Introduction
Patients with pulmonary hypertension (PH) have a
5-year survival ~65% (1) and is
defined by an increase in the mean pulmonary artery pressure >25
mmHg at rest (1–4). Based on various clinical and
hemodynamic features, PH is classified into several subgroups,
which share similar symptom, such as increased breathlessness,
worsening right heart failure and eventually death (1,4) and
similar pathological characteristics, including sustained
vasoconstriction and progressive pulmonary vascular remodeling
(5). Previous studies demonstrated
that vascular cells, such as endothelial, smooth muscle and
adventitial fibroblast cells and the extracellular matrix (ECM) had
an important role in vasculopathy, including intimal and medial
thickening, plexiform lesions, fibrosis and ECM deposition
(5,6). Therefore, strategies that target
vasoconstriction and vascular remodeling and preserve the structure
and function of the pulmonary vasculature may potentially be used
to attenuate the pathological progression of PH.
Angiotensin II (AngII) is considered to contribute
to pathological vascular remodeling. Notably, expression of
angiotensin converting enzyme and AngII type 1 (AT1) receptor are
increased in pulmonary vasculature of PH models (7–9).
Previous studies have demonstrated that losartan (10–12)
and telmisartan (13) attenuate
pulmonary hypertension in humans and animal models, however other
studies failed to demonstrate beneficial effects of angiotensin
receptor blockers (ARBs) on PH (14). These contradictory results caused
by various factors, including differing drug performances and
dosages of the ARBs. The majority available data regarding
valsartan is predominantly derived from hypertensive studies,
however, there is evidence that valsartan has other beneficial
effects on heart failure, including regression in ventricular
remodeling and improvement of systemic vascular resistance
(15–17). Therefore, based on the observed
protective effects of valsartan against inflammation, reactive
oxygen species (ROS) production and tissue remodeling, the present
study hypothesized that valsartan may be effective in attenuating
PH development. Two animal models were used to evaluate the
efficacy of valsartan on PH.
Materials and methods
Ethics
The present study was approved by the Animal Care
and Use Committee of Tongji University (Shanghai, China) and
conducted in accordance with the National Institutes of Health
Guide for the Care and Use of Laboratory Animals (18). Measurements of hemodynamics were
performed under anesthesia and all efforts were made to minimize
the animal suffering and distress.
Animal models and experimental
design
To examine the effects of valsartan on PH, the
present study used two animal models: Monocrotaline (MCT)-induced
PH model (drug and toxin induced pulmonary arterial hypertension)
and a hypoxia-induced PH model (pulmonary hypertension due to lung
diseases and/or hypoxia) (2). Male
Sprague-Dawley rats (110–130 g) and male C57BL/6 mice (22–26 g)
were purchased from Shanghai Slac Laboratory Animal Co., Ltd
(Shanghai, Chain) and housed in standard cages given water and
normal diet under room temperature in controlled light conditions
(a 12 h light/dark cycle).
As previously described, MCT (Oakwood Products Inc.,
SC, USA) was dissolved in 0.5 N HCl and was adjusted to pH 7.4 with
0.5 N NaOH (19). The solution was
administered as a single subcutaneous injection (40 mg/kg) to male
rats to induce PH (19). Rats
received a single injection of MCT or saline (control group, n=6).
Following MCT injection, animals were treated with valsartan (MCT +
valsartan 20 mg/kg group, n=7; MCT + valsartan 40 mg/kg group, n=6)
or vehicle (an equal volume of saline, MCT group, n=8) once daily
by gavage for 3 weeks. Valsartan was purchased from Novartis China
(Beijing, China).
Male mice were exposed to hypobaric hypoxia as
described previously (20).
Briefly, the pressure chamber was decreased progressively from 0.8
atm (16.9% O2) on day 1 to 0.5 atm (10.5% O2)
following day 7 and was maintained at 10.5% O2 for a
further 3 weeks. The chamber was opened once every week for
cleaning and feeding. Mice exposed to hypobaric hypoxia were
randomly divided into 2 groups for oral administration of valsartan
(hypoxia + 20 mg/kg valsartan group, n=10; hypoxia + 40 mg/kg
valsartan group, n=10) or vehicle (an equal volume of saline,
hypoxia group, n=19). Control mice were maintained in normobaric
conditions (n=8) (20).
Measurements of aortic pressure and
right ventricular (RV) hemodynamics
Following anaesthetization and tracheotomy of rats,
a polyethylene catheter was introduced via the right common carotid
artery for measurement of systemic arterial pressure 3 weeks after
MCT injection. A right heart catheter was then introduced into the
right external jugular vein and the tip advanced into the right
ventricle until a typical right ventricular pressure wave pattern
appeared, as described previously (19,20).
Hemodynamic parameters were assessed by a polygraph system
(PowerLab 8/30; ADInstruments, Bella Vista, NSW, Australia).
After 4 weeks of hypobaric hypoxia treatment, mice
were anesthetized and intubated with a 20-gauge Teflon tube
attached to a MiniVent type 845 mouse ventilator (Hugo Sachs
Elektronik GmbH, Germany). A pressure catheter was introduced via
the right common carotid artery into the ascending aorta for
measurement of systolic and diastolic blood pressures and left
ventricular (LV) hemodynamics were measured as previously described
(21). For RV hemodynamics,
open-chest RV catheterization was performed during anesthesia with
1.5% isoflurane. Data were collected when a steady state was
reached (19,20).
Sample preparation
Following hemodynamic assessment at the
aforementioned indicated time point, all rats and mice were
euthanized by exsanguination and the heart, lung and other major
organs were harvested. Lung weight was measured and the left lung
was frozen in liquid nitrogen until biochemical analysis. The
airways of the top right lobe were perfused with and fixed in 10%
buffered formalin for histological analysis. The RV free wall was
dissected from the left ventricular septum (LV + S) and weighed
separately. The ratio of RV/(LV + S; g/g) and RV/body weight (BW;
mg/g) were calculated as an index of RV hypertrophy (19,20).
Histological analysis
Following hemodynamic measurements, rat lung tissue
was fixed in 10% formalin for >24 h, dehydrated, embedded in
paraffin and subsequently sectioned at 5 µm for morphometric
analyses. Pulmonary vascular muscularization was determined by
hematoxylin and eosin staining, using Olympus Inverted Microscope
IX83 (Olympus Corporation, Tokyo, Japan). Briefly, the medial wall
thickness of the arteries with a diameter of 50–100 µm was
calculated, according to the following formula: Medial thickness
(%) = 2 × medial wall thickness/arterial external diameter × 100.
Additionally, a total of 60 intra-acinar arteries were examined in
each rat and categorized as non-muscular, partially muscular or
fully muscular arteries (19) and
the relative percentage were calculated. Lung fibrosis was detected
using Masson's Trichrome Stain kit (cat. no. BB-44222-1; Bestbio,
Shanghai, China, http://www.bestbio.com.cn/).
Immunohistochemical analysis
In order to evaluate proliferating cells,
proliferating cell nuclear antigen (PCNA, cat. no. sc-25280; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA) staining was performed
in rat lung tissue sections. Slides (5 µm) were dry-heated at 65°C
for 1 h, deparaffinized with xylene, and rehydrated in serial
dilutions of ethanol. Antigen retrieval was performed by incubation
in citrate buffer for 20 min at 95–100°C, followed by washing in
PBS. The sections were incubated with 3% H2O2
in PBS for 20 min followed by 10% normal goat serum blocking
solution (cat. no. 50062Z; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) for 30 min at room temperature. Sections were then
incubated with a mouse monoclonal primary antibody against PCNA
(1:200) overnight at 4°C, followed by incubation with rabbit
anti-mouse biotinylated horseradish peroxidase-conjugated secondary
antibody (cat. no. GP016129; Gene Tech Biotechnology, Shanghai,
China) at the dilution of 1:200 for 1 h at room temperature. The
percentage of PCNA-positive cells were calculated in 10 randomly
chosen fields of each section using an inverted microscope IX83 at
×400 magnification (22,23).
Western blot analysis
Rat lung tissues were homogenized using a cell lysis
buffer supplemented with protease inhibitor (cat nos. 9803 and
5871; Cell Signaling Technology, Billerica, MA, USA) using a tissue
homogenizer and the protein concentrations were assessed using a
BCA Protein Assay kit (cat. no. 23225; Thermo Fisher Scientific,
Inc.). Protein samples (40 µg total protein/well) were separated
with 10% SDS-polyacrylamide gel electrophoresis and transferred to
a nitrocellulose membrane. Membranes were blocked with 5% BSA (cat.
no. 97061-416; VWR International, Radnor, PA, USA) in 0.1% TBS
Tween-20 for 1 h at room temperature prior to 4oC overnight
incubation with the indicated primary antibodies, followed by
incubation with goat anti-mouse and donkey anti-rabbit fluorescent
secondary antibodies (cat. nos. 926-32220 and 926-32213; LiCor
Biosciences, Lincoln, TN, USA) at the dilution of 1:10,000 for 1 h
in the dark at room temperature. The membranes were visualized
using the Odyssey system (LiCor Biosciences) and the relative
protein levels were quantified using Image J version 1.4.3.67
(imagej.nih.gov/ij/). Antibodies for
total and phosphorylated extracellular regulated kinase (ERK) 1/2
(cat. nos. 9102 and 4370), c-Jun N-terminal kinase (JNK) 1/2 (cat.
nos. 9252 and 9255), p38 (cat. nos. 9212 and 4511), cyclin D1 (cat.
no. 2978), histone H3 (cat. no. 4499) and β-actin (cat. no. 3700),
where total and phosphorylated ERK1/2, JNK1/2, p38, cyclin D1 and
histone H3 were diluted at 1:1,000 and β-actin was diluted at
1:2,000 and purchased from Cell Signaling Technology, Inc. The
antibodies for PCNA (cat. no. sc-25280; 1:300) and transforming
growth factor β1 (TGF-β1, cat. no. sc-146; 1:1,000) were purchased
from Santa Cruz Biotechnology, Inc. Antibodies for matrix
metalloproteinases (MMP)-2 (cat. no. ab37150; 1:500) and MMP-9
(cat. no. ab38898; 1:800) were purchased from Abcam (MA, USA).
Statistical analysis
All values are expressed as the mean ± standard
error. Differences among multiple groups were analyzed by one-way
analysis of variance, followed Bonferroni's correction as a
post-hoc analysis. Statistical analysis was performed using SPSS
version 13.0 (SPSS Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Valsartan significantly attenuates
MCT-induced increase of RV pressure and RV hypertrophy in rats
The MCT group developed severe PH at day 21 with
increased RV systolic pressure (RVSP; a marker of systolic
pulmonary pressure) compared with control group (Fig. 1A and Table I). Valsartan treatment
dose-dependently suppressed development of PH in the low and
high-dose valsartan groups (a marked reduction in RVSP by 13 and
21%, respectively, compared with the MCT group; P=0.15 and P=0.01).
In addition, valsartan at the concentration of 40 mg/kg reversed
MCT-induced increase of RV hypertrophy, as indicated by increased
RV, RV/BW, and RV/(LV+S) (Fig.
1B-D and Table I). These
findings demonstrated that 40 mg/kg valsartan treatment
significantly suppressed MCT-induced increase of RVSP and RV
hypertrophy. It is of note that the mean systemic arterial pressure
was significantly decreased in the MCT group (95±5 mmHg) compared
with control group (109±2 mmHg; P<0.01). However, valsartan did
not affect systemic arterial pressure in low and high-dose groups
at day 21 (92±8 and 90±8 mmHg, respectively) compared with MCT
group.
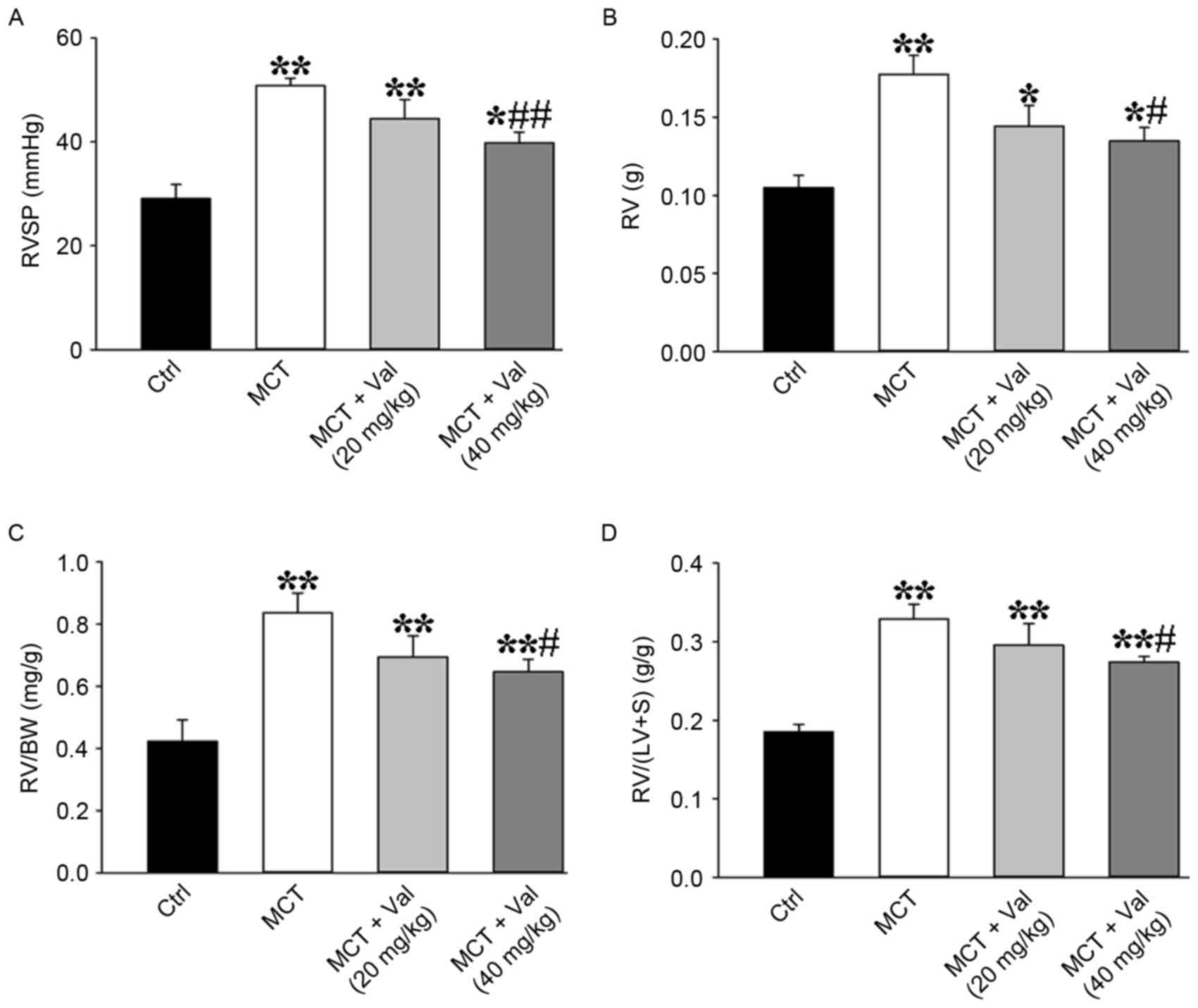 | Figure 1.Valsartan reduces MCT-induced
increased RVSP and RV hypertrophy index. Valsartan treatment
significantly attenuated MCT-induced increases of (A) RVSP, RV
hypertrophy as indicated by (B) RV weight, (C) the ratio of RV
weight to BW and (D) the ratio of RV weight to (LV+S) weight (Ctrl
group, n=6; MCT group, n=8; MCT+Val (20 mg/kg) group n=7; MCT+Val
(40 mg/kg) group, n=6). Results are expressed as the mean ±
standard error. *P<0.05, **P<0.01 vs. Ctrl;
#P<0.05, ##P<0.01 vs. MCT group. BW,
body weight; Ctrl, control; LV, left ventricle; MCT, monocrotaline;
RV, right ventricle; RVSP, right ventricular systolic pressure; S,
septum; Val, valsartan. |
 | Table I.Morphometric parameters of rats at
sacrifice. |
Table I.
Morphometric parameters of rats at
sacrifice.
|
| Group |
|---|
|
| Group |
|---|
| Feature | Control | MCT | MCT + valsartan (20
mg/kg) | MCT + valsartan (40
mg/kg) |
|---|
| No. of rats | 6 | 8 | 7 | 6 |
| BW (g) | 251.86±4.13 |
213.75±5.46b | 208.43±3.57 | 208.83±1.78 |
| RV (g) |
0.11±0.01 |
0.18±0.01b |
0.14±0.01 |
0.14±0.01c |
| LV+S (g) |
0.56±0.02 |
0.54±0.01 |
0.49±0.03 |
0.49±0.03 |
| Ventricle (g) |
0.67±0.03 |
0.71±0.02 |
0.63±0.03 |
0.63±0.03 |
| RV/(LV+S)
(g/g) |
0.19±0.01 |
0.33±0.02b |
0.30±0.03 |
0.27±0.01c |
| RV/BW (mg/g) |
0.42±0.03 |
0.84±0.06b |
0.69±0.07 |
0.65±0.04c |
| RA (g)a |
0.10±0.01 |
0.13±0.02 |
0.13±0.02 |
0.12±0.01 |
| LA (g)a |
0.14±0.01 |
0.15±0.02 |
0.16±0.02 |
0.13±0.02 |
| RA/LA
(g/g)a |
0.67±0.08 |
0.87±0.04 |
0.88±0.17 |
0.96±0.12 |
Valsartan ameliorated hypoxia-induced
increase of RV pressure and RV hypertrophy in mice
To confirm the effects of valsartan on PH, a
hypoxia-induced PH mice model was used in the present study.
Consistent with the results in MCT-induced PH rat model, mice
treated with high-dose valsartan exhibited significant decreases in
hypoxia-induced increase of RVSP (Fig.
2A). Additionally, high-dose valsartan significantly reduced RV
hypertrophy, such as RV weight from 0.032±0.001 g in the hypoxia
group to 0.028±0.001 g in hypoxia+valsartan (40 mg/kg) group
(P<0.05; Fig. 2B and Table II), RV/BW from 1.30±0.04 mg/g in
the hypoxia group decreasing to 1.15±0.04 mg/g in hypoxia+valsartan
(40 mg/kg) group, (P<0.05; Fig.
2C and Table II), and RV/(LV
+ S) from 0.41±0.01 g/g in hypoxia group decreasing to 0.37±0.01
g/g in hypoxia+valsartan (40 mg/kg) group (P<0.05; Fig. 2D and Table II). The data therefore suggested
that 40 mg/kg valsartan treatment effectively attenuated the
development of PH in the mouse hypoxia and rat MCT models.
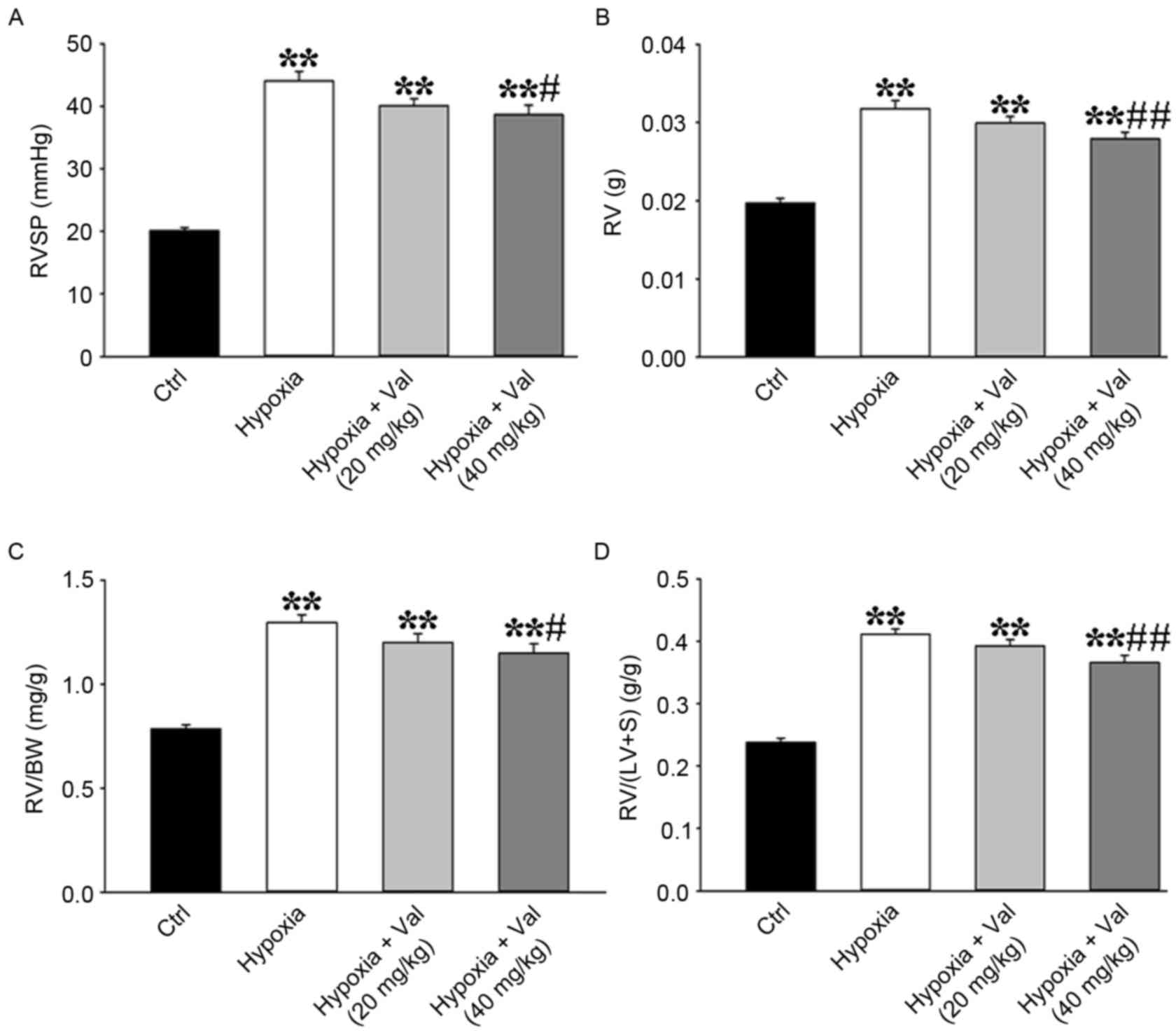 | Figure 2.Valsartan inhibits increased RVSP and
RV hypertrophy index induced by hypoxia. Valsartan treatment in
mice attenuated hypoxia-induced increases of (A) RVSP, RV
hypertrophy as indicated by (B) RV weight, (C) the ratio of RV
weight to BW and (D) the ratio of RV weight to (LV + S) weight
(Ctrl group, n=8; hypoxia group, n=19; hypoxia+Val (20 mg/kg)
group, n=10; hypoxia+Val (40 mg/kg) group, n=10). Results are
expressed as the mean ± standard error. **P<0.01 vs. Ctrl;
#P<0.05, ##P<0.01 vs. hypoxia group.
BW, body weight; Ctrl, control; LV, left ventricular; RV, right
ventricle; RVSP, right ventricular systolic pressure; S, septum;
Val, valsartan. |
| Table II.Anatomic data of control, hypoxia and
hypoxia + valsartan groups. |
Table II.
Anatomic data of control, hypoxia and
hypoxia + valsartan groups.
|
| Experimental
group |
|---|
|
|
|
|---|
| Feature | Control | Hypoxia | Hypoxia + valsartan
(20 mg/kg) | Hypoxia + valsartan
(40 mg/kg) |
|---|
| No. of mice | 8 | 19 | 10 | 10 |
| BW (g) | 25.08±0.63 | 24.50±0.32 | 24.99±0.36 | 24.38±0.55 |
| Heart (g) | 0.1085±0.0042 | 0.1160±0.0025 | 0.1132±0.0017 | 0.1107±0.0017 |
| Lung (g) | 0.1322±0.0056 |
0.1874±0.0024a | 0.1852±0.0031 |
0.1741±0.0040b |
| Liver (g) | 0.8833±0.0412 | 0.8829±0.0114 | 0.8939±0.0277 | 0.8787±0.0218 |
| Kidney (g) | 0.2668±0.0050 | 0.2724±0.0051 | 0.2669±0.0064 | 0.2596±0.0069 |
| Spleen (g) | 0.0679±0.0023 | 0.0676±0.0016 | 0.0670±0.0013 | 0.0651±0.0013 |
| RA (g) | 0.0028±0.0001 |
0.0044±0.0001a | 0.0042±0.0002 | 0.0039±0.0003 |
| LA (g) | 0.0028±0.0003 | 0.0027±0.0001 | 0.0028±0.0001 | 0.0027±0.0001 |
| RV (g) | 0.0197±0.0006 |
0.0318±0.0011a | 0.0299±0.0008 |
0.0279±0.0009c |
| LV+S (g) | 0.0832±0.0035 | 0.0770±0.0016 | 0.0763±0.0010 | 0.0762±0.0011 |
| RV/(LV+S)
(g/g) | 0.2381±0.0067 |
0.4115±0.0084a | 0.3925±0.0101 |
0.3662±0.0115c |
| RV/BW (mg/g) | 0.7858±0.0189 |
1.2962±0.0378a | 1.2006±0.0418 |
1.1490±0.0449b |
Valsartan attenuated MCT-induced
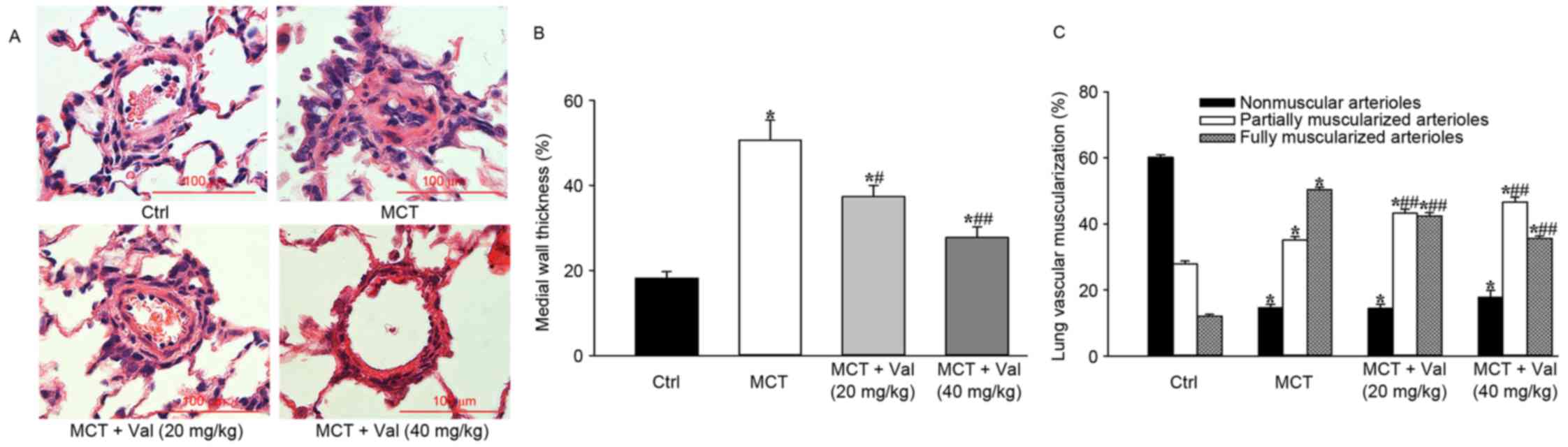
pulmonary vascular remodeling and lung fibrosis in rats
Histological and biochemical analysis of lung
tissues was performed to examine whether valsartan influences
pulmonary vascular remodeling in the MCT-induced PH model. In
comparison with vehicle treated MCT model rats, valsartan treatment
significantly reduced lung vessel medial wall thickness of arteries
with an external diameter of 50–100 µm (P<0.05) and the ratio of
fully muscularized distal pulmonary arteries to total number of
arteries (P<0.01; Fig. 3).
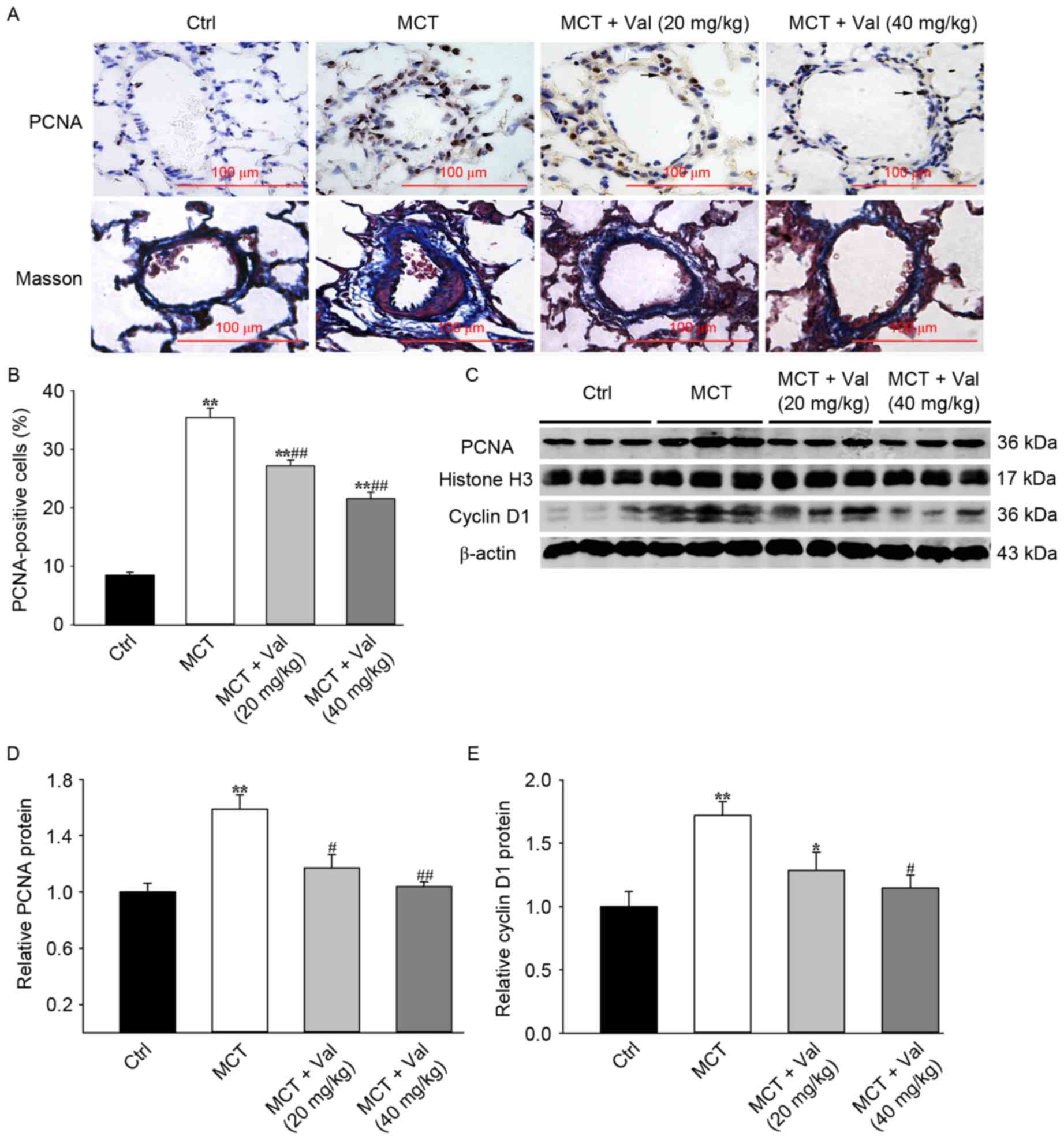
PCNA expression is a marker for cell proliferation.
Immunohistochemical analysis of lung tissue from control and
valsartan treated animals revealed that valsartan treatment
significantly reduced the percentage of PCNA-positive cells in the
lung vessels compared with MCT group (P<0.01; Fig. 4A and B). Increased cell
proliferation is indicative of increased progression through the
cell cycle and western blot analysis indicated that valsartan
significantly reduced cyclin D1 and PCNA protein expression
(Fig. 4C-E).
Perivascular fibrosis is a common indicator of
vascular inflammation and tissue remodeling that occurs in PH.
Masson's trichrome staining revealed that valsartan treatment
reduced the number of dense focal collagen deposits induced by MCT
treatment, compared with vehicle treated animals (Fig. 4A).
Valsartan attenuates MCT-induced
increase of lung mitogen-activated protein kinase (MAPK) signaling
pathways in rats
MAPK signaling is important in cell growth and
proliferation. Therefore, the present study determined if valsartan
influenced MAPK activation in the lung of MCT-treated rats. Among
the four groups, differences in the total protein expression of
p38, JNK1/2, ERK1/2 failed to reach statistical significance
(Fig. 5). However, there was a
significant increase in the phosphorylation of p38, JNK1/2 and
ERK1/2 in the MCT group compared with control group (P<0.05).
Valsartan treatment reduced the MCT-induced phosphorylation of p38,
JNK1/2 and ERK1/2 (P<0.05).
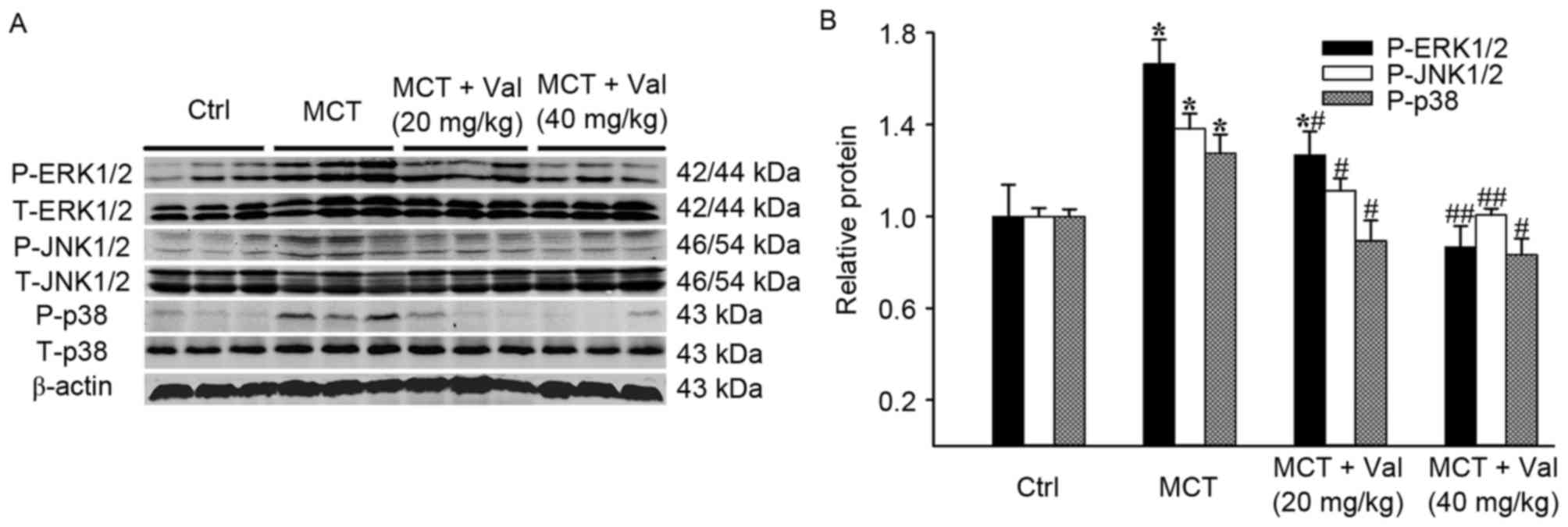 | Figure 5.Effects of valsartan on MCT-induced
phosphorylation of p38, JNK and ERK in lung tissue. (A)
Representative blot of total and phosphorylated protein expression
levels of p38, JNK and ERK detected by western blot analysis (n=5
rats per group). (B) Quantification of phosphorylated protein
expression levels of p38, JNK and ERK relative to total p38, JNK
and ERK. Results are expressed as the mean ± standard error.
*P<0.05 vs. Ctrl; #P<0.05, ##P<0.01
vs. MCT group. Ctrl, control; ERK, extracellular regulated kinase;
JNK, c-Jun N-terminal kinase; MCT, monocrotaline; P,
phosphorylated; T, total; Val, valsartan. |
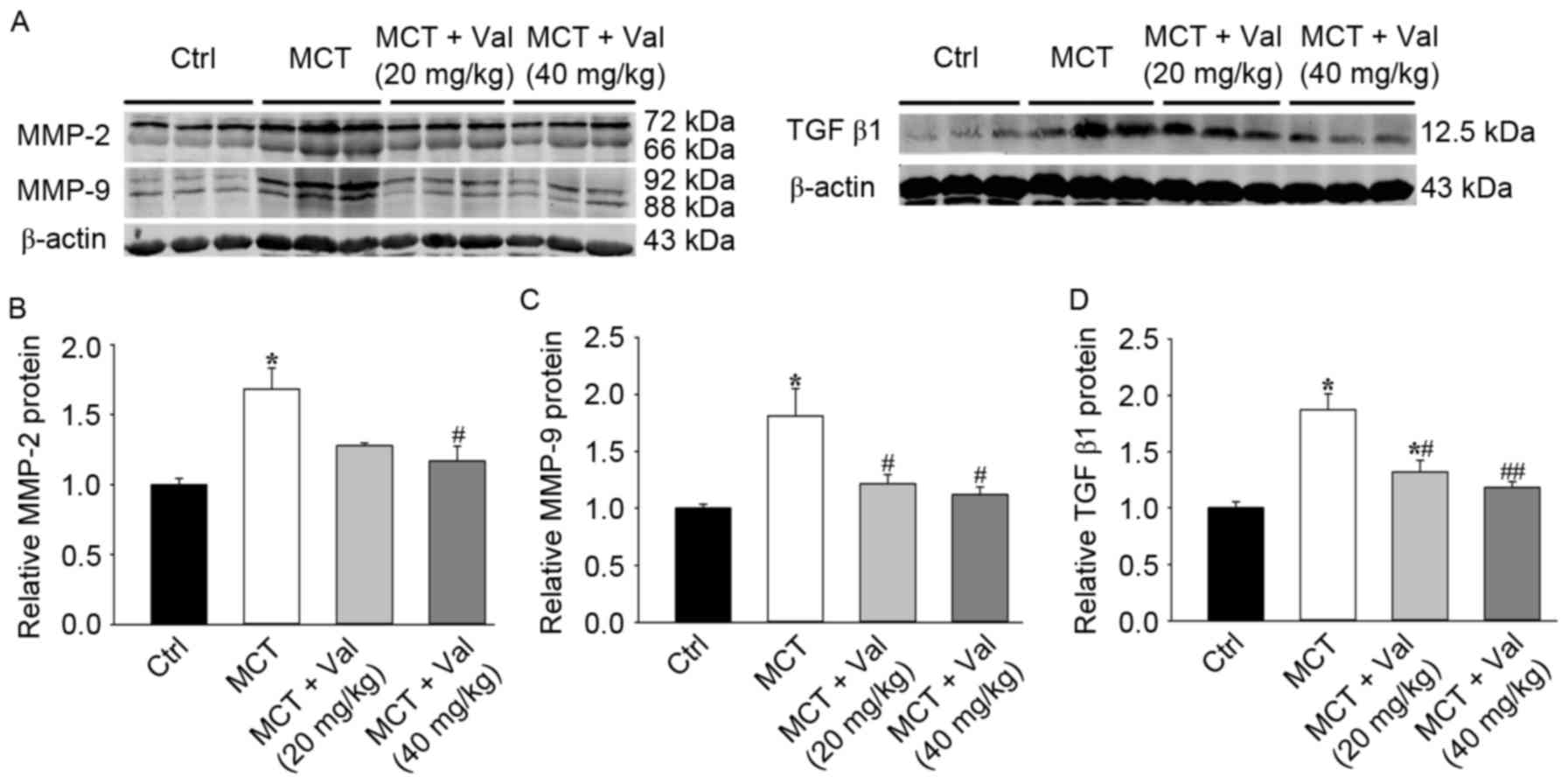
Valsartan attenuated MCT-induced
expression of lung MMPs and TGF-β1 in rats
MMP-2 and −9 degrade ECM components and are
important in smooth muscle cell migration, tissue remodeling and PH
development (13). Consistent with
increased tissue remodeling in the PH model, MCT resulted in a
significant upregulation of lung MMP-2 and −9 expression. The
increase in MMP-2 and −9 expression however, was significantly
attenuated by valsartan treatment (Fig. 6). Valsartan significantly
attenuated lung MMP-2 and −9 activity, as indicated by reduced
cleavage to the lower molecular weight active form (Fig. 6A-C). TGF-β1 is an established
tissue remodeling factor that promotes fibrosis and smooth muscle
cell proliferation in PH (20).
MCT resulted in a significant increase of lung TGF-β1 protein
content, which was significantly diminished by valsartan treatment
(Fig. 6A and D). These data
indicated that valsartan reduced expression of tissue remodeling
factors in PH.
Discussion
The present study demonstrated that valsartan
significantly attenuated the development of PH and right
ventricular hypertrophy in two PH animal models. The protective
effects were modulated, in part, by the inhibition of MAPK
signaling. Furthermore, valsartan reduced expression of lung MMP-2,
−9 and TGF-β1 in rats. These findings suggested that valsartan
suppressed pulmonary vascular remodeling and attenuated development
of PH in the rodents.
Previous studies have indicated that the
renin-angiotensin system is involved in development of PH (7–9).
Angiotensin and the AT1 receptor have been demonstrated to exhibit
an increase in PH patients (9),
and beneficial effects of ARBs, including losartan and telmisartan,
have been confirmed in cases of PH (10–13).
However, further studies have demonstrated that losartan does not
attenuate PH in rodents (24).
There may be various reasons for these inconsistent results from
the ARB on PH. Firstly, these cardioprotective effects may only be
mediated in part via the AT1 receptor signaling pathway.
Additionally, differing doses of ARBs may result in varying effects
on PH. However, valsartan has a 20,000-fold greater affinity for
AT1 compared with the AngII-type 2 receptor (25). The Valsartan in Heart Failure Trial
(Val-HeFT) study demonstrated that the beneficial effects of the
drug are maintained in patients with renal dysfunction at baseline
and those experiencing early worsening of renal function (26). Furthermore, Katada et al
(27) suggested that valsartan
exerts greater positive effects on cognitive functions than
amlodipine in elderly hypertensive patients, independent of the
antihypertensive effects. In addition, valsartan reduces levels of
inflammatory factors, ROS and expression of tissue plasminogen
activator (28,29). The present study demonstrated that
valsartan reduced vascular remodeling, RVSP and RV hypertrophy in a
dose-dependent manner. It is of note, that valsartan at 40 mg/kg
revealed increased efficacy compared with PH rodents, whereas 20
mg/kg demonstrated only a marginal beneficial effect in attenuating
PH.
Accumulating evidence indicates that impairment of
pulmonary vascular homeostasis is important in the pathobiology of
PH (30). AngII stimulates
expression of MMPs (31,32) in vascular smooth muscle cells,
which are important for ECM breakdown and cell migration. The
matrix degrading enzymes MMP-2 and −9 are essential in mediating
structural alterations in tissue growth, however increased MMP-2 or
−9 activity may contribute to pathological lung vascular remodeling
by promoting smooth muscle cell migration (33) and inflammatory cell infiltration
(34). Previous studies have
demonstrated that MMP-2 and −9 expression and activity are
increased in the lung vasculature of PH rodents (35). MMP inhibition limits pulmonary
vascular remodeling in MCT-induced (36) and hypoxia-induced PH (37). Conversely, Vieillard-Baron et
al (36) reported that MMP
inhibition increases pulmonary vascular remodeling in
hypoxia-induced PH. The present study demonstrated that valsartan
decreased vascular remodeling induced by MCT. Valsartan
additionally suppressed MCT-induced increase of MMP-2 and −9
expression. MMPs may therefore exhibit various effects in differing
PH models at any different time point.
Additionally, valsartan suppressed TGF-β1 expression
in MCT-induced PH. TGF-β1 is a cytokine that regulates numerous
cellular responses. Increased TGF-β1 expression has been identified
as a maladaptive factor in PH (38), in part via activation of
activin-like kinase 5, which promotes vascular smooth muscle cell
proliferation (39). The
repression of TGF-β1 expression by valsartan may explain how it
reduces smooth muscle cell proliferation in MCT-induced PH. Another
mechanism by which valsartan may reduce vascular smooth muscle cell
proliferation is via inhibition of p42/44 ERK/MAPK activation. The
ERK/MAPK signaling pathway is important for cell cycle progression
in numerous cell types. AngII promotes p42/44 ERK/MAPK dependent
proliferation of pulmonary artery smooth muscle cells via the AT1
receptor (40). The findings
demonstrated that valsartan treatment suppressed MCT-induced p42/44
MAPK activation. The additional observation of decreased PCNA and
cyclin D1 expression levels, suggested an additional mechanism by
which valsartan prevents smooth muscle cell proliferation and the
maladaptive vascular remodeling that drives PH. However, the
present study is not able to exclude further potential mechanisms
by which vascular remodeling may be modulated by valsartan.
In conclusion, valsartan suppressed pulmonary
vascular remodeling and RV hypertrophy in MCT and hypoxia-induced
PH animal models. These beneficial effects of valsartan may have
occurred via inhibition of MAPK signaling and MMP expression. These
findings suggest that valsartan may act as a valuable therapeutic
approach for the treatment of PH in the future.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81270194 to D.X).
The authors would like to thank Professor John Fassett from the
Lillehei Heart Institute and the Cardiovascular Division,
University of Minnesota (Minneapolis, MN, USA) for critical
comments and suggestions.
References
|
1
|
Farber HW, Miller DP, Poms AD, Badesch DB,
Frost AE, Muros-Le Rouzic E, Romero AJ, Benton WW, Elliott CG,
McGoon MD and Benza RL: Five-Year outcomes of patients enrolled in
the REVEAL Registry. Chest. 148:1043–1054. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Simonneau G, Gatzoulis MA, Adatia I,
Celermajer D, Denton C, Ghofrani A, Sanchez MA Gomez, Kumar R
Krishna, Landzberg M, Machado RF, et al: Updated clinical
classification of pulmonary hypertension. J Am Coll Cardiol. 62 25
Suppl:D34–D41. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Opitz C, Rosenkranz S, Ghofrani HA, Grunig
E, Klose H, Olschewski H and Hoeper M: ESC guidelines 2015
pulmonary hypertension: Diagnosis and treatment. Dtsch Med
Wochenschr. 141:1764–1769. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hurdman J, Condliffe R, Elliot CA, Davies
C, Hill C, Wild JM, Capener D, Sephton P, Hamilton N, Armstrong IJ,
et al: ASPIRE registry: Assessing the spectrum of pulmonary
hypertension identified at a REferral centre. Eur Respir J.
39:945–955. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thompson AA and Lawrie A: Targeting
Vascular remodeling to treat pulmonary arterial hypertension.
Trends Mol Med. 23:31–45. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chelladurai P, Seeger W and Pullamsetti
SS: Matrix metalloproteinases and their inhibitors in pulmonary
hypertension. Eur Respir J. 40:766–782. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Morrell NW, Atochina EN, Morris KG,
Danilov SM and Stenmark KR: Angiotensin converting enzyme
expression is increased in small pulmonary arteries of rats with
hypoxia-induced pulmonary hypertension. J Clin Invest.
96:1823–1833. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Orte C, Polak JM, Haworth SG, Yacoub MH
and Morrell NW: Expression of pulmonary vascular
angiotensin-converting enzyme in primary and secondary plexiform
pulmonary hypertension. J Pathol. 192:379–384. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
de Man FS, Tu L, Handoko ML, Rain S,
Ruiter G, Francois C, Schalij I, Dorfmuller P, Simonneau G, Fadel
E, et al: Dysregulated renin-angiotensin-aldosterone system
contributes to pulmonary arterial hypertension. Am J Respir Crit
Care Med. 186:780–789. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Morrell NW, Higham MA, Phillips PG, Shakur
BH, Robinson PJ and Beddoes RJ: Pilot study of losartan for
pulmonary hypertension in chronic obstructive pulmonary disease.
Respir Res. 6:882005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rondelet B, Kerbaul F, van Beneden R,
Hubloue I, Huez S, Fesler P, Remmelink M, Brimioulle S, Salmon I
and Naeije R: Prevention of pulmonary vascular remodeling and of
decreased BMPR-2 expression by losartan therapy in shunt-induced
pulmonary hypertension. Am J Physiol Heart Circ Physiol.
289:H2319–H2324. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xie L, Lin P, Xie H and Xu C: Effects of
atorvastatin and losartan on monocrotaline-induced pulmonary artery
remodeling in rats. Clin Exp Hypertens. 32:547–554. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Okada M, Harada T, Kikuzuki R, Yamawaki H
and Hara Y: Effects of telmisartan on right ventricular remodeling
induced by monocrotaline in rats. J Pharmacol Sci. 111:193–200.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Saygili E, Rana OR, Saygili E, Reuter H,
Frank K, Schwinger RH, Muller-Ehmsen J and Zobel C: Losartan
prevents stretch-induced electrical remodeling in cultured atrial
neonatal myocytes. Am J Physiol Heart Circ Physiol.
292:H2898–H2905. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ridker PM, Danielson E, Rifai N and Glynn
RJ: Val-MARC Investigators: Valsartan, blood pressure reduction and
C-reactive protein: Primary report of the Val-MARC trial.
Hypertension. 48:73–79. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Anand IS, Kuskowski MA, Rector TS, Florea
VG, Glazer RD, Hester A, Chiang YT, Aknay N, Maggioni AP, Opasich
C, et al: Anemia and change in hemoglobin over time related to
mortality and morbidity in patients with chronic heart failure:
Results from Val-HeFT. Circulation. 112:1121–1127. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Croom KF and Keating GM: Valsartan: A
review of its use in patients with heart failure and/or left
ventricular systolic dysfunction after myocardial infarction. Am J
Cardiovasc Drugs. 4:395–404. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
National Research Council (US) Committee
for the update of the guide for the care and use of laboratory
animals: Guide for the care and use of laboratory animals. 8th.
National Academies Press; Washington, DC: 2011
|
|
19
|
Xu D, Guo H, Xu X, Lu Z, Fassett J, Hu X,
Xu Y, Tang Q, Hu D, Somani A, et al: Exacerbated pulmonary arterial
hypertension and right ventricular hypertrophy in animals with loss
of function of extracellular superoxide dismutase. Hypertension.
58:303–309. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen Y, Guo H, Xu D, Xu X, Wang H, Hu X,
Lu Z, Kwak D, Xu Y, Gunther R, et al: Left ventricular failure
produces profound lung remodeling and pulmonary hypertension in
mice: Heart failure causes severe lung disease. Hypertension.
59:1170–1178. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pacher P, Nagayama T, Mukhopadhyay P,
Bátkai S and Kass DA: Measurement of cardiac function using
pressure-volume conductance catheter technique in mice and rats.
Nat Protoc. 3:1422–1434. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fan YF, Zhang R, Jiang X, Wen L, Wu DC,
Liu D, Yuan P, Wang YL and Jing ZC: The phosphodiesterase-5
inhibitor vardenafil reduces oxidative stress while reversing
pulmonary arterial hypertension. Cardiovasc Res. 99:395–403. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Abe K, Shimokawa H, Morikawa K, Uwatoku T,
Oi K, Matsumoto Y, Hattori T, Nakashima Y, Kaibuchi K, Sueishi K
and Takeshit A: Long-term treatment with a Rho-kinase inhibitor
improves monocrotaline-induced fatal pulmonary hypertension in
rats. Circ Res. 94:385–393. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cassis LA, Rippetoe PE, Soltis EE, Painter
DJ, Fitz R and Gillespie MN: Angiotensin II and
monocrotaline-induced pulmonary hypertension: Effect of losartan
(DuP 753), a nonpeptide angiotensin type 1 receptor antagonist. J
Pharmacol Exp Ther. 262:1168–1172. 1992.PubMed/NCBI
|
|
25
|
Destro M, Cagnoni F, D'Ospina A, Ricci AR,
Demichele E, Peros E, Zaninelli A and Preti P: Role of valsartan,
amlodipine and hydrochlorothiazide fixed combination in blood
pressure control: An update. Vasc Health Risk Manag. 6:253–260.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lesogor A, Cohn JN, Latini R, Tognoni G,
Krum H, Massie B, Zalewski A, Kandra A, Hua TA and Gimpelewicz C:
Interaction between baseline and early worsening of renal function
and efficacy of renin-angiotensin-aldosterone system blockade in
patients with heart failure: Insights from the Val-HeFT study. Eur
J Heart Fail. 15:1236–1244. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Katada E, Uematsu N, Takuma Y and
Matsukawa N: Comparison of effects of valsartan and amlodipine on
cognitive functions and auditory p300 event-related potentials in
elderly hypertensive patients. Clin Neuropharmacol. 37:129–132.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Manabe S, Okura T, Watanabe S, Fukuoka T
and Higaki J: Effects of angiotensin II receptor blockade with
valsartan on pro-inflammatory cytokines in patients with essential
hypertension. J Cardiovasc Pharmacol. 46:735–739. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang J, Jiang H, Yang J, Ding JW, Chen LH,
Li S and Zhang XD: Valsartan preconditioning protects against
myocardial ischemia-reperfusion injury through TLR4/NF-kappaB
signaling pathway. Mol Cell Biochem. 330:39–46. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schraufnagel DE and Schmid A: Pulmonary
capillary density in rats given monocrotaline. A cast corrosion
study. Am Rev Respir Dis. 140:1405–1409. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kopaliani I, Martin M, Zatschler B,
Bortlik K, Müller B and Deussen A: Cell-specific and
endothelium-dependent regulations of matrix metalloproteinase-2 in
rat aorta. Basic Res Cardiol. 109:4192014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guo RW, Yang LX, Wang H, Liu B and Wang L:
Angiotensin II induces matrix metalloproteinase-9 expression via a
nuclear factor-kappaB-dependent pathway in vascular smooth muscle
cells. Regul Pept. 147:37–44. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pullamsetti S, Krick S, Yilmaz H, Ghofrani
HA, Schudt C, Weissmann N, Fuchs B, Seeger W, Grimminger F and
Schermuly RT: Inhaled tolafentrine reverses pulmonary vascular
remodeling via inhibition of smooth muscle cell migration. Respir
Res. 6:1282005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
George J and D'Armiento J: Transgenic
expression of human matrix metalloproteinase-9 augments
monocrotaline-induced pulmonary arterial hypertension in mice. J
Hypertens. 29:299–308. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Frisdal E, Gest V, Vieillard-Baron A,
Levame M, Lepetit H, Eddahibi S, Lafuma C, Harf A, Adnot S and
Dortho MP: Gelatinase expression in pulmonary arteries during
experimental pulmonary hypertension. Eur Respir J. 18:838–845.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vieillard-Baron A, Frisdal E, Raffestin B,
Baker AH, Eddahibi S, Adnot S and D'Ortho MP: Inhibition of matrix
metalloproteinases by lung TIMP-1 gene transfer limits
monocrotaline-induced pulmonary vascular remodeling in rats. Hum
Gene Ther. 14:861–869. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zaidi SH, You XM, Ciura S, Husain M and
Rabinovitch M: Overexpression of the serine elastase inhibitor
elafin protects transgenic mice from hypoxic pulmonary
hypertension. Circulation. 105:516–521. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Long L, Crosby A, Yang X, Southwood M,
Upton PD, Kim DK and Morrell NW: Altered bone morphogenetic protein
and transforming growth factor-beta signaling in rat models of
pulmonary hypertension: Potential for activin receptor-like
kinase-5 inhibition in prevention and progression of disease.
Circulation. 119:566–576. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Thomas M, Docx C, Holmes AM, Beach S,
Duggan N, England K, Leblanc C, Lebret C, Schindler F, Raza F, et
al: Activin-like kinase 5 (ALK5) mediates abnormal proliferation of
vascular smooth muscle cells from patients with familial pulmonary
arterial hypertension and is involved in the progression of
experimental pulmonary arterial hypertension induced by
monocrotaline. Am J Pathol. 174:380–389. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Morrell NW, Upton PD, Kotecha S, Huntley
A, Yacoub MH, Polak JM and Wharton J: Angiotensin II activates MAPK
and stimulates growth of human pulmonary artery smooth muscle via
AT1 receptors. Am J Physiol. 277:L440–L448. 1999.PubMed/NCBI
|