Introduction
Hypoxic-ischemic pulmonary injury is a common type
of pathological damage, and investigations of anesthetics for lung
protection have predominantly focused on the perioperative stage.
Propofol (2,6-diisopropylphenol) is an intravenous anesthetic,
which is used widely in general anesthesia and for sedation in
intensive care units (1). Several
studies have focused on the protective effect of propofol on organs
in vivo (2–4). Propofol prevents lung injury by
inhibiting the expression of CD14 and Toll-like receptor 4, and
activation of the nuclear factor erythroid 2-related factor 2
pathway (3,5). It exerts anti-inflammatory effects by
inhibiting the phosphorylation of p38 mitogen-activated protein
kinase, stress activated protein kinase/c-Jun N-terminal kinase,
activating transcription factor 2 and c-jun (6), and can exert anti-apoptotic effects
by decreasing the accumulation of hypoxia-inducible factor 1α (HIF
1α), B cell lymphoma-2 (Bcl-2) interacting protein 3 (Bnip3) and
cytokines in alveolar epithelial cells (7). Propofol has also been reported to
exert an antioxidative effect via its inhibition of inducible
nitric oxide synthase in lung L2 cells (8). However, these studies did not
distinguish alveolar type I (ATI) from alveolar type II (ATII),
particularly in terms of physiological function. ATII cells can
synthesize and secrete alveolar surface-active substance, transport
water and electrolytes across the membrane, and are involved in
oxidative metabolism (9,10). When ATI cells are injured, ATII can
proliferate and transform into ATI cells (11). The results of a previous study
demonstrated that ATII cells were crucial in the maintenance of
physiologic function and the repair of impaired lung tissue
(12). Considering the significant
differences in physiologic function between ATI and ATII cells, it
is important to perform specific respective investigations. Our
previous study showed that propofol alleviated hypoxia-induced
apoptosis and increased the viability of ATII cells (13). Thus, further protective effects of
propofol against pathological stimuli in ATII cells require further
investigation.
Autophagy, also termed type II programmed cell
death, often coexists with apoptosis and necrosis during hypoxia
and ischemia. Under normal conditions, a degree of autophagy
contributes to the maintenance of cell homeostasis through the
removal of denatured proteins and aging organelles, and through the
provision of substrate and raw materials for different types of
biochemical reactions in cells (14). In the case of adverse pathological
stimuli, including hypoxia, autophagic activity increases rapidly
to adapt to the changes (15).
However, the over-activation of autophagy leads to autophagic cell
death. The involvement of autophagy in apoptosis remains
controversial (16). It has been
reported that apoptosis can be inhibited by the autophagy signaling
pathway in lung epithelial cells (7,17).
By contrast, autophagy may activate apoptosis indirectly due to the
inducible effects on apoptotic signal activation by the
nonselective degradation of autophagy (18). Taken together, how to modulate
autophagy and subsequently exert promoting effects on cell survival
requires elucidation.
It has been demonstrated that autophagy is involved
in programmed cell death by regulating the p53-mediated pathway;
propofol attenuates cell death through autophagic mechanisms in the
rat hippocampus (19). The
expression levels of proteins associated with autophagy were found
to be induced by propofol treatment in a cellular hypoxia
reoxygenation model of human umbilical vein endothelial cells,
which revealed the effect of propofol on the autophagy signaling
network (20). In addition, our
previous study reported the survival-promoting effects of propofol
via the inhibition of ATII cell apoptosis (13). At present, the primary mechanism
underlying the effect of propofol on ATII cell autophagy remains to
be elucidated. The aim of the present study was to investigate the
protective effect of propofol on ATII cell autophagy under hypoxia,
and examine the underlying mechanisms.
Materials and methods
Animals
A total of 3 male Sprague-Dawley rats (weight,
150–200 g; age, 3–4 months) were purchased from the Animal
Experimental Centre of the Chinese Academy of Sciences (Shanghai,
China). They were housed in a cage with an atmosphere of 50%
humidity and a 12-h light/dark cycle at room temperature with an
adequate supply of food and water. The present study was approved
by the Ethics Committee of Changzheng Hospital, The Second Military
Medical University (Shanghai, China).
Reagents
Propofol, DNase, trypsin, heparin sodium, the SDS
Gel Preparation kit and N-acetyl-L-cysteine were all purchased from
Sigma-Aldrich; Merck Millipore (Darmstadt, Germany). Pentobarbital
sodium was from Merck Millipore; DMEM and fetal bovine serum (FBS)
were purchased from Gibco; Thermo Fisher Scientific, Inc. (Waltham,
MA, USA). Rabbit primary microtubule-associated protein 1 light
chain 3 (LC3) polyclonal antibody (cat. no. ab48394), rabbit
primary BNIP3 polyclonal antibody (cat. no. ab38621), rabbit
primary GABARAP polyclonal antibody (cat. no. ab1398) and mouse
primary GAPDH monoclonal antibody (cat. no. ab8245) were all
purchased from Abcam (Cambridge, MA, USA). Horseradish peroxidase
(HRP)-conjugated anti-rat immunoglobulin G (IgG; cat. no. sc-2450)
was obtained from MR Biotech (Santa Cruz, CA, USA). Protein A/G
Agarose and RIPA lysis buffer were from Beyotime Institute of
Biotechnology (Haimen, China). The electrophoresis apparatus used
in western blot analysis was from Invitrogen; Thermo Fisher
Scientific, Inc. 3-MA was purchased from Sigma-Aldrich; Merck
Millipore and rapamycin was purchased from Sangon Biotech Co., Ltd.
(Shanghai, China).
Isolation and culture of primary ATII
cells
The ATII cells were isolated from the rats as
described in a previous study (21) with modifications. Briefly, heparin
sodium (400 IU/kg) and pentobarbital (60 mg/kg) were used for rat
anesthesia. Under sterile conditions, open pleuroperitoneal cavity
surgery of the rats was performed, and the lungs were then flushed
with saline and ventilated until they become pale. Bronchoalveolar
lavage was performed to perfuse the lungs. Following removal from
the body and incubated with trypsin for 30 sec, the perfused lungs
were incubated with 10 ml of trypsin at 37°C for 5 min, followed by
the addition of 5 ml trypsin every 5 min three times. Subsequently,
the enzyme reaction was blocked by immersion into DNase containing
FBS, and the lungs were removed and sliced into sections of 1 mm3.
The digested tissue was agitated at 37°C for 5 min on a shaker and
filtered using stainless steel cells strainers. The obtained
filtrate was centrifuged at 4°C for 10 min (800 × g). Following
re-suspension in DMEM containing 0.25% of DNase, 1×106 ATII cells
were incubated with rat IgG at 37°C, which was pre-coated on
culture bottles. After 1 h, non-adherent cells were collected,
suspended in DMEM containing 20% FBS, and cultured at 37°C. The
primary culture of ATII cells entered the logarithmic phase on the
second day, and cells cultured for 24 h were used for subsequent
experiments.
Propofol treatment
The cells were divided into four groups according to
the different treatment regimens: Cells in the control group were
cultured under normoxic conditions; cells in the propofol group
were pre-treated with propofol (10 or 20 µmol/l) for 1 h under
normoxic condition, followed by culturing in normoxic conditions
for 24 h; cells in the hypoxia group were cultured in normoxic
conditions for 1 h and in 5% oxygen for 24 h; cells in the
hypoxia-propofol group were pre-treated with propofol (10 or 20
µmol/l) for 1 h under normoxic conditions, followed by stimulation
under hypoxia (5% oxygen) for 24 h.
HIF 1α siRNA transfection
When the cells were grown to 60–80% confluence,
Lipofectamine 2000 transfection reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) was used for transient transfection according to
the manufacturer's protocol. The cells were divided into a control
group (transfected with scrambled siRNA) and HIF 1α siRNA group. At
48 h post-transfection, 5×105 cells were incubated in normoxia or
hypoxia for 24 h, with or without pre-treatment with propofol (20
µmol/l), as described above. Scrambled siRNA (forward,
5′-GATCCGCTGATGACCGGAACTTG-3′; reverse,
5′-GCTTTTCCAAAAACTGTGCCAGG-3′) and HIF 1α siRNA (forward,
5′-AGAGGUGGAUAUGUGUGGGDTDT-3′; reverse,
5′-GGATCACACACTGTGTCCAGTTT-3′) were synthesized by Invitrogen;
Thermo Fisher Scientific, Inc.
Western blot analysis
The expression levels of proteins associated with
autophagy and apoptosis were determined using western blot
analysis. The cell contents were extracted with an extraction kit
(KGP250; Nanjing Keygen Biotech. Co., Nanjing, China) and incubated
for 30 min at 4°C, followed by sonication and centrifugation (5,000
× g for 30 min at 4°C). The supernatants were collected immediately
and stored at −70°C for later experiments. Protein concentrations
were determined using a Takara Bicinchoninic Protein Assay kit
(cat. no. T9300A; Takara Biotechnology, Co., Ltd., Dalian, China),
according to the manufacturer's instructions. Proteins (20 µg) were
isolated on a 6% SDS-PAGE gel and transferred onto nitrocellulose
membranes (Bio-Rad Laboratories, Inc., Hercules, CA, USA) for
sample analysis. The membranes were blocked with 5% BSA and
incubated with the primary antibodies (1:1,000; all described in
the ‘Reagents’ section) for 16 h at 4°C, followed by incubation
with HRP-conjugated secondary antibodies (1:10,000) for 1 h at room
temperature. ECL reagents were used to detect the blotting
signals.
Co-immunoprecipitation assay
For co-immunoprecipitation, the protein samples (600
µg) were mixed with rabbit anti-rat IgG (1 µg powder; cat. no.
A3231) and Protein A/G Agarose (20 µl), and incubated for 1.5 hat
4°C. Following centrifugation at 2,500 × g for 5 min at 4°C, 1 µg
anti-Bcl-2 primary antibody (1:500; cat. no. 15071; Cell Signaling
Technology, Inc., Danvers, MA, USA) was added to the supernatant
and incubated overnight at 4°C, following which incubation was
continued with another 40 µl Protein A/G Agarose for 2 h at 4°C.
The supernatant was removed carefully following centrifugation at
9,500 × g for 15 min at 4°C. The protein-bead was then washed with
washing solution I containing 50 mM Tris, 150 mM NaCl and 0.1%
NP-40 (pH 7.5) and washing solution II comprising 10 mM Tris (pH
7.5) three times, respectively. The sediments were collected and
boiled in loading buffer for 5 min, prior to loading 20 µg total
proteins/lane for separation using 10% SDS-PAGE.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from the ATII cells using
TRIzol reagent (Promega Corp., Madison, WI, USA) according to the
manufacturer's instruction. The RT-qPCR analysis was performed in
the 7500 Fast Real Time PCR system (Applied Biosystems; Thermo
Fisher Scientific, Inc.). Quantification of gene expression was
performed using the 2−∆∆Cq method (22), with β-actin as an endogenous
control. The total reaction system (final reaction volume, 25 µl;
cat. no. RR036Q; Takara Biotechnology, Co., Ltd.) of RT-qPCR
included 2 µl of the cDNA template, 0.5 µl of the upstream primer,
0.5 µl of the downstream primer, 12.5 µl of the SYBR green super
mix, 0.5 µl of Takara Ex Taq HS (5 U/µH), 0.5 µl of PrimeScript RT
Enzyme Mix II and 8.5 µl of RNase Free dH2O. The primer
sequences used in the present study were as follows: β-actin,
forward 5′-CTATCGGCAATGAGCGGTTC-3′ and reverse
5′-GATCTTGATCTTCATGGTGCTAGG-3′. HIF 1α, forward
5′-AACAAGCCGGGGGAGGACGA-3′ and reverse 5′-GCCACACTGCGGCTGGTCT-3′.
The PCR program was as follows: 50°C for 2 min, 95°C for 5 min,
followed by 40 cycles at 95°C for 15 sec and 60°C for 45 sec.
FITC-conjugated AV/PI staining
Apoptosis was detected using FITC-conjugated AV/PI
staining. The cells were stained with PI solution (20 µg/ml) and
mixed with FITC-conjugated AV for 15 min at room temperature in the
dark. The cells were then washed with 400 µl ice-cold PBS. The
samples were measured within 1 h using a FACScan flow cytometer (BD
Biosciences, Franklin Lakes, NJ, USA) equipped with CellQuest Pro
software version 5.1. For each sample 10,000 events were
collected.
Results
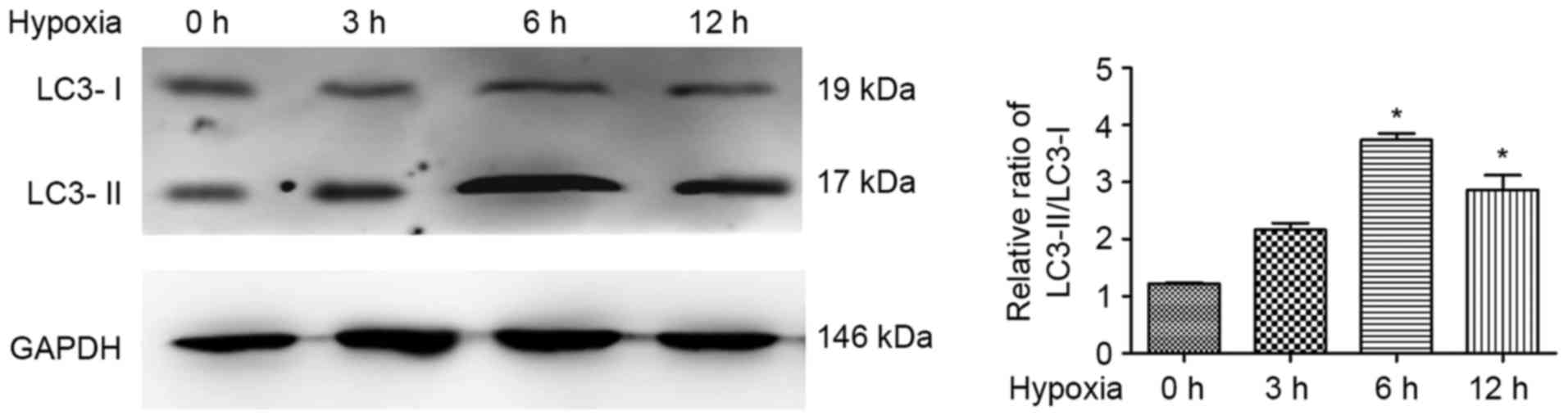
Hypoxia induces autophagy in ATII
cells in a time-dependent manner
As previous investigations revealed that cell
autophagy was induced by hypoxia (23), the present study investigated the
expression levels of LC3 in response to hypoxic exposure in ATII
cells. The results of the western blot analysis revealed that
LC3-II accumulated and the LC3-II/LC3-I ratio was significantly
increased in the hypoxia-treated ATII cells, compared with that in
the control cells, exhibiting a peak at 6 h of hypoxia treatment
(Fig. 1). This indicated that the
induced autophagy was caused by hypoxia treatment in the ATII cells
in a time-dependent manner.
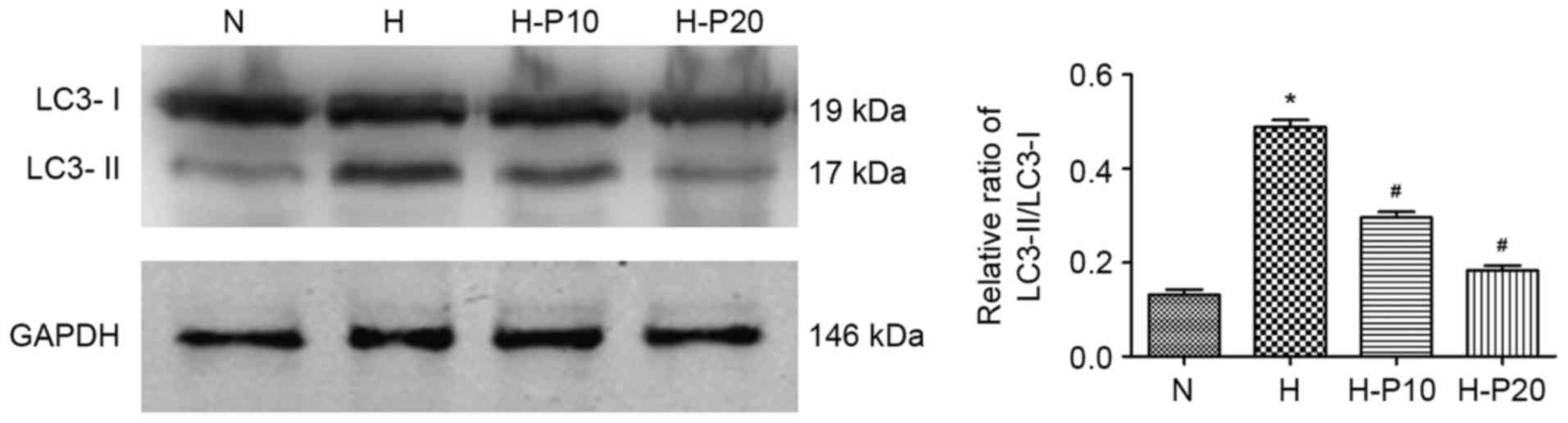
Propofol dose-dependently attenuates
hypoxia-induced autophagy in ATII cells
To examine the effect of propofol on hypoxia-induced
autophagy in ATII cells, the present study evaluated the
accumulation of LC3 in cells with or without pre-treatment with
propofol using western blot analysis. As shown in Fig. 2, hypoxia exposure significantly
enhanced the accumulation of LC3-II and increased the LC3-II/LC3-I
ratio, whereas the enhanced accumulation was significantly reduced
when the cells were pre-treated with propofol, particularly at 20
µM. These results indicated that propofol prohibited
hypoxia-induced autophagy of ATII cells in a dose-dependent
manner.
Effect of propofol on the expression
of autophagy-associated proteins under hypoxic conditions
Autophagy is predominantly regulated by two types of
complex: mammalian target of rapamycin (mTOR) and Beclin-1. To
examine how propofol regulates hypoxia-induced autophagy, the
present study analyzed the expression of several
autophagy-associated proteins involved in these pathways in the
ATII cells. As shown in Fig. 3A,
the expression levels of HIF 1α and Bnip3 were increased in the
cells under hypoxic conditions. In addition, when the cells were
pre-treated with hypoxia, the levels of p-mTOR and Beclin-1 were
significantly reduced. However, pre-treatment with propofol in the
hypoxia-treated ATII cells significantly inhibited the
hypoxia-induced expression of HIF 1α and Bnip3, and upregulated the
expression of p-mTOR. The results of the immunoprecipitation assay
revealed that hypoxia markedly decreased the interaction between
Beclin-1 and Bcl-2, whereas the decreased interaction between
Beclin-1 and Bcl-2 induced by hypoxia was markedly attenuated by
propofol (Fig. 3B).
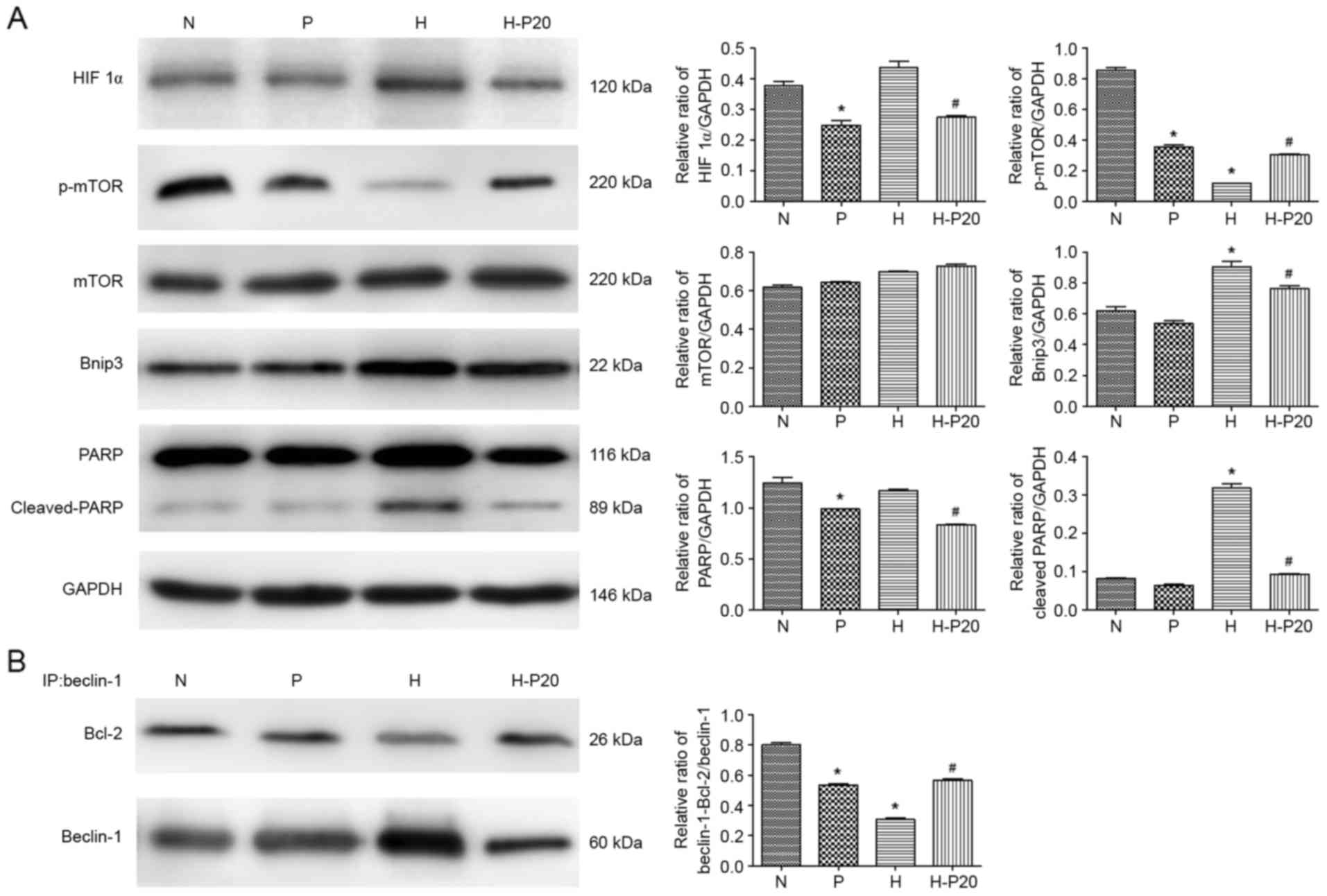 | Figure 3.Expression levels of proteins
associated with autophagy under hypoxia. (A) Expression levels of
autophagy-associated proteins in alveolar epithelial type II cells
were detected using western blot analysis with specific antibodies
against each protein. (B) Immunoprecipitation was used to detect
the Bcl-2/Beclin-1 interaction. Total cell lysates were incubated
with anti-Beclin-1 antibodies. The Bcl-2/Beclin-1 interaction and
total input of Beclin-1were detected using western blot analysis
using anti-Bcl-2 and anti-Beclin-1 antibodies, respectively.
*P<0.05, compared with the control (N) #P<0.05,
compared with the control (H). N, normoxia; P, pre-treated with
propofol (20 µM); H, hypoxia (5% O2); H-P20, pre-treated
with propofol (20 µM) and hypoxia (5% O2). ATII, LC3,
microtubule-associated protein 1 light chain 3; HIF 1α,
hypoxia-inducible factor 1α; mTOR, mammalian target of rapamycin;
p-mTOR, phosphorylated mTOR; Bcl-2, B-cell lymphoma-2; Bnip3, Bcl-2
interacting protein 3; PARP, poly ADP-ribose polymerase. |
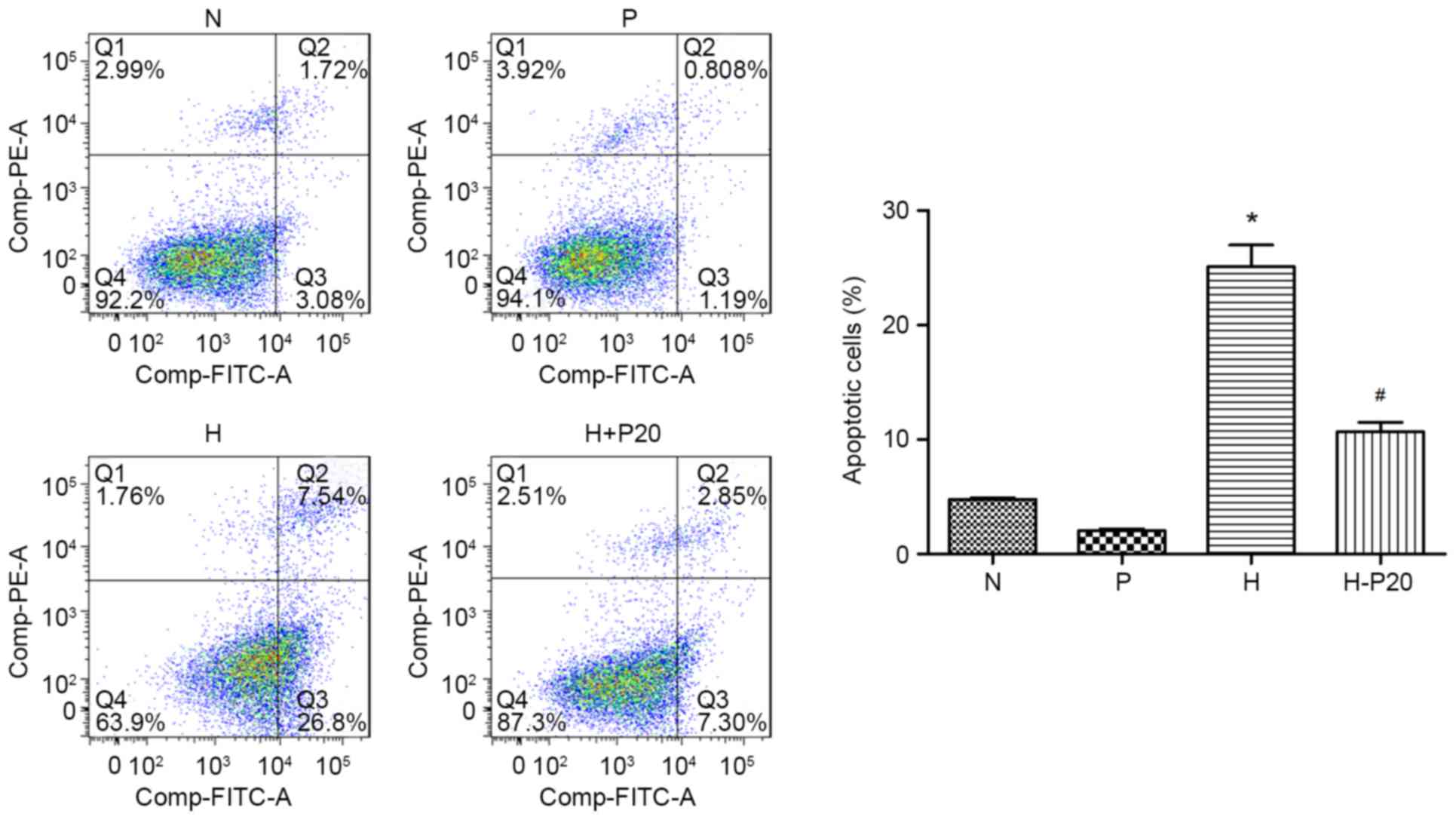
Effect of propofol on hypoxia-induced
apoptosis in ATII cells
It was previously reported that propofol can reduce
hypoxia-induced apoptosis (13).
Therefore, the present study examined cell apoptosis and the
expression of cleaved-poly ADP-ribose polymerase (PARP) in the
different groups. As shown in Fig.
3A, the expression level of cleaved-PARP was markedly increased
when the cells were under hypoxic conditions, however, the
increased expression of cleaved-PARP was reduced when the cells
were pre-treated with propofol. These results were consistent with
changes in autophagy-associated proteins in response to diverse
conditions. The results of the flow cytometry supported the
conclusion that hypoxic condition caused cell apoptosis. Compared
with cells in normal conditions, hypoxia notably promoted the
apoptotic cell ratio, whereas pre-treatment with propofol reversed
the hypoxia-induced apoptosis (Fig.
4).
Knockdown of HIF 1α inhibits
hypoxia-induced autophagy in ATII cells
To further verify whether HIF 1α and Bnip3 regulate
autophagyin ATII cells under hypoxia, specific siRNA targeting HIF
1α was constructed. As shown in Fig.
5A and B, the mRNA and protein expression levels of HIF 1α were
markedly decreased. The expression levels of Bnip3 and LC3-II were
also evaluated in cells treated with hypoxia. The results of the
western blot analysis showed that the hypoxia-induced increase in
the expression levels of Bnip3 and LC3-II was suppressed by the
inhibition of HIF 1α (Fig. 5C).
Consequently, hypoxia-induced autophagy was ameliorated by HIF 1α
gene deletion. These data suggested that the hypoxia-induced
autophagy and apoptosis were reduced by pre-treatment with
propofol, which was partially dependent on the expression level of
HIF 1α.
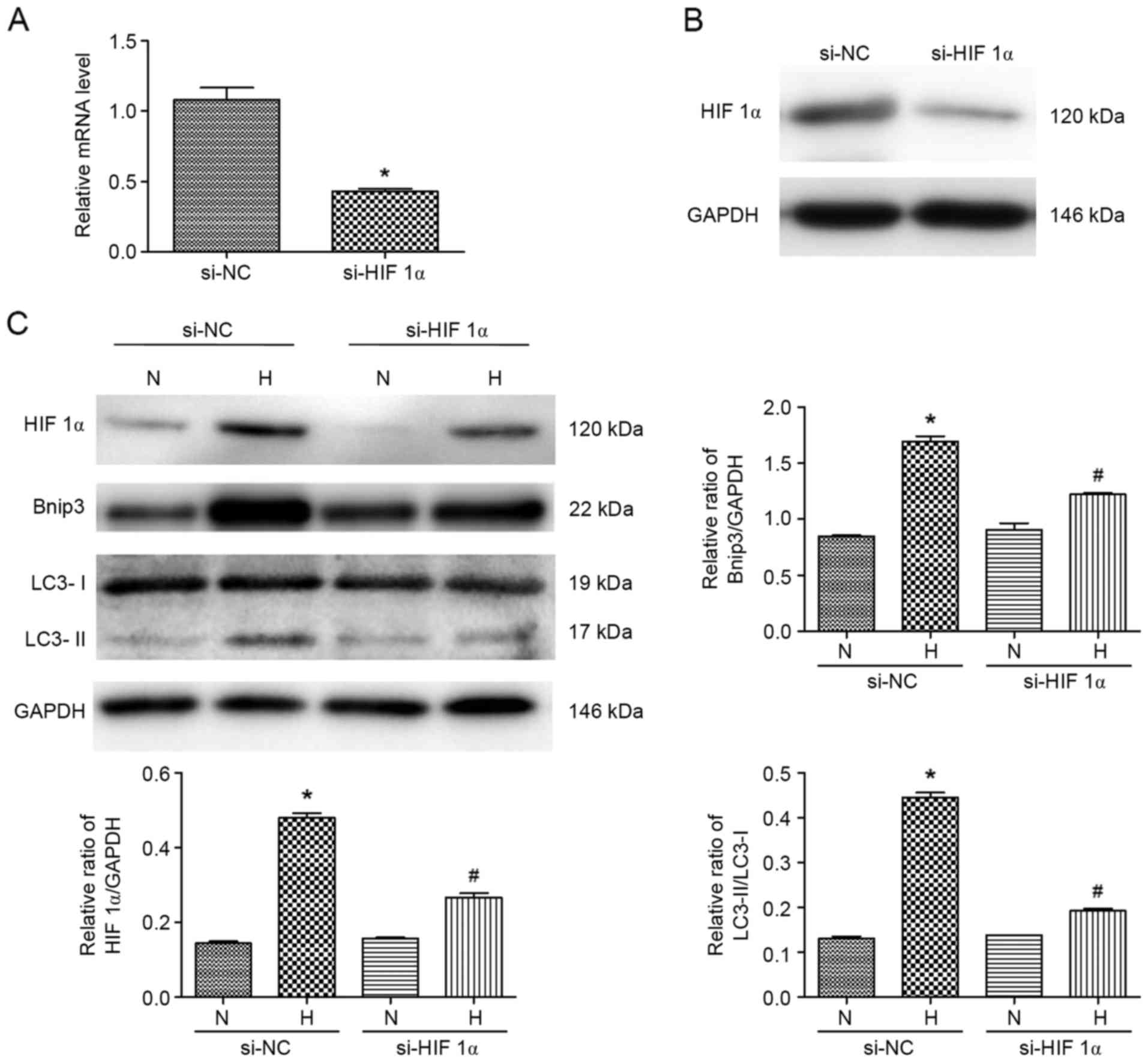 | Figure 5.Specific HIF 1α siRNA target
attenuates hypoxia-induced effects in ATII cells. (A) Transfection
efficiency of si-HIF 1α was verified using reverse
transcription-quantitative polymerase chain reaction analysis.
*P<0.05, compared with control si-NC. (B) Transfection
efficiency of si-HIF 1α was verified using western blot analysis.
(C) Expression levels of Bnip3 and LC3 in ATII cells pre-treated
with si-HIF 1α transfection and exposed to hypoxia were detected
using western blot analysis. *P<0.05, compared with the control
si-NC (N); #P<0.05, compared with the control si-NC
(H). N, normoxia; P, pre-treated with propofol (20 µM); H, hypoxia
(5% O2); H-P20, pre-treated with propofol (20 µM) and
hypoxia (5% O2). ATII, alveolar epithelial type II; LC3,
microtubule-associated protein 1 light chain 3; HIF 1α,
hypoxia-inducible factor1α; Bnip3, Bcl-2 interacting protein 3;
si-, small interfering RNA; NC, negative control. |
Discussion
LC3, which is necessary for the formation of the
autophagosome, is modified by the yeast autophagy associated gene 8
modification system. Following the synthesis of LC3, there are two
forms, termed LC3-I and LC3-II. Among these, LC3-II is the final
form of LC3 and is involved in the formation of the autophagosome.
Therefore, the level of LC3-II and the LC3-II/LC3-I ratio can be
detected to indicate autophagic activity (24,25).
In the present study, LC3-II was regarded as an autophagy molecular
marker and it was found that propofol attenuated the
hypoxia-induced accumulation of LC3-II in ATII cells. The
anti-apoptotic effects of propofol on ATII cells under hypoxic
exposure have been reported previously (13), and these findings were supported by
those of the present study using flow cytometry. To further
investigate whether propofol promotes cell viability associated
with the regulation of autophagy, the present study also detected
the expression of the apoptosis-associated protein, PARP, in
parallel with autophagy-associated proteins. As shown in Fig. 3, the hypoxia-induced upregulation
of cleaved-PARP was markedly reduced by propofol, which occurred
with the inhibition of autophagy. These results demonstrated that
propofol increased the cell survival rate and may rely on the
regulation of autophagy.
Cells respond to hypoxia by activating HIF 1α
(26). As the target gene of HIF
1α, Bnip3 has been implicated in the induction of autophagy and may
be regulated by hypoxia (27).
Autophagy is predominantly regulated by two types of complexes
upstream of ATG, the mTOR complex and the Beclin-1 complex.
Previous studies have shown that the phosphorylation of mTOR
prevented formation of the autophagosome, thus, negatively
mediating autophagy (28,29). Rheb, a Ras-related small GTPase, is
a key upstream activator of mTOR. Bnip3, which is induced by
hypoxia, can directly bind to Rheb (30). As a consequence, Bnip3 leads to the
negative regulation of mTOR. Beclin-1 directly interacts with class
III phosphatidylinositol 3-kinase (PI3KC3) to induce autophagy, and
the inhibition or loss of lipid kinase components inhibits
autophagy (29,31). Beclin-1 also interacts with Bcl-2
family proteins, including Bcl-2. The binding of Bcl-2 with
Beclin-1 prevents the formation of the Beclin-1-PI3KC3 complex and
eventually inhibits autophagy (29). In the present study, it was found
that hypoxia-induced autophagy was associated with the accumulation
of HIF 1α and Bnip3, the deactivation of m-TOR, and attenuation of
the interaction between Beclin-1 and Bcl-2. However, pre-treatment
with propofol altered this status, increasing the level of p-mTOR,
promoting the interaction between Beclin-1 and Bcl-2, and reducing
the levels of HIF 1α and Bnip3 (Fig.
3A and B). These results suggested that propofol negatively
regulated hypoxia-induced autophagy via Bnip3 through the
m-TOR-dependent and Beclin-1-dependent pathways, and that HIF 1α
may act as a regulator in this process.
HIF 1α is important in the regulation of cell
proliferation, apoptosis, autophagy and glucose metabolism under
hypoxic conditions. Previously, suppressing the expression of HIF
1α by silencing HIF 1α in glioblastoma U87 cells was shown to
inhibit the proliferation of U87 cells (32). Glucose metabolism is also promoted
by HIF 1α under hypoxic conditions, which protects liver cells from
damage (33). HIF 1α regulates
apoptosis and autophagy via HIF 1α-microRNA feedback in response to
hypoxia (15,34), and it has been reported that HIF 1α
is essential to the pro-apoptotic response of ATII cells upon
hypoxia (35). In the present
study, HIF 1α was knocked down using siRNA, and it was found that
the hypoxia-induced increase in the expression levels of Bnip3 and
LC3-II was inhibited in these cells (Fig. 5C), which indicated that HIF 1α may
modulate autophagic activity in ATII cells under hypoxia by
regulating the downstream gene, Bnip3.
In conclusion, the results of the present study
demonstrated that propofol was important in the inhibition of the
hypoxia-induced autophagy. In addition, HIF 1α was a crucial
regulator of autophagy in ATII cells via Bnip3, through
m-TOR-dependent and Beclin-1-dependent pathways. These results
provide novel understanding of the effect of propofol in modulating
autophagic cell death via HIF 1α-Bnip3 in hypoxia, and revealed the
potential clinical role of propofol in the treatment of
hypoxic-ischemic pulmonary injury.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81100049), the Shanghai
Municipal Science and Technology Commission medical guide project
(grant no. 15411966000) and the Shanghai Pujiang Program (grant no.
15PJD003).
References
|
1
|
Li C, Xu M, Wu Y, Li YS, Huang WQ and Liu
KX: Limb remote ischemic preconditioning attenuates lung injury
after pulmonary resection under propofol-remifentanil anesthesia: A
randomized controlled study. Anesthesiology. 121:249–259. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bae HB, Li M, Lee SH, Jeong CW, Kim SJ,
Kim HS, Chung SS and Kwak SH: Propofol attenuates pulmonary injury
induced by collapse and reventilation of lung in rabbits.
Inflammation. 36:680–688. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yao W, Luo G, Zhu G, Chi X, Zhang A, Xia Z
and Hei Z: Propofol activation of the Nrf2 pathway is associated
with amelioration of acute lung injury in a rat liver
transplantation model. Oxid Med Cell Longev. 2014:2585672014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hatakeyama N and Matsuda N: Alert cell
strategy: Mechanisms of inflammatory response and organ protection.
Curr Pharm Des. 20:5766–5778. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ma L, Wu XY, Zhang LH, Chen WM, Uchiyama
A, Mashimo T and Fujino Y: Propofol exerts anti-inflammatory
effects in rats with lipopolysaccharide-induced acute lung injury
by inhibition of CD14 and TLR4 expression. Braz J Med Biol Res.
46:299–305. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wei L, Matsumoto H and Yamaguchi H:
Propofol attenuates lipopolysaccharide-induced monocyte
chemoattractant protein-1 production through p38 MAPK and SAPK/JNK
in alveolar epithelial cells. J Anesth. 27:366–373. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yeh CH, Cho W, So EC, Chu CC, Lin MC, Wang
JJ and Hsing CH: Propofol inhibits lipopolysaccharide-induced lung
epithelial cell injury by reducing hypoxia-inducible factor-1alpha
expression. Br J Anaesth. 106:590–599. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tsai YC, Huang CC, Chu LM and Liu YC:
Differential influence of propofol on different cell types in terms
of the expression of various oxidative stress-related enzymes in an
experimental endotoxemia model. Acta Anaesthesiol Taiwan.
50:159–166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Heberlein W, Wodopia R, Bärtsch P and
Mairbäurl H: Possible role of ROS as mediators of hypoxia-induced
ion transport inhibition of alveolar epithelial cells. Am J Physiol
Lung Cell Mol Physiol. 278:L640–L648. 2000.PubMed/NCBI
|
|
10
|
Matthay MA, Folkesson HG and Clerici C:
Lung epithelial fluid transport and the resolution of pulmonary
edema. Physiol Rev. 82:569–600. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Clegg GR, Tyrrell C, McKechnie SR, Beers
MF, Harrison D and McElroy MC: Coexpression of RTI40 with alveolar
epithelial type II cell proteins in lungs following injury:
Identification of alveolar intermediate cell types. Am J Physiol
Lung Cell Mol Physiol. 289:L382–L390. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ghosh MC, Gorantla V, Makena PS, Luellen
C, Sinclair SE, Schwingshackl A and Waters CM: Insulin-like growth
factor-I stimulates differentiation of ATII cells to ATI-like cells
through activation of Wnt5a. Am J Physiol Lung Cell Mol Physiol.
305:L222–L228. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
He XY, Shi XY, Yuan HB, Xu HT, Li YK and
Zou Z: Propofol attenuates hypoxia-induced apoptosis in alveolar
epithelial type II cells through down-regulating hypoxia-inducible
factor-1α. Injury. 43:279–283. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rabinowitz JD and White E: Autophagy and
metabolism. Science. 330:1344–1348. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sun Y, Xing X, Liu Q, Wang Z, Xin Y, Zhang
P, Hu C and Liu Y: Hypoxia-induced autophagy reduces
radiosensitivity by the HIF-1α/miR-210/Bcl-2 pathway in colon
cancer cells. Int J Oncol. 46:750–756. 2015.PubMed/NCBI
|
|
16
|
Yousefi S and Simon HU: Apoptosis
regulation by autophagy gene 5. Crit Rev Oncol Hematol. 63:241–244.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo XG, Ji TX, Xia Y and Ma YY: Autophagy
protects type II alveolar epithelial cells from Mycobacterium
tuberculosis infection. Biochem Biophys Res Commun. 432:308–313.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Thorburn J, Horita H, Redzic J, Hansen K,
Frankel AE and Thorburn A: Autophagy regulates selective HMGB1
release in tumor cells that are destined to die. Cell Death Differ.
16:175–183. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cui DR, Wang L, Jiang W, Qi AH, Zhou QH
and Zhang XL: Propofol prevents cerebral ischemia-triggered
autophagy activation and cell death in the rat hippocampus through
the NF-κB/p53 signaling pathway. Neuroscience. 246:117–132. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen Z, Hu Z, Lu Z, Cai S, Gu X, Zhuang H,
Ruan Z, Xia Z, Irwin MG, Feng D and Zhang L: Differential microRNA
profiling in a cellular hypoxia reoxygenation model upon
posthypoxic propofol treatment reveals alterations in autophagy
signaling network. Oxid Med Cell Longev. 2013:3784842013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dobbs LG, Gonzalez R and Williams MC: An
improved method for isolating type II cells in high yield and
purity. Am Rev Respir Dis. 134:141–145. 1986.PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu YL, Jahangiri A, De Lay M and Aghi MK:
Hypoxia-induced tumor cell autophagy mediates resistance to
anti-angiogenic therapy. Autophagy. 8:979–981. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mizushima N: Methods for monitoring
autophagy. Int J Biochem Cell Biol. 36:2491–2502. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bellot G, Garcia-Medina R, Gounon P,
Chiche J, Roux D, Pouysségur J and Mazure NM: Hypoxia-induced
autophagy is mediated through hypoxia-inducible factor induction of
BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol.
29:2570–2581. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang J and Ney PA: Role of BNIP3 and NIX
in cell death, autophagy, and mitophagy. Cell Death Differ.
16:939–946. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM,
Cao J, Kundu M and Kim DH: ULK-Atg13-FIP200 complexes mediate mTOR
signaling to the autophagy machinery. Mol Biol Cell. 20:1992–2003.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Noh HS, Shin IW, Ha JH, Hah YS, Baek SM
and Kim DR: Propofol protects the autophagic cell death induced by
the ischemia/reperfusion injury in rats. Mol Cells. 30:455–460.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li Y, Wang Y, Kim E, Beemiller P, Wang CY,
Swanson J, You M and Guan KL: Bnip3 mediates the hypoxia-induced
inhibition on mammalian target of rapamycin by interacting with
Rheb. J Biol Chem. 282:35803–35813. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wirth M, Joachim J and Tooze SA:
Autophagosome formation-the role of ULK1 and Beclin1-PI3KC3
complexes in setting the stage. Semin Cancer Biol. 23:301–309.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shen SH, Kwan AL, Chen YY and Wang ZX:
Effect of silencing HIF-1α on proliferation, invasion and migration
of glioblastoma U87 cells. Neurol Sci. 34:365–371. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhuonan Z, Sen G, Zhipeng J, Maoyou Z,
Linglan Y, Gangping W, Cheng J, Zhongliang M, Tian J, Peijian Z and
Kesen X: Hypoxia preconditioning induced HIF-1α promotes glucose
metabolism and protects mitochondria in liver I/R injury. Clin Res
Hepatol Gastroenterol. 39:610–619. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu Y, Nie H, Zhang K, Ma D, Yang G, Zheng
Z, Liu K, Yu B, Zhai C and Yang S: A feedback regulatory loop
between HIF-1α and miR-21 in response to hypoxia in cardiomyocytes.
FEBS Lett. 588:3137–3146. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Krick S, Eul BG, Hänze J, Savai R,
Grimminger F, Seeger W and Rose F: Role of hypoxia-inducible
factor-1alpha in hypoxia-induced apoptosis of primary alveolar
epithelial type II cells. Am J Respir Cell Mol Biol. 32:395–403.
2005. View Article : Google Scholar : PubMed/NCBI
|