Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive
and fatal neurodegenerative disease, characterized by selective
degeneration and death of motor neurons in the motor cortex,
brainstem and spinal cord. A total of ~90% of ALS cases are
sporadic (sALS), while 10% are familial (fALS) (1). The clinical presentation and
pathology of sALS and fALS are similar. The superoxide dismutase 1
(SOD1) gene, encoding Cu/Zn superoxide dismutase, was the first
identified ALS risk gene. Mutations in SOD1 account for ~20% of
fALS and ~3% of sALS cases (2).
However, the precise mechanisms causing motor neuron degeneration
in ALS remain unknown. Several mechanisms have been investigated,
including oxidative stress, mitochondrial dysfunction,
excitotoxicity, endoplasmic reticulum stress and neuroinflammation
(3). In addition, the involvement
of blood-brain barrier (BBB) and blood-spinal cord barrier (BSCB)
dysfunction in ALS has been a focus of research.
The BBB and BSCB have important roles in controlling
the homeostasis of water and other substances in the central
nervous system (CNS), by transporting substances selectively
through the systemic compartment and preventing passive diffusion
of harmful blood solutes. A number of studies have discovered that
the BSCB is impaired patients with fALS and sALS (4–7). In
addition, similar BSCB impairment was observed in SOD1 mutant ALS
animal models. Previous studies demonstrated capillary
ultrastructure alterations and BSCB damage in SOD1G93A transgenic
mice (mice carrying a mutant human SOD1 cDNA inserted randomly into
the mouse genome) at the early and late stages of the disease
(8,9); the direct effects of the SOD1
mutation on APQ protein function are currently unknown. Recently, a
study demonstrated that restoration of BSCB integrity at an early
disease stage delayed the onset of motor neuron impairment and
degeneration in ALS (10).
Astrocytes are important components of the CNS. It
has been demonstrated that astrocytes are associated with disease
onset and progression in ALS (11). Astrocytic endfeet wrap around the
blood vessel wall and form parts of the BSCB. In addition,
astrocytes serve primary roles in maintaining the homeostasis of
water, glutamate and ions in the CNS, depending on specific
molecules expressed in astrocytes, including water channel protein
aquaporin-4 (AQP4), glutamate transporter 1 (GLT-1 in rodents,
EAAT2 in humans) and potassium inwardly-rectifying channel,
Kir4.1.
AQP4 water channels are the most abundant AQPs
expressed in the CNS, permitting passive and bidirectional water
diffusion. Several studies have demonstrated that AQP4 is involved
in BBB integrity and perturbation (12). In ovariectomized animals, AQP4
expression was demonstrated to be increased following disruption of
the BBB by intraparenchymal injection of lipopolysaccharide
(13). A recent study demonstrated
that remote ischemic post-conditioning was able to alleviate brain
edema and BBB permeability by downregulating AQP4 (14). In addition, accumulating evidence
indicates that AQP4 serves an important role in neurological
disorders, including cerebral ischemia (14), epilepsy (15), traumatic brain injury (16), Alzheimer's disease (17) and spinal cord injury (18).
AQP4 overexpression has been observed in the brain
and spinal cord of SOD1G93A ALS models rodent (19–21),
suggesting that AQP4 may contribute to motor neuron degeneration in
ALS. The function of AQP4 is dependent upon perivascular AQP4
polarization, which refers to that AQP4 that is localized primarily
in perivascular astrocytic endfeet domains. The function of AQP4
overexpression in ALS has not been elucidated. Specifically, it
remains to be investigated whether ALS is associated with a loss of
AQP4 polarization. In the present study, alterations in AQP4
distribution were investigated in ALS animal models.
Materials and methods
Animals
Transgenic mice carrying human G93A mSOD1 [strain
B6SJL-Tg (SOD1-G93A) 1Gur/J] were obtained from the Jackson
Laboratory (Ben Harbor, ME, USA) and crossed with female mice with
a B6SJL/F1 background for ≥4 generations. Transgenic offspring were
genotyped using polymerase chain reaction analysis of DNA obtained
from tail biopsies, as described previously (22). According to the scoring system used
to evaluate signs of motor deficit in SOD1G93A mice, animals were
divided into three stages: i) Presymptomatic stage (60 days) with
no sign of motor dysfunction; ii) disease onset stage with hind
limb tremors observed when suspended by the tail, followed by gait
abnormalities; iii) end-stage, animal unable to right itself within
30 sec (23).
A total of 20 adult male mice (weight, 18–25 g; age,
8–10 weeks) were housed in a 12/12 h light-dark cycle and a
controlled temperature (23±2°C), with free access to food and
water. All procedures performed on the animals were approved by the
Animal Ethical Committee at Sun Yat-sen University (Guangzhou,
China), and were in accordance with the Guidelines for the Care and
Use of Laboratory Animals of the National Institutes of Health
(Bethesda, MA, USA; publication no. 80–23; revised in 1996).
Immunofluorescence
Transgenic and wild type (WT) mice were deeply
anesthetized with 1% pentobarbital (50 mg/kg intraperitoneally) and
transcardially perfused with 0.9% normal saline, followed by 4%
paraformaldehyde in 0.1 mol/l PBS (pH 7.4). The spinal cords were
dissected to identify the cervical and lumbar segments, further
post-fixed in 4% paraformaldehyde at 4°C overnight, and immersed in
20 and 30% sucrose for 2 days at 4°C. Tissues were cut into 20 µm
frozen sections. Antigen retrieval was conducted on the selected
sections, which were heated in the microwave for 7 mins in 0.01
mol/l citric acid solution (pH 6.0). Following washing with 0.01 M
PBS for 5 mins, the sections were blocked with Immunol Staining
Blocking Buffer (cat. no. P0102; Beyotime Institute of
Biotechnology, Shanghai, China) for 1 h at room temperature. The
sections were then washed three times with 0.01 M PBS for 5 min in
an orbital shaker prior to incubation with primary antibodies.
Primary antibodies included goat anti-mouse choline
O-acetyltransferase (ChAT) polyclonal antibody (cat. no. AB144P;
EMD Millipore, Billerica, MA, USA) at 1:400 dilution; mouse
anti-mouse glial fibrillary acidic protein (GFAP) monoclonal
antibody (cat. no. 3670; Cell Signaling Technology, Inc., Danvers,
MA, USA) at 1:500 dilution; rabbit anti-mouse AQP4 polyclonal
antibody (cat. no. AQP-014; Alomone Labs, Jerusalem, Israel) at
1:400 dilution; and rabbit anti-mouse GLT-1 polyclonal antibody
(cat. no. ab106289; Abcam, Cambridge, UK) at 1:300 dilution,
overnight at 4°C. The sections were washed three times with 0.01
mol/l PBS for 5 min in an orbital shaker, and incubated for 1 h at
room temperature with the appropriate species-specific
fluorophore-conjugated secondary antibodies: Donkey anti-goat IgG
Alexa Fluor® 594 (cat. not. A-11058; Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA; 1:400) for ChAT; goat
anti-mouse IgG Alexa Fluor® 555 (cat. no. 4409; Cell Signaling
Technology, Inc.; 1:400) for GFAP; anti-rabbit IgG Alexa Fluor® 488
(cat. no. 4412; Cell Signaling Technology, Inc.; 1:400) for AQP4
and GLT-1. Following washing with 0.01 mol/l PBS, the sections were
mounted using Fluoroshield Mounting Medium with DAPI
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany).
Analysis of motor neuron survival in
the spinal cord
In order to analyze the survival of motor neurons in
the cervical and lumbar spinal cord, the total number of
ChAT-expressing cells was counted in every fourth section (in 5
animals/group). ChAT-expressing cells were counted if they
exhibited a nucleolus that was located in the ventral horns of
immunoreacted sections. The number of motor neurons in the ventral
horn of the WT animal was used as the control value. The number of
surviving motor neurons in the ventral horn of SOD1G93A mice was
described quantitatively as percentages of the control value.
Imaging was conducted at ×20 objective power using a fluorescence
microscope (Olympus DP70; Olympus Corporation, Tokyo, Japan).
Evaluation of GFAP and AQP4 expression
and localization
Free-floating immunofluorescence staining of 20 µm
fixed cross-spinal sections was performed to evaluate the protein
expression patterns of AQP4 and GFAP. Imaging was conducted at a
low magnification (x20 objective power) and at a high magnification
(x63 oil-immersion objective power using a laser scanning confocal
microscope; SP5; Leica Microsystems, Inc., Buffalo Grove, IL, USA).
In order to measure regional GFAP expression, regions were
uniformly thresholded and the area coverage of GFAP
immunoreactivity (as a percentage of the whole region area) was
measured. In order to evaluate global AQP4 expression levels, the
mean AQP4 immunofluorescence intensity was measured within each
region. In order to measure AQP4 polarity, the area of the image
with a pixel intensity greater than or equal to that of the
perivascular endfeet was measured (value expressed as a percentage
of total field of view), according to a previous study (22). AQP4 polarization was characterized
within the astrocytic endfeet associated with the soma in the
spinal cord. A total of five randomly selected sections/animal in
each group were analyzed in a blinded manner by two investigators
using ImageJ version 1.50b bundled with Java 1.8.0_60 software
(National Institutes of Health).
Evaluation of GLT-1 expression in the
lumbar spinal cord
In order to measure the alteration in GLT-1
expression in the ventral horn of the lumbar spinal cord between WT
and SOD1G93A mice at the end stage, imaging was conducted at ×20
objective power using the fluorescence microscope. The area of
GLT-1 immunoreactivity was expressed as a percentage of the overall
field of view. A total of five randomly selected sections/animal in
each group were analyzed in a blinded manner by two investigators
using ImageJ software.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. Comparisons between different groups were performed using
one-way analysis of variance followed by Bonferroni's post hoc
test, and using a Student's t-test between two groups. All data
were processed using SPSS software (version 20.0; IBM Corp.,
Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
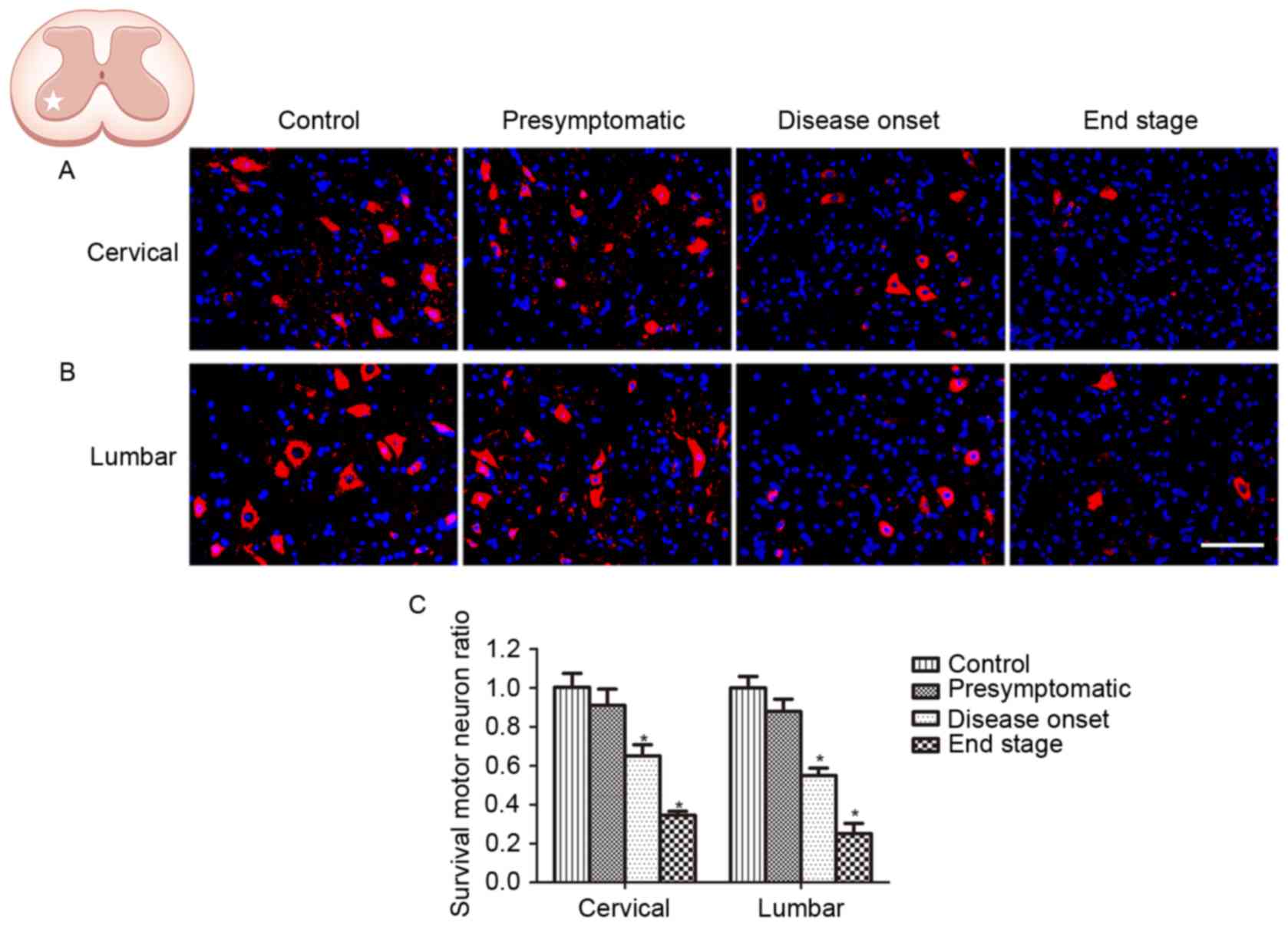
Motor neuron degeneration in SOD1G93A
mice
ChAT is considered to be a marker of motor neurons.
In order to verify the onset and progression of spinal motor neuron
loss in ALS, the survival of ChAT-positive neurons located in the
ventral horn of SOD1G93A mice was investigated at the
presymptomatic, disease onset and end stages. In order to assess
disease progression, motor neuron loss was measured in the cervical
and lumbar spinal cord at the same stage. No difference in the
number of ChAT-positive cells was observed in the spinal and lumbar
ventral horn between WT control mice and SOD1G93A mice at the
presymptomatic stage (Fig. 1). The
number of ChAT-positive neurons in the cervical and lumbar spinal
cord of SOD1G93A mice was decreased at disease onset, when motor
deficits presented; 67.7% ChAT-positive neurons survived in the
cervical ventral horn, and 54.7% ChAT-positive neurons survived in
the lumbar ventral horn, compared with the control (Fig. 1). The number of ChAT-positive
neurons was observed to be further decreased at the end stage, with
34.9 and 25.0% ChAT-positive neurons in the spinal and lumbar
ventral horn, respectively (Fig.
1). Although SOD1G93A mice exhibit motor deficits primarily in
the hind limbs, the results of the present study demonstrated that
motor neuron loss occurred synchronously in the cervical and lumbar
spinal cord, indicating that the pathological condition was
widespread in the ALS model animals.
AQP4 overexpression and depolarization
in SOD1G93A mice
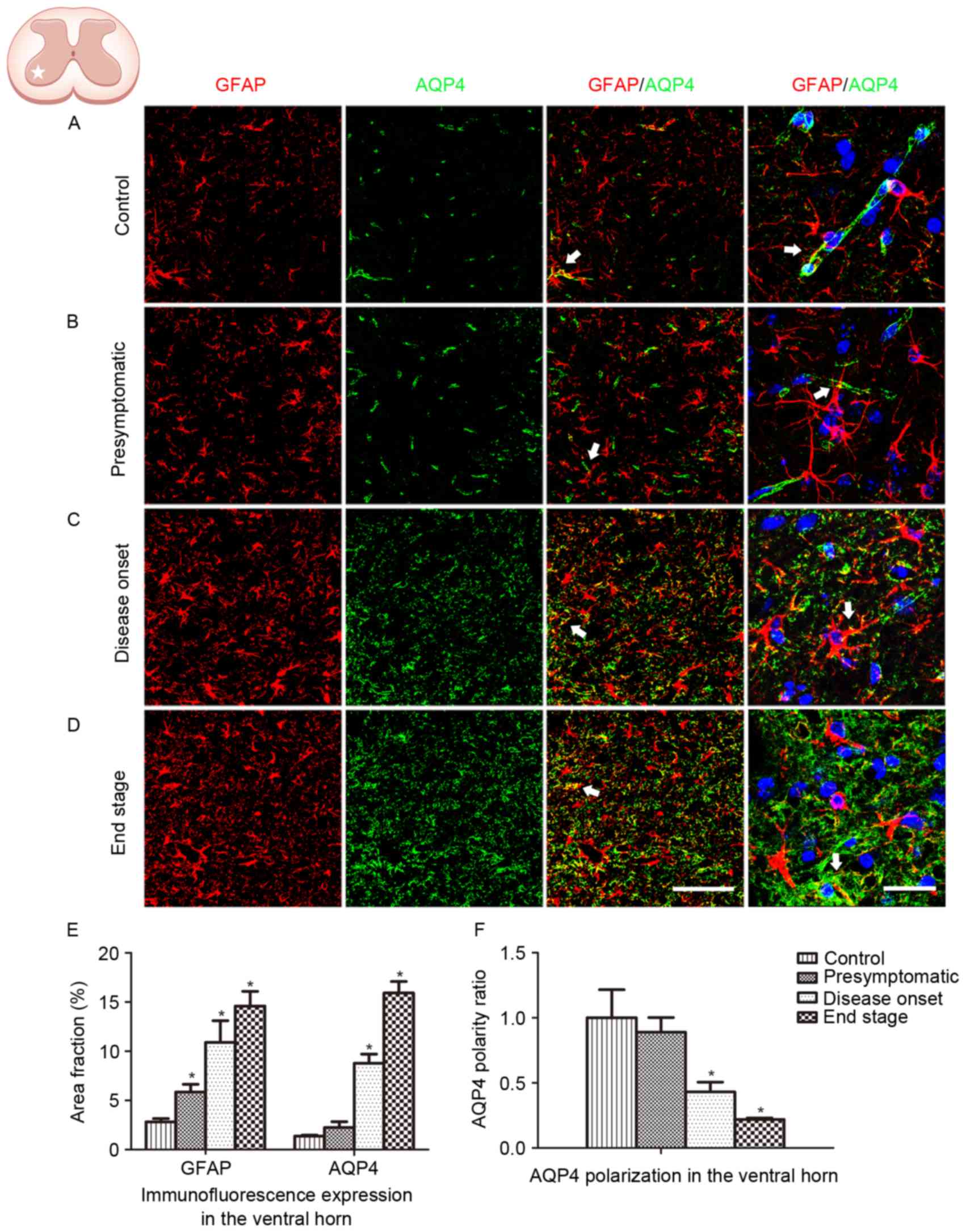
GFAP and AQP4 expression in the lumbar ventral horn
was analyzed in the present study (Fig. 2). An increased number of
GFAP-positive cells, coupled with swelling cell morphology,
indicates astrocyte activation. In the present study, no astrocyte
activation was observed in WT control animals (Fig. 2A). Astrocyte activation was
detected at the presymptomatic stage, and increased in the
subsequent disease onset and end stages in the ALS model animals
(Fig. 2B-D). Persistent reactive
astrocytes with swollen cell bodies were observed at the disease
onset and end stages. Global AQP4 expression was increased as the
disease progressed. Co-localization of GFAP labeling with AQP4
labeling was observed (Fig. 2E).
AQP4 expression in WT animals and SOD1G93A mice at the
presymptomatic stage was predominantly located in the astrocytic
endfeet, which exhibited a cord-like shape (Fig. 2A and B). However, as the disease
progressed in the SOD1G93A mice, AQP4 expression was not restricted
to astrocytic endfeet domains. At high magnification, AQP4
expression was observed to shift to the membranes of swollen
astrocytic soma at the disease onset stage and the end stage,
indicating a loss of AQP4 polarization (Fig. 2C, D and F).
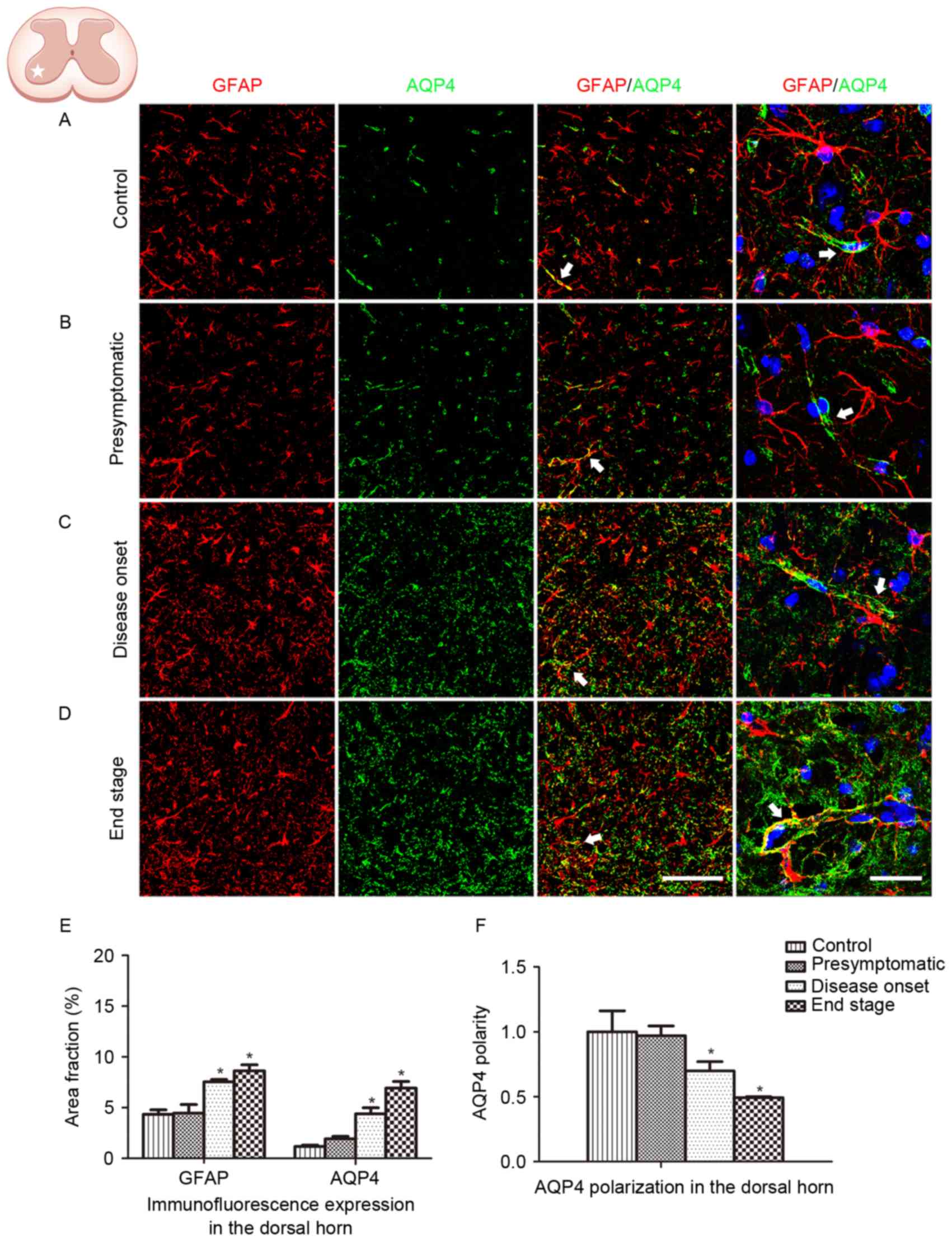
The spinal ventral horn, where motor neurons are
located, is the most affected area in ALS. In order to further
depict the alteration in GFAP and AQP4 expression in ALS, protein
expression in the dorsal horn was additionally analyzed. Similar
GFAP and AQP4 expression profiles were observed in the dorsal horn
of the lumbar spinal cord, with AQP4 expression increasing and
shifting to the membranes of astrocytic soma as the disease
progressed (Fig. 3). AQP4
overexpression and depolarization were additionally observed in the
cervical spinal cord of SOD1G93A mice (data not presented),
indicating that the loss of AQP4 polarization existed throughout
the spinal cord in the ALS animals.
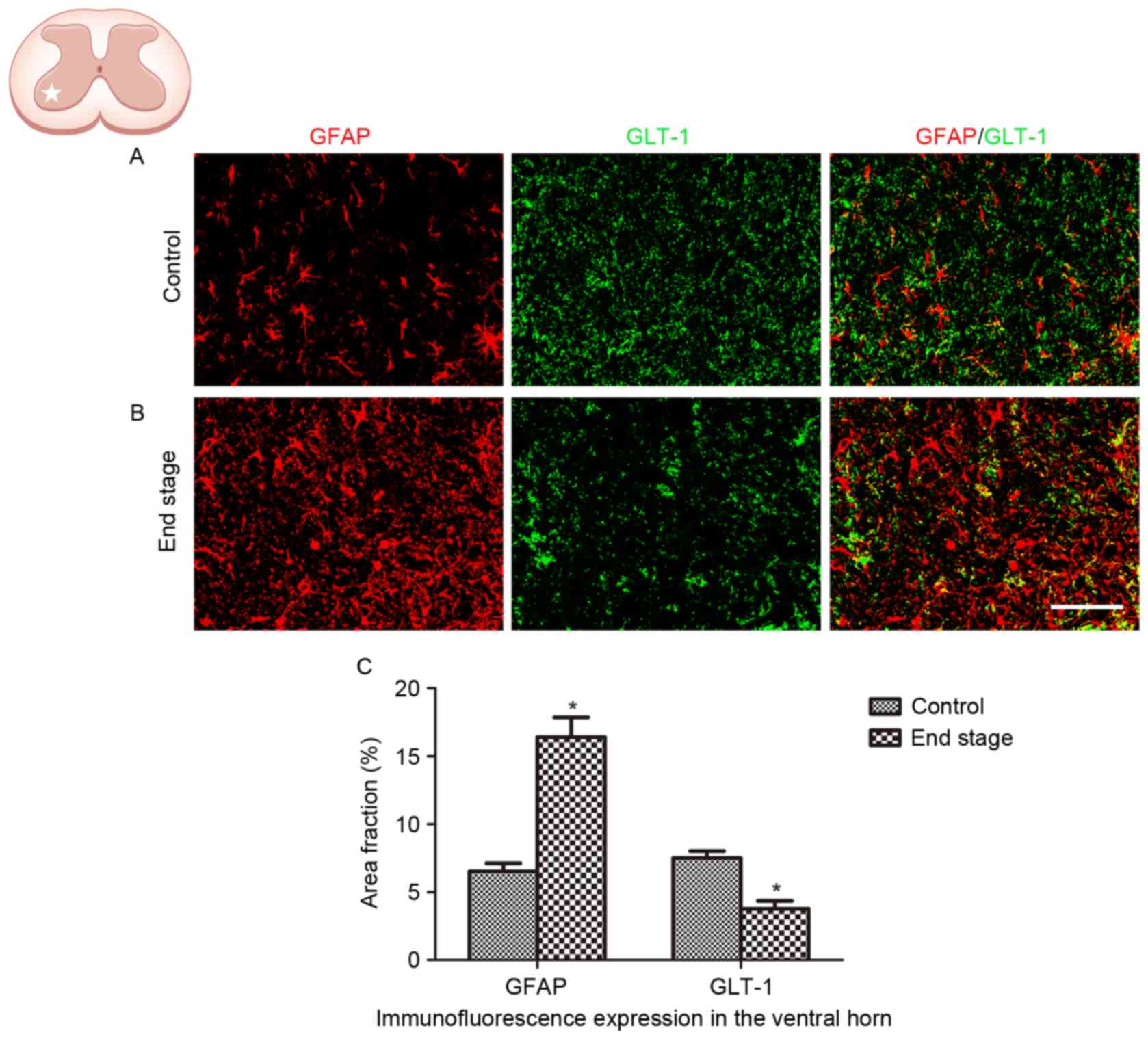
Decreased GLT-1 expression in SOD1G93A
mice
The alteration in GLT-1 expression in ALS was
analyzed in the present study. A significant decrease in GLT-1
immunoreactivity was observed in the lumbar ventral horn of
SOD1G93A mice at the end stage, compared with the WT control
(Fig. 4; P<0.01).
Discussion
The homeostasis of the CNS and the stability of its
function are dependent on the integrity of the BBB and the balance
of water, glutamate and ions. A major contributing factor to the
maintenance of these homeostatic conditions in the vicinity of
active neurons is the expression of AQP4, GLT-1 and Kir4.1, which
are co-expressed in the membrane of astrocytes. The association
between BBB and BSCB disruption, and astrocyte dysfunction in ALS,
has been demonstrated by numerous studies (4–10).
In the present study, using SOD1G93A mutant mice, the association
between motor neuron survival, astrocyte activation, AQP4
dysregulation and the alteration in GLT1 expression in ALS was
investigated.
Astrocyte activation accompanied by motor neuron
degeneration was observed in the spinal cord of SOD1G93A mice.
Astrocyte activation was observed at the presymptomatic stage when
motor deficits were not detected, suggesting that the glial
pathophysiological response in ALS is initiated prior to the onset
of clinical symptoms. The present study demonstrated the
overexpression of AQP4, and additionally demonstrated the loss of
AQP4 polarization in SOD1G93A mice. In physiological conditions,
AQP4 exhibits a polarized distribution, with increased density in
perivascular astrocytic endfeet membranes compared with other
membrane domains. AQP4 polarization may be an important facilitator
of the function of AQP4 in maintaining water homeostasis; a
hypothesis that was supported by a previous study using
α-syntrophin knockout mice. The protein α-syntrophin is required in
order for AQP4 to anchor at the perivascular endfeet membrane. In
α-syntrophin knockout mice, AQP4 localization was shifted away from
the endfeet domains, while overall AQP4 expression was not altered,
which resulted in impaired AQP4-dependent K+ clearance
(24) and cerebral edema following
cerebral ischemia (25). A
previous study using AQP4 knockout mice demonstrated that
perivascular AQP4 served a role in regulating extracellular volume
dynamics by controlling water flux in the mammalian brain (26).
Therefore, the loss of perivascular AQP4 may
initiate a number of pathophysiological processes. Redistribution
of AQP4 was observed to counteract early edema formation in an
animal model of brain infarction (27). In an animal model of mesial
temporal lobe epilepsy, the depolarization of perivascular AQP4 was
demonstrated to precede the development of chronic seizures
(15). A loss of AQP4 polarization
has additionally been observed in a mouse model of Alzheimer's
disease (28).
Due to the physiological role of AQP4, it has been
demonstrated to serve as an important component of the paravascular
pathway that assist in the clearance of β-amyloid, and other
molecules, from the brain parenchyma (29). Additionally, AQP4 depolarization
has been observed in the aging brain, with impaired paravascular
clearance (30). In an animal
model of traumatic brain injury, an elevated global level of AQP4
protein with a redistribution manner of depolarization was observed
in the acute stage of injury, leading to the formation and
resolution of cerebral edema (31). However, the sustained loss of AQP4
perivascular polarization impaired the paravascular pathway
clearance of interstitial solutes from the brain, contributing to
microtubule-associated protein tau aggregation and
neurodegeneration, in the chronic phase following traumatic brain
injury (16). It is therefore
likely that polarized AQP4 serves a role in clearance of
interstitial solutes from the brain.
In the present study, perivascular localization of
AQP4 in astrocytic endfeet was observed in the spinal cord prior to
disease onset, which is consistent with previous studies (19,20,32).
Depolarized AQP4 expression was observed to occur in line with the
progression of ALS. AQP4 depolarization may be a pathological
factor associated with the onset and progression of ALS. Sustained
depolarization of AQP4 impairs the function of maintaining water
balance in the spinal cord, leading to swelling and malformation of
astrocytes and interfering with neuronal function in ALS; however,
the aggregation of misfolded mutant SOD1 proteins is known to be a
cause of ALS. It is hypothesized that the loss of AQP4 polarization
has a negative impact on the clearance of interstitial solutes,
including mutant SOD1 proteins, from the spinal cord, and thereby
contributes to the aggregation of misfolded mutant SOD1 proteins in
ALS. Therefore, further investigation is required to investigate
the association between the loss of AQP4 polarization and the
aggregation of misfolded mutant SOD1 proteins.
An additional molecule expressed in astrocytes is
GLT-1, a glutamate transporter that is responsible for the majority
of functional glutamate uptake in the CNS (33). It has been demonstrated that one of
the major causes of motor neuron degeneration in ALS is
excitotoxicity due to dysregulation of extracellular glutamate
homeostasis (34). Compromised
glutamate uptake, due to decreased expression and aberrant
functioning of GLT-1, has been observed in astrocytes of the spinal
cord and motor cortex in patients with ALS (35,36)
and SOD1G93A rodents (37). In the
present study, decreased expression of GLT-1 in the spinal cord in
SOD1G93A mice was observed, which is consistent with the previous
studies (38). Downregulated GLT-1
expression in astrocytes and impaired uptake of glutamate were
observed in AQP4-deficient mice (38), indicating that AQP4 serves a role
in regulating the expression and function of GLT-1. Glutamate has
been demonstrated to increase AQP4 water permeability in astrocytes
(39). A number of studies into
various neurological disorders have demonstrated that AQP4 and
GLT-1 exist in astrocytic membranes as a functional complex
(40–43). It is hypothesized that the
overexpression and depolarization of AQP4 leads to abnormal
functioning, which may therefore lead to the decreased expression
and aberrant functioning of GLT-1. The association between AQP4 and
GLT-1 may result in impairment of the balance between glutamate and
water transport, dysregulation of neuronal activity, and
excitotoxic neuronal dysfunction. Further studies are required to
elucidate the interaction between AQP4 and GLT-1.
In the present study, SOD1G93A mice were used as a
model to investigate the role of AQP4 in the pathological
development of ALS. Global overexpression, with decreased polarized
astrocytic localization of AQP4, was observed in the spinal cord,
indicating an impaired function of AQP4 in ALS. The results of the
present study demonstrated that AQP4 depolarization is a widespread
pathological condition and may contribute to motor neuron
degeneration in ALS.
Acknowledgements
The present study was supported by grants from the
Guangzhou Science and Technology Project (grant no. 2014J4500031),
the Guangdong Science and Technology Project (grant nos.
2013B021800274, 2014B030301035 and 2015B050501003), the Major
Cultivation and Interdisciplinary Project of Sun Yat-Sen University
(grant no. 15ykjc15b) and the Macau Science and Technology
Development Fund (grant no. 063/2015/A2).
References
|
1
|
Kiernan MC, Vucic S, Cheah BC, Turner MR,
Eisen A, Hardiman O, Burrell JR and Zoing MC: Amyotrophic lateral
sclerosis. Lancet. 377:942–955. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rosen DR, Siddique T, Patterson D,
Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP,
Deng HX, et al: Mutations in Cu/Zn superoxide dismutase gene are
associated with familial amyotrophic lateral sclerosis. Nature.
362:59–62. 1993. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mancuso R and Navarro X: Amyotrophic
lateral sclerosis: Current perspectives from basic research to the
clinic. Prog Neurobiol. 133:1–26. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Garbuzova-Davis S, Hernandez-Ontiveros DG,
Rodrigues MC, Haller E, Frisina-Deyo A, Mirtyl S, Sallot S, Saporta
S, Borlongan CV and Sanberg PR: Impaired blood-brain/spinal cord
barrier in ALS patients. Brain Res. 1469:114–128. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Winkler EA, Sengillo JD, Sullivan JS,
Henkel JS, Appel SH and Zlokovic BV: Blood-spinal cord barrier
breakdown and pericyte reductions in amyotrophic lateral sclerosis.
Acta Neuropathol. 125:111–120. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yamadera M, Fujimura H, Inoue K, Toyooka
K, Mori C, Hirano H and Sakoda S: Microvascular disturbance with
decreased pericyte coverage is prominent in the ventral horn of
patients with amyotrophic lateral sclerosis. Amyotroph Lateral
Scler Frontotemporal Degener. 16:393–401. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sasaki S: Alterations of the blood-spinal
cord barrier in sporadic amyotrophic lateral sclerosis.
Neuropathology. 35:518–528. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Garbuzova-Davis S, Haller E, Saporta S,
Kolomey I, Nicosia SV and Sanberg PR: Ultrastructure of blood-brain
barrier and blood-spinal cord barrier in SOD1 mice modeling ALS.
Brain Res. 1157:126–137. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Garbuzova-Davis S, Saporta S, Haller E,
Kolomey I, Bennett SP, Potter H and Sanberg PR: Evidence of
compromised blood-spinal cord barrier in early and late symptomatic
SOD1 mice modeling ALS. PLoS One. 2:e12052007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Winkler EA, Sengillo JD, Sagare AP, Zhao
Z, Ma Q, Zuniga E, Wang Y, Zhong Z, Sullivan JS, Griffin JH, et al:
Blood-spinal cord barrier disruption contributes to early
motor-neuron degeneration in ALS-model mice. Proc Natl Acad Sci
USA. 111:E1035–E1042. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Blackburn D, Sargsyan S, Monk PN and Shaw
PJ: Astrocyte function and role in motor neuron disease: A future
therapeutic target? Glia. 57:1251–1264. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nicchia GP, Nico B, Camassa LM, Mola MG,
Loh N, Dermietzel R, Spray DC, Svelto M and Frigeri A: The role of
aquaporin-4 in the blood-brain barrier development and integrity:
Studies in animal and cell culture models. Neuroscience.
129:935–945. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tomás-Camardiel M, Venero JL, Herrera AJ,
de Pablos RM, Pintor-Toro JA, Machado A and Cano J: Blood-brain
barrier disruption highly induces aquaporin-4 mRNA and protein in
perivascular and parenchymal astrocytes: Protective effect by
estradiol treatment in ovariectomized animals. J Neurosci Res.
80:235–246. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li S, Hu X, Zhang M, Zhou F, Lin N, Xia Q,
Zhou Y, Qi W, Zong Y, Yang H and Wang T: Remote ischemic
post-conditioning improves neurological function by AQP4
down-regulation in astrocytes. Behav Brain Res. 289:1–8. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Alvestad S, Hammer J, Hoddevik EH, Skare
Ø, Sonnewald U, Amiry-Moghaddam M and Ottersen OP: Mislocalization
of AQP4 precedes chronic seizures in the kainate model of temporal
lobe epilepsy. Epilepsy Res. 105:30–41. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM,
Soltero M, Yang L, Singh I, Deane R and Nedergaard M: Impairment of
glymphatic pathway function promotes tau pathology after traumatic
brain injury. J Neurosci. 34:16180–16193. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu Z, Xiao N, Chen Y, Huang H, Marshall C,
Gao J, Cai Z, Wu T, Hu G and Xiao M: Deletion of aquaporin-4 in
APP/PS1 mice exacerbates brain Aβ accumulation and memory deficits.
Mol Neurodegener. 10:582015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nesic O, Lee J, Ye Z, Unabia GC, Rafati D,
Hulsebosch CE and Perez-Polo JR: Acute and chronic changes in
aquaporin 4 expression after spinal cord injury. Neuroscience.
143:779–792. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nicaise C, Soyfoo MS, Authelet M, de
Decker R, Bataveljic D, Delporte C and Pochet R: Aquaporin-4
overexpression in rat ALS model. Anat Rec (Hoboken). 292:207–213.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bataveljic D, Nikolić L, Milosević M,
Todorović N and Andjus PR: Changes in the astrocytic aquaporin-4
and inwardly rectifying potassium channel expression in the brain
of the amyotrophic lateral sclerosis SOD1(G93A) rat model. Glia.
60:1991–2003. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cui Y, Masaki K, Yamasaki R, Imamura S,
Suzuki SO, Hayashi S, Sato S, Nagara Y, Kawamura MF and Kira J:
Extensive dysregulations of oligodendrocytic and astrocytic
connexins are associated with disease progression in an amyotrophic
lateral sclerosis mouse model. J Neuroinflammation. 11:422014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gurney ME, Pu H, Chiu AY, Dal Canto MC,
Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX,
et al: Motor neuron degeneration in mice that express a human Cu,
Zn superoxide dismutase mutation. Science. 264:1772–1775. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Weydt P, Hong SY, Kliot M and Möller T:
Assessing disease onset and progression in the SOD1 mouse model of
ALS. Neuroreport. 14:1051–1054. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Amiry-Moghaddam M, Williamson A, Palomba
M, Eid T, de Lanerolle NC, Nagelhus EA, Adams ME, Froehner SC, Agre
P and Ottersen OP: Delayed K+ clearance associated with aquaporin-4
mislocalization: Phenotypic defects in brains of
alpha-syntrophin-null mice. Proc Natl Acad Sci USA.
100:13615–13620. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Amiry-Moghaddam M, Otsuka T, Hurn PD,
Traystman RJ, Haug FM, Froehner SC, Adams ME, Neely JD, Agre P,
Ottersen OP and Bhardwaj A: An alpha-syntrophin-dependent pool of
AQP4 in astroglial end-feet confers bidirectional water flow
between blood and brain. Proc Natl Acad Sci USA. 100:2106–2111.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Haj-Yasein NN, Jensen V, Østby I, Omholt
SW, Voipio J, Kaila K, Ottersen OP, Hvalby Ø and Nagelhus EA:
Aquaporin-4 regulates extracellular space volume dynamics during
high-frequency synaptic stimulation: A gene deletion study in mouse
hippocampus. Glia. 60:867–874. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Steiner E, Enzmann GU, Lin S, Ghavampour
S, Hannocks MJ, Zuber B, Rüegg MA, Sorokin L and Engelhardt B: Loss
of astrocyte polarization upon transient focal brain ischemia as a
possible mechanism to counteract early edema formation. Glia.
60:1646–1659. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang J, Lunde LK, Nuntagij P, Oguchi T,
Camassa LM, Nilsson LN, Lannfelt L, Xu Y, Amiry-Moghaddam M,
Ottersen OP and Torp R: Loss of astrocyte polarization in the
tg-ArcSwe mouse model of Alzheimer's disease. J Alzheimers Dis.
27:711–722. 2011.PubMed/NCBI
|
|
29
|
Iliff JJ, Wang M, Liao Y, Plogg BA, Peng
W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, et
al: A paravascular pathway facilitates CSF flow through the brain
parenchyma and the clearance of interstitial solutes, including
amyloid β. Sci Transl Med. 4:147ra1112012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kress BT, Iliff JJ, Xia M, Wang M, Wei HS,
Zeppenfeld D, Xie L, Kang H, Xu Q, Liew JA, et al: Impairment of
paravascular clearance pathways in the aging brain. Ann Neurol.
76:845–861. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ren Z, Iliff JJ, Yang L, Yang J, Chen X,
Chen MJ, Giese RN, Wang B, Shi X and Nedergaard M: ‘Hit & Run’
model of closed-skull traumatic brain injury (TBI) reveals complex
patterns of post-traumatic AQP4 dysregulation. J Cereb Blood Flow
Metab. 33:834–845. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Oklinski MK, Lim JS, Choi HJ, Oklinska P,
Skowronski MT and Kwon TH: Immunolocalization of water channel
proteins AQP1 and AQP4 in rat spinal cord. J Histochem Cytochem.
62:598–611. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Maragakis NJ and Rothstein JD: Glutamate
transporters: Animal models to neurologic disease. Neurobiol Dis.
15:461–473. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Maragakis NJ and Rothstein JD: Mechanisms
of disease: Astrocytes in neurodegenerative disease. Nat Clin Pract
Neurol. 2:679–689. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rothstein JD, Martin LJ and Kuncl RW:
Decreased glutamate transport by the brain and spinal cord in
amyotrophic lateral sclerosis. N Engl J Med. 326:1464–1468. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rothstein JD, van Kammen M, Levey AI,
Martin LJ and Kuncl RW: Selective loss of glial glutamate
transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol.
38:73–84. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Howland DS, Liu J, She Y, Goad B,
Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, et
al: Focal loss of the glutamate transporter EAAT2 in a transgenic
rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis
(ALS). Proc Natl Acad Sci USA. 99:1604–1609. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zeng XN, Sun XL, Gao L, Fan Y, Ding JH and
Hu G: Aquaporin-4 deficiency down-regulates glutamate uptake and
GLT-1 expression in astrocytes. Mol Cell Neurosci. 34:34–39. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gunnarson E, Zelenina M, Axehult G, Song
Y, Bondar A, Krieger P, Brismar H, Zelenin S and Aperia A:
Identification of a molecular target for glutamate regulation of
astrocyte water permeability. Glia. 56:587–596. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hinson SR, Roemer SF, Lucchinetti CF,
Fryer JP, Kryzer TJ, Chamberlain JL, Howe CL, Pittock SJ and Lennon
VA: Aquaporin-4-binding autoantibodies in patients with
neuromyelitis optica impair glutamate transport by down-regulating
EAAT2. J Exp Med. 205:2473–2481. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang J, Li MX, Luo Y, Chen T, Liu J, Fang
P, Jiang B, Hu ZL, Jin Y, Chen JG and Wang F: Chronic ceftriaxone
treatment rescues hippocampal memory deficit in AQP4 knockout mice
via activation of GLT-1. Neuropharmacology. 75:213–222. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mogoanta L, Ciurea M, Pirici I,
Margaritescu C, Simionescu C, Ion DA and Pirici D: Different
dynamics of aquaporin 4 and glutamate transporter-1 distribution in
the perineuronal and perivascular compartments during ischemic
stroke. Brain Pathol. 24:475–493. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Geis C, Ritter C, Ruschil C, Weishaupt A,
Grünewald B, Stoll G, Holmoy T, Misu T, Fujihara K, Hemmer B, et
al: The intrinsic pathogenic role of autoantibodies to aquaporin 4
mediating spinal cord disease in a rat passive-transfer model. Exp
Neurol. 265:8–21. 2015. View Article : Google Scholar : PubMed/NCBI
|