JH:
Ca2+/Calmodulin-dependent protein kinase II δ mediates myocardial
ischemia/reperfusion injury through nuclear factor-κB. Circ Res.
112:935–944. 2013. View Article : Google Scholar : PubMed/NCBI
|