Introduction
Acute lung injury (ALI), a clinical complication
associated with respiratory dysfunctions usually a consequence of
sepsis and a systemic inflammatory response (1,2). The
most severe form of ALI can lead to acute respiratory distress
syndrome, respiratory failure, increasing susceptibility to
multi-organ dysfunction, and ultimately death with high rates of
morbidity and mortality (3). The
physiological hallmarks of ALI are a disruption of the
alveolar-capillary membrane barrier resulting in non-cardiogenic
pulmonary edema, neutrophil and macrophage accumulation,
endothelial and epithelial injury, and severe inflammatory response
in the lungs and neutrophilic alveolitis (4,5).
Despite considerable research, the underlying molecular mechanisms
involved in the pathogenesis of acute lung injury and appropriate
treatment approaches are still unknown. However, an effective
treatment of this life-threatening disease requires a better
understanding of the molecular and cellular pathophysiology of
ALI.
Lipopolysaccharides (LPS), a pathogenic component of
endotoxin released from the cell wall of Gram-negative bacteria,
are widely used to induce animal model of ALI (6,7). LPS
may induce neutrophil infiltration and the accumulation of
pro-inflammatory cytokines, in order to amplify acute lung
inflammation (5). A recent study
indicated that the binding of LPS to Toll-like receptor 4 caused
IκB-α phosphorylation and degradation, activated nuclear factor
(NF)-κB, and subsequently led to the uncontrolled and excessive
production of pro-inflammatory mediators, such as tumor necrosis
factor (TNF)-α, interleukin (IL)-1β, IL-6 (8,9). In
addition, mitochondrial dysfunction and cell apoptosis participated
in the pathological process of LPS-induced ALI, and inhibition of
apoptosis decreased lung damage (10). More recently, upregulation
expression of IL-17, including IL-17A and IL-17F, were observed in
the lung tissues, BALF and serum of ALI rats (11). It is generally believed that IL-17
as a pro-inflammatory cytokine serve a crucial role in triggering
inflammatory responses.
The IL-17 family including six members, designated
IL-17A-F, is a pleiotropic pro-inflammatory cytokine (12,13).
Growing evidences suggested that IL-17 serve an essential role in a
multitude of autoimmune diseases, inflammation and cancer (14–16).
Recent evidence indicated that IL-17 may promote epithelial cells
to secrete IL-6, GCSF and GM-CSF, which, in turn, recruits
neutrophils to the airways (17).
Notably, it is reported that TNF-α, IL-1β, IL-6 and IL-8 are the
downstream target genes of IL-17 (18). However, the potential role and
underlying mechanism of IL-17 in development of LPS-induced ALI and
pulmonary inflammation are currently unknown. Therefore, the
purpose of the present study was to investigate the expression of
IL-17 and possible molecular mechanisms involved in inflammatory
response in an ALI rat model by LPS intratracheal instillation.
Materials and methods
Animals
Male Sprague-Dawley rats, 8-weeks-old and (weight,
200–220 g), were obtained from Vital River Laboratories Co., Ltd.
(Beijing, China). All animals used for experiments were allowed
free access to food and water, and housed under specific
pathogen-free conditions with a 12 h light/dark cycle in a
controlled temperature (20–25°C) and humidity (50±5%) environment.
All experimental protocols were approved by the Laboratory Animal
Committee of Hebei Medical University, (Shijiazhuang, China). The
rats were sacrificed under anesthesia with chloral hydrate (350
mg/kg i.p.), followed by cervical dislocation. All efforts were
made to minimize suffering.
Experimental protocol and LPS-induced
ALI model
A total of 90 rats were randomly divided into three
groups (n=12/group): (1) Control
group: Received saline by intratracheal instillation (5 ml/kg)
under anesthesia using inhaled isoflurane (Boston Biochem, Inc.,
Cambridge, MA, USA); (2) ALI
group: ALI was induced by intratracheal instillation of LPS to
induce acute lung injury model in vivo, as previously described
(5,19). In brief, rats were anaesthetized
and injected intravenously with LPS (5 mg/kg, Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany). (3)
IL-17 mAb group: Rats were injected intraperitoneally with IL-17
monoclonal antibody (100 µg/ml, IL-17 mAb, Clone # 50101; R&D
Systems, Inc., Minneapolis, MN, USA). Rats in all groups were
sacrificed respectively at 3, 6 and 24 h following injection, 10
rats per group per time point.
Lung wet/dry (W/D) weight ratio
At 6 h following LPS or saline challenge, the lung
W/D weight ratio was calculated to evaluate the extent of acute
pulmonary edema, Five rats per group were anesthetized, harvested
and cleaned from blood, weighed to obtain the wet weight, and then
placed in an oven for 48 h at 80°C for the measurement of the dry
weight. The ratio of the wet weight to dry weight was then
calculated (20).
Histopathological evaluation
Morphological changes in the lungs were examined by
hematoxylin and eosin (H&E) staining. At 6 h and 24 h following
the instillation of LPS, rats were euthanized for histological
assessment. The right upper lobes were collected, fixed in 10%
formalin for 48 h and embedded in paraffin. Sections of fixed
embedded tissues (5 µm thick) were cut on a Leica model 2165 rotary
microtome (Leica Microsystems GmbH, Wetzlar, Germany), placed on
glass slides, deparaffinized and sequentially stained with H&E
(Thermo Fisher Scientific, Inc., Waltham, MA, USA) for examination
under an optical microscope (BX51; Olympus Corporation, Tokyo,
Japan). The extents of histological injury were evaluated by two
blinded experienced investigators. The degree of lung injury was
graded using a scoring system based on histological features,
including edema, congestion and hyperemia, tissue infiltration and
neutrophil margination, and the presence of intra-alveolar
hemorrhage, debris and cellular hyperplasia. Each feature was
graded as either absent=0, mild =1, moderate=2, or severe=3. The
total score was calculated for histopathological evaluation of each
rat.
Bronchoalveolar lavage fluid (BALF)
collection
BALF was collected by intratracheal intubation as
previously described (5). Briefly,
after mice were anesthetized, the trachea was exposed and
cannulated with a sterile catheter. BAL was performed with a 5 ml
0.9% saline solution. The lavage was repeated twice with saline to
recover a total volume of 4–5 ml. Lavage samples were centrifuged
at 1,000 × g at 4°C for 10 min. All samples were stored at −80°C to
measure chemokine levels by using ELISA kits.
ELISA
The concentrations of TNF-α, IL-6, IL-1β and IL-17
in BALF were detected using cytokine specific Quantikine ELISA kits
(TNF-α, SEA133Ra; IL-6, SEA079Ra; IL-1β, SEA073Ra; IL-17, SEA063Ra;
Wuhan USCN Business Co., Ltd., Wuhan, China) according to the
manufacturer's instructions. The absorbance was read at 490 nm on
an ELISA plate scanner (Molecular Devices, LLC, Sunnyvale, CA,
USA). All experiments are performed at least in triplicate samples
and results are presented as the mean value. A standard curve using
recombinant cytokine was generated for each assay.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
Total RNA was extracted from rat lung tissues with
Tripure Isolation reagent (Thermo Fisher Scientific, Inc.). The
qPCR kits (Takara Biotechnology Co., Ltd., Dalian, China) were used
for the qPCR experiment. cDNA was synthesized from total RNA using
a SuperScript Reverse Transcriptase kit (Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. The primers of
PCR were as follows: Rat IL-17 forward,
5′-ATCCCTCAAAGTTCAGTGTGTCC-3′ and reverse,
5′-GGACAATAGAGGAAACGCAGGT-3′; GAPDH forward,
5′-CAAAGTTGTCATGGATGACC-3′ and reverse, 5′-CCATGGAGAAGGCTGGGG-3′.
RT-qPCR was performed on a Real-Time PCR System (Applied
Biosystems; Thermo Fisher Scientific, Inc.; ABI Prism 7300)
instrument with SYBR Premix Ex Taq (Takara Bio, Inc., Otsu, Japan),
starting with 1 ng reverse-transcribed total RNA. PCR was performed
under the following conditions: 95°C for 10 min, 40 cycles of 95°C
for 5 sec and 60°C for 30 sec. GAPDH was used as an internal
control. The relative mRNA expression levels were calculated by the
2−ΔΔCq method (21).
Western blot analysis
Lung tissues were lysed in total protein cell lysis
buffer (Thermo Fisher Scientific, Inc.) supplemented with 5%
Proteinase Inhibitor cocktail (Sigma-Aldrich; Merck KGaA),
incubated on ice for 30 min, and centrifuged at 15,000 × g for 15
min. Protein concentrations were determined with the bicinchoninic
acid protein assay reagents (Nanjing Jiancheng Bioengineering
Institute, Nanjing, China). Total protein (50 µg) was loaded in
each lane, separated using 10% SDS-PAGE and transferred onto
polyvinylidene fluoride membranes using a wet transfer method at
room temperature. Nonspecific binding sites were blocked with 5%
BSA for 1 h, then incubated with rabbit anti-rat IL-17 (1:1,000;
SC-52567), nuclear factor (NF)-κB (1:1,000; SC-514451),
p-extracellular signal-regulated kinase (ERK) 1/2 (1:1,000;
SC-136521) and β-actin polyclonal antibody (1:1,000; SC-7210) (all
from Santa Cruz Biotechnology, Inc., Dallas, TX, USA) overnight at
4°C. The following day, the membranes were incubated with incubated
with horseradish peroxidase conjugated secondary antibody (1:5,000,
#7074S; Cell Signaling Technology, Inc., Danvers, MA, USA). The
immunoreactive bands were visualized with an enhanced
chemiluminescence (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
reagent. Blots were scanned by densitometry, and integrated density
of pixels was quantified using Image Quant software (version, 5.2;
Molecular Devices, LLC, Sunnyvale, CA, USA).
Statistical analysis
All data in the tests and figures were presented as
means ± standard deviation. The statistical software SPSS software
(version, 13.0; SPSS, Inc., Chicago, IL, USA) was used for data
analysis. One-way analysis of variance (ANOVA) followed by Tukey's
post hoc test, or a two-tailed unpaired Student's t-test was
applied to evaluate statistical significances. Measurements in the
intergroup differences at single timepoints were analyzed by an
ANOVA, and, if they demonstrated significance, they were further
analyzed by the two-tailed unpaired Student's t-test. P<0.05 was
considered statistically significant.
Results
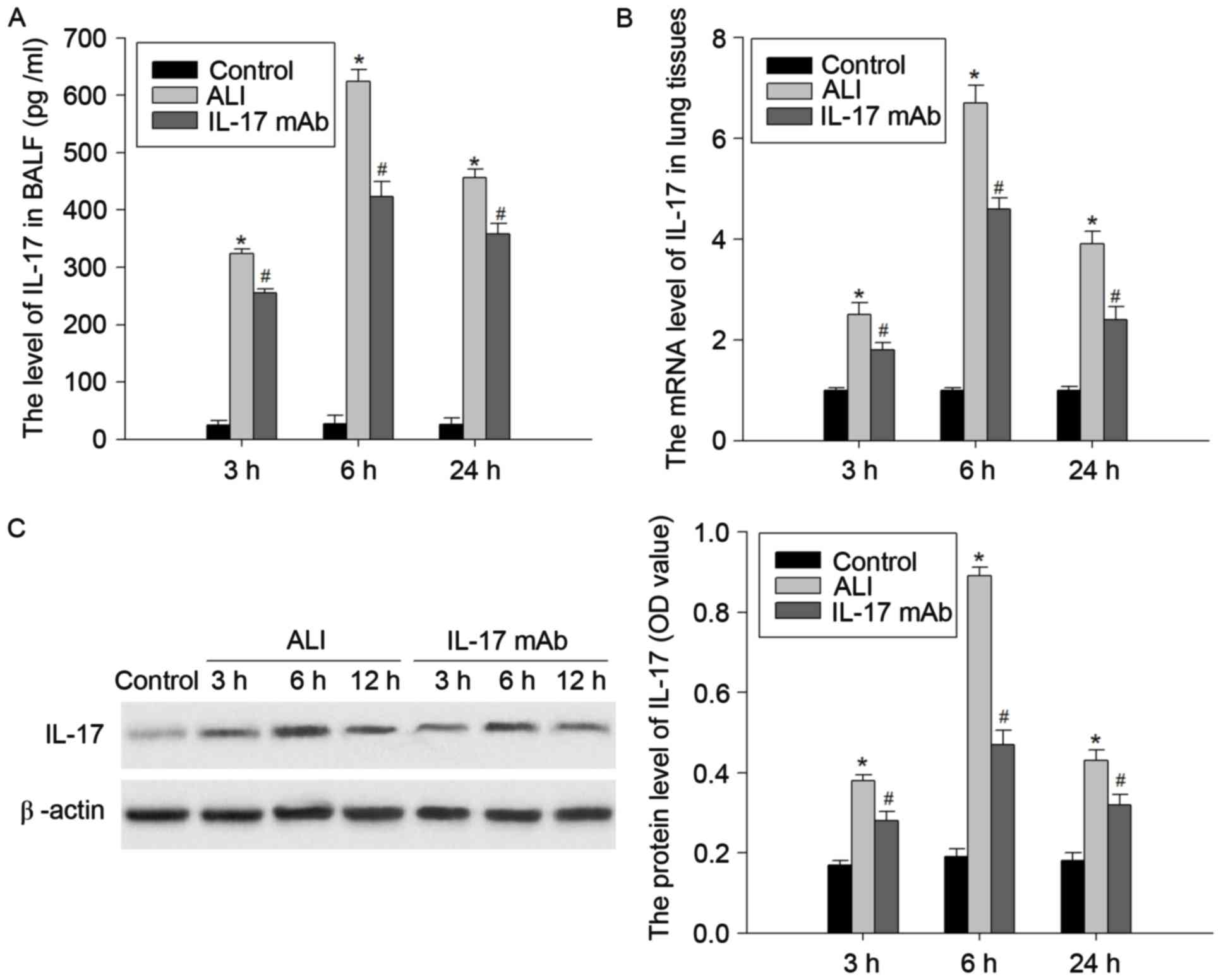
Expression of IL-17 in the lung
tissues of ALI rats induced by LPS
In order to confirm that the expression of IL-17 is
involved in pathophysiologic process of ALI, the authors detected
the mRNA and protein levels of IL-17 in the BALF and lung tissues
of a LPS-induced ALI rat model at 3, 6 and 24 h following LPS
challenge. As presented in Fig. 1,
the protein levels of IL-17 in the BALF and lung tissues were
markedly elevated at 3, 6 and 24 h following LPS injection.
Similarly, the mRNA expression of IL-17 in the lung tissues of
LPS-treated rats was enhanced. The mRNA and protein levels of L-17
peaked at 6 h, and prolonged the lag time of 24 h. These results
indicated that the expression of IL-17 may be closely correlated
with development of LPS-induced lung injury.
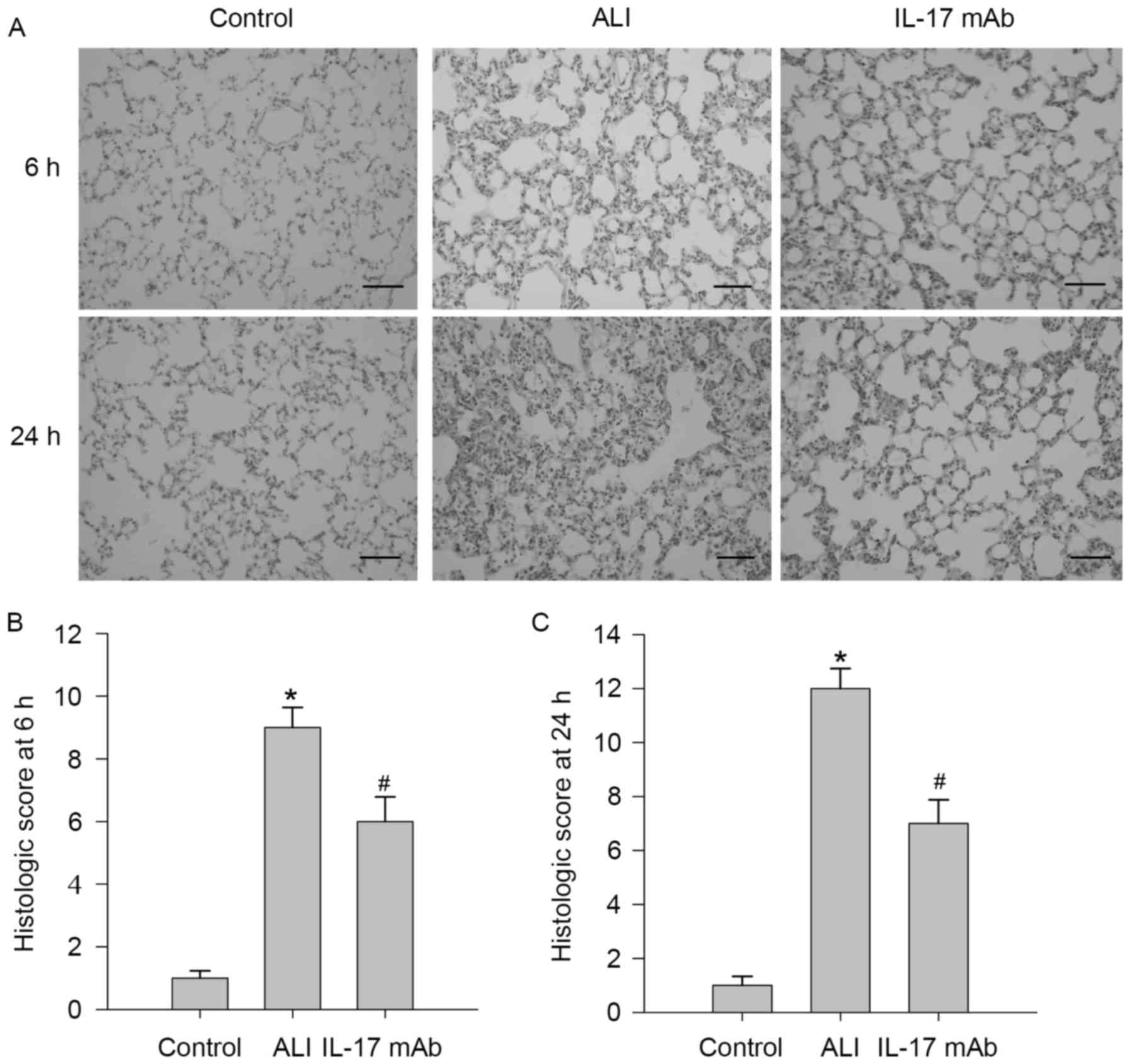
Blockade of IL-17 alleviated the
severity of lung lesion
To determine the roles of IL-17 in LPS-induced lung
injury, the authors examined the effects of IL-17 mAb on lung
injury. The lung histopathology changes at 6 h and 24 h post-ALI
are presented in Fig. 2. To
observe characteristics of LPS-induced lung injury in rats, LPS
treated rats exhibited severe lung injury characterized by
inflammation, cell infiltration, interstitial edema, alveolar
structural damages and alveolar wall thickening, which was markedly
improved by IL-17 mAb treatment, while those phenomena could not be
observed in the saline group. A scoring system was used to grade
the degree of lung injury, and rats exposed to LPS presented a
remarkable increase in lung histologic scores, but the histological
scores of IL-17 mAb treated rats were lower than those of ALI group
at 6 h (Fig. 2B) and 24 h
(Fig. 2C).
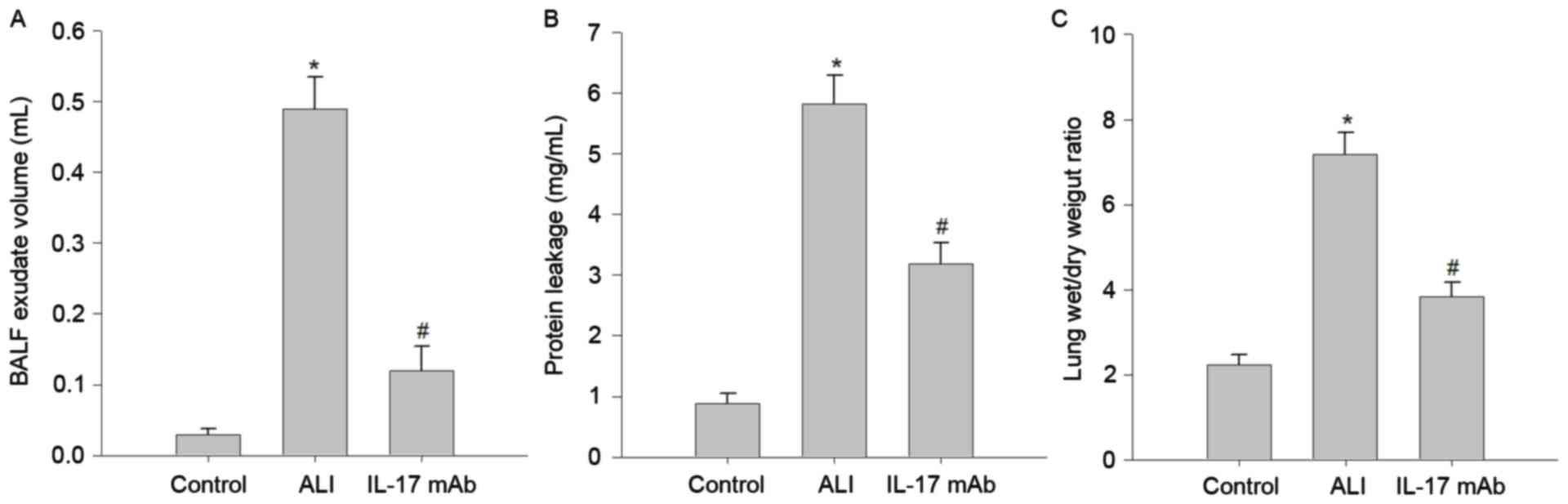
Blockade of IL-17 attenuated the
severity of pulmonary edema
Several well-known hallmarks of ALI-induced
pulmonary edema, such as BALF exudate volume (Fig. 3A), protein leakage in BALF
(Fig. 3B), and lung W/D weight
ratio (Fig. 3C) were remarkably
increased in LPS challenge rats compared with those in the control
group. Importantly, inhibition of IL-17 using IL-17 mAb,
significantly alleviated LPS-induced BALF exudate volume, protein
leakage and lung W/D weight ratio. These results demonstrated that
IL-17 mAb treatment may improve pulmonary edema in LPS-challenged
rats.
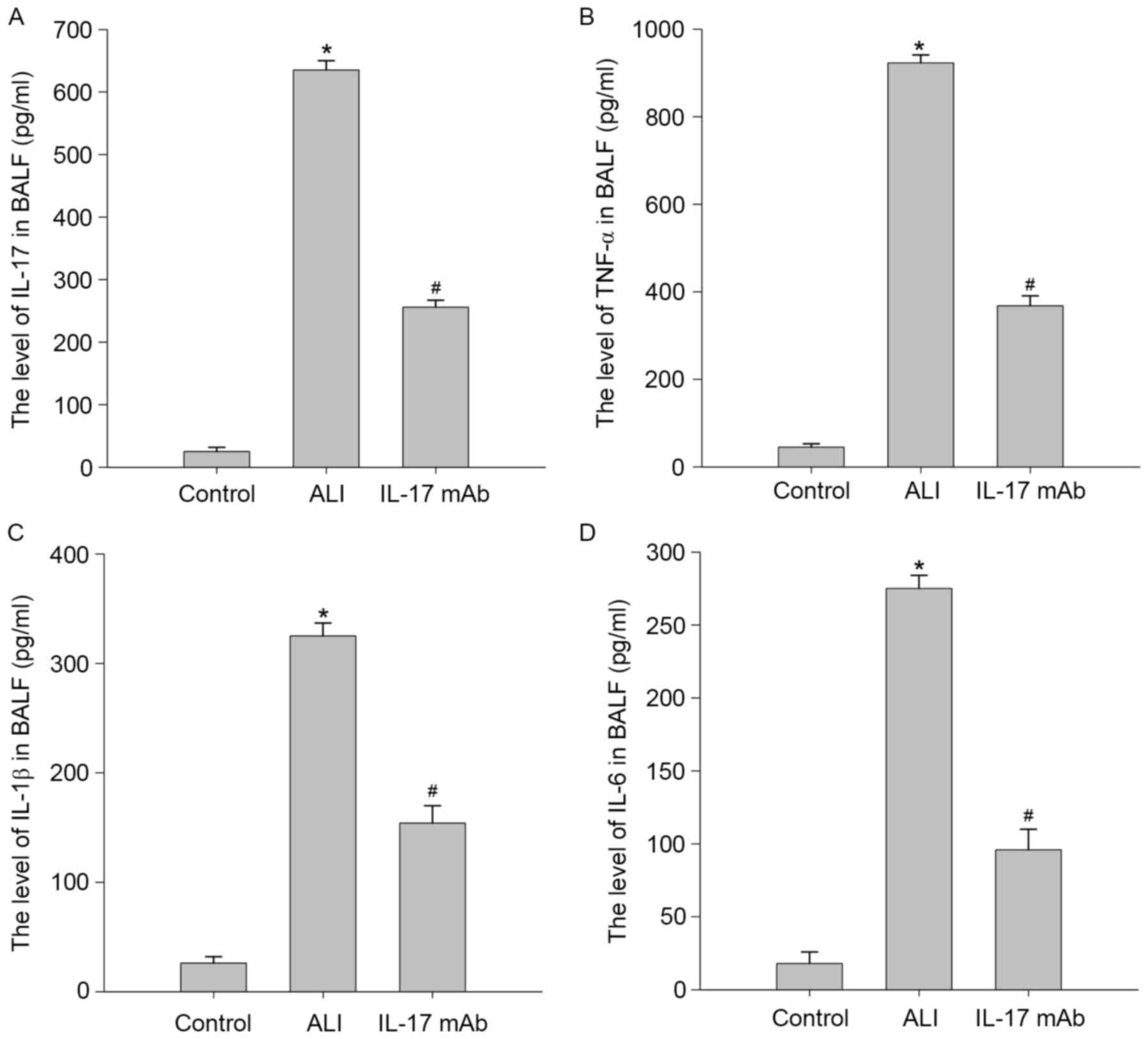
Blockade of IL-17 ameliorated
LPS-induced pulmonary inflammation
To confirm the effects of IL-17 mAb treatment on
LPS-stimulated expression and release of proinflammatory cytokines
and chemokines, the authors measured the levels of IL-17, TNF-α,
IL-1β and IL-6 in the BALF of rat by ELISA. As demonstrated in
Fig. 4, proinflammatory cytokines
including TNF-α, IL-1β and IL-6 were all significantly elevated in
BALF in response to LPS treatment. Consistent with this, levels of
these proinflammatory factors were dramatically decreased compared
with LPS-treated group. These results suggested that IL-17 mAb
administration significantly prevented the upregulation of
proinflammatory cytokines and chemokines in BALF of LPS challenged
rats.
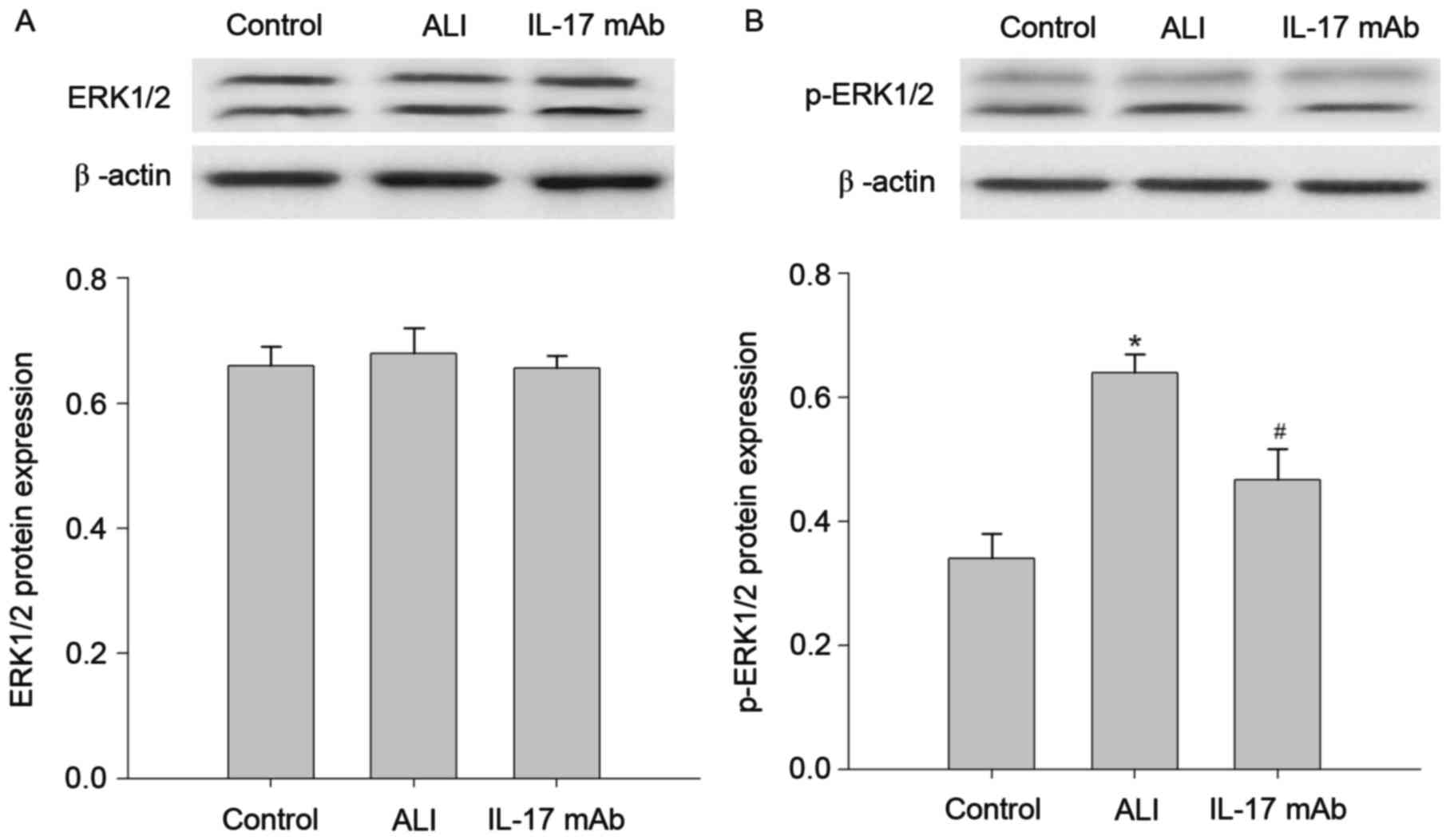
Blockade of IL-17 inhibited
LPS-induced ERK1/2 pathway activation
Given the crucial role of IL-17 in the pulmonary
inflammation, the authors next sought to investigate the underlying
mechanisms of IL-17 involved in the development of ALI. The
expression of ERK1/2, p-ERK1/2 in the lung tissues of rats
suffering from LPS stimulation was investigated. As demonstrated in
Fig. 5, the LPS-treated group
presented significant increases in the expression of the p-ERK1/2
protein when compared with the control group. Moreover, the
upregulation of p-ERK1/2 protein was reversed by IL-17 mAb
treatment. However, the expression of the ERK1/2 protein presented
no significant difference between each group. These findings
suggested that effects of IL-17 mAb on the features of LPS-induced
lung inflammation primarily through inhibition of ERK1/2 pathway
activity.
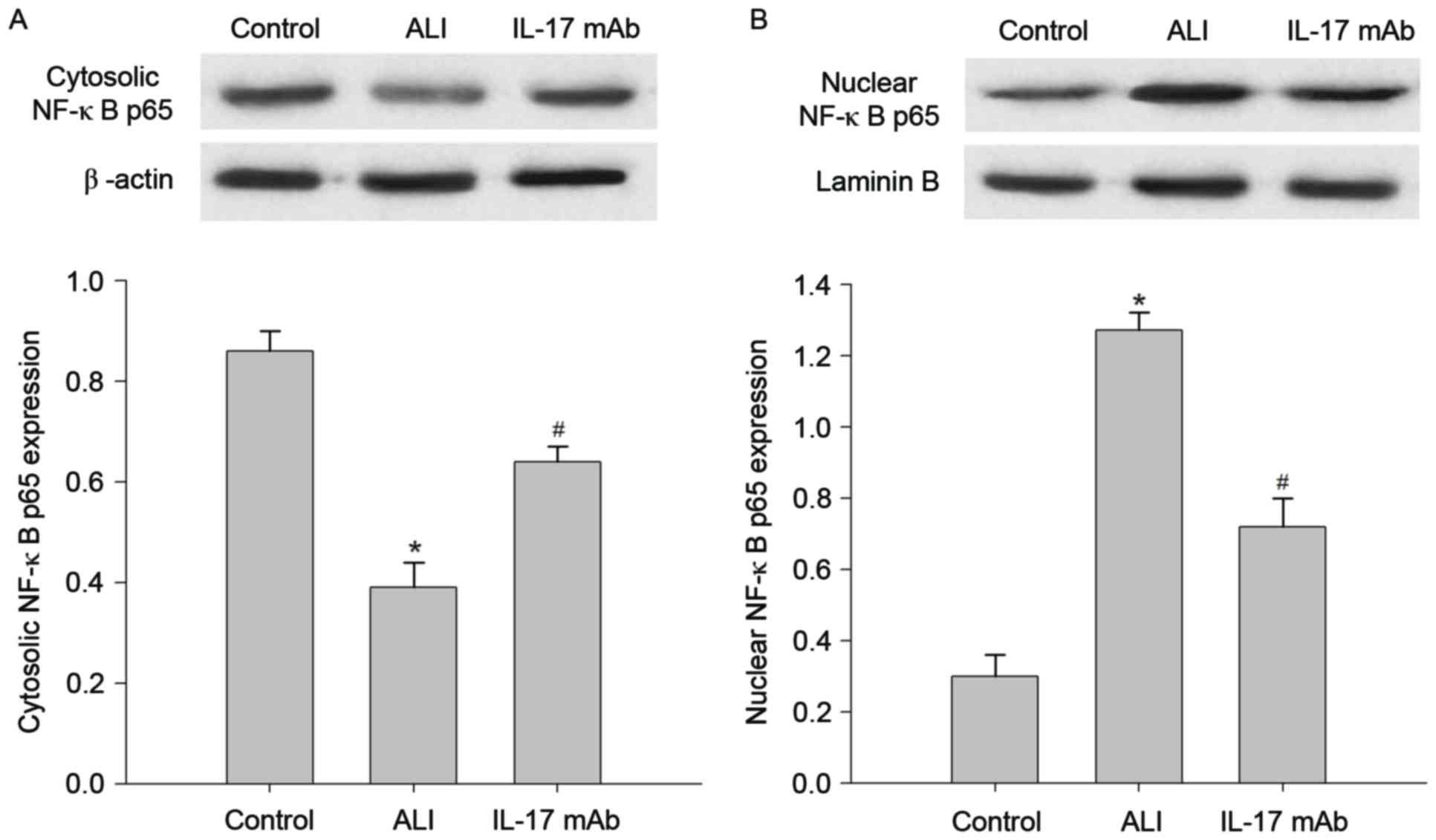
Blockade of IL-17 suppressed
LPS-induced NF-κB pathway activation
Since NF-κB is known to be a critical transcription
factor for inflammation, the molecular mechanisms by which IL-17
mAb administration ameliorates LPS-induced lung inflammation we
explored. This was conducted by measuring the levels of NF-κB p65
in the cytoplasm and nucleus. As presented in Fig. 6, a basal level of NF-κB p65 was
observed in the cytoplasm and nuclei of lung samples in the control
group. The level of NF-κB p65 in nuclear extracts of lung samples
was significantly increased following the instillation of LPS
compared with control group, this increase was reduced by
administration of IL-17 mAb. In contrast, cytosolic NF-κB p65
expression was decreased in the ALI group in comparison with
control group, and IL-17 mAb treatment could reversed the decrease
in NF-κB p65 protein. Additionally, western blot analyses revealed
that the LPS-induced nuclear translocation of NF-κB p65 was
restored by administration of IL-17 mAb.
 | Figure 6.Blockade of IL-17 inhibits the
activation of the ERK1/2 pathway in the lung of LPS-induced ALI.
Western blotting was used to analyze the expression of ERK1/2 and
p-ERK1/2 protein in the lung tissues at 6 h following ALI. (A)
Relative protein band densities of ERK1/2 expression in the lungs,
and the respective densitometric analysis of ERK1/2 bands,
normalized against β-actin. (B) Relative protein band densities of
nuclear p-ERK1/2 expression, and the respective densitometric
analysis of p-ERK1/2 bands, normalized against laminin B. Data are
presented as the mean ± standard deviation of eight independent
experiments. *P<0.05 vs. the Control group,
#P<0.05 vs. the ALI group. IL, interleukin; ERK,
extracellular signal-regulated kinase; ALI, acute lung injury; mAb,
monoclonal antibody; NF, nuclear factor. |
Discussion
ALI and acute respiratory distress syndrome (ARDS)
are the two most severe clinical diseases that account for
extensive morbidity and mortality; this is due to lacking effective
therapy strategies. However, looking at decades of extensive
investigation, the early diagnostic pathogenetic factors,
pathogenesis and specific treatment options of ALI remain
undefined. Thus, searching and identifying novel therapeutic
strategies with protective effect against ALI are an urgent
requirement. ALI is primarily characterized by a severe acute
inflammatory response in the lungs and neutrophilic alveolitis
(4). Particularly, growing
evidence suggests that IL-17 may induce the excessive expression
and release of pro-inflammatory cytokines from various cells
including Th17 cells, macrophages, and natural killer cells
(22). In addition, IL-17 has been
reported to be necessary for LPS-induced airway neutrophilia
(23).
The present study aimed to evaluate the role of
IL-17 in LPS-induced ALI, especially focusing on the associated
with the ERK1/2 and NF-κB signaling pathway. LPS-induced lung
injury in rats is frequently used as a model for studying ALI.
Firstly, the authors demonstrated that the instillation of LPS
induced pathological damage of lung tissues including alveolar
distortion, neutrophil recruitment, interstitial edema and
disruption of epithelial integrity. Following this, LPS treatment
induction of pulmonary edema was observed, as evidenced by the
changes in BALF exudate volume, protein leakage and lung W/D weight
ratio. In addition, levels of pro-inflammatory cytokines in BALF,
such as TNF-α, IL-1β and IL-6 were significantly increased in
LPS-induced ALI. Furthermore, levels of IL-17 mRNA and protein were
elevated in the BALF and lung tissues. Interestingly, treating a
rat model of LPS-induced ALI with IL-17 neutralizing antibody
markedly reduced these typical manifestations of lung injury
including histologic changes, pulmonary edema and lung
inflammation. These findings suggested an involvement of IL-17 in
the pathophysiological process of LPS-induced lung injury in rats.
Herein, the present study provided evidence demonstrating
correlations between the increased in IL-17 levels and the severity
of lung injury.
More importantly, the authors further explored
whether IL-17-induced the expression of cytokines and chemokines
are associated with the ERK1/2 and NF-κB signaling pathway in a rat
model of ALI. NF-κB, a critical transcription factor is involved in
the regulation of gene expression in numerous pro-inflammatory
mediators (24,25). Furthermore, the activated NF-κB
may, in turn, activate and control inflammatory responses and
regulate gene expression of various enzymes involved in
inflammation amplification and maintenance (26). These results suggested that the
activation of NF-κB signal was important in the early inflammatory
response. In the present study, the blockade of IL-17 markedly
suppressed nuclear translocation of NF-κB p65, further inhibiting
the excessive release of inflammatory factors. Additionally, the
elevated p-ERK1/2 in lung tissues of ALI rats was reversed by the
IL-17 neutralizing antibody. It is reported that activation of the
ERK1/2 signal could stabilize the mRNAs of the IL-17 downstream
target genes (27,28). These findings demonstrated that
IL-17 drives the ERK1/2 and NF-κB signaling pathway to trigger and
stabilize the transcription of downstream target genes
respectively, such as TNF-α, IL-1β and IL-6.
In summary, the authors identified the potential
role of IL-17 in the development/maintenance of LPS-induced lung
inflammation/injury. Elevated IL-17 expression closely correlated
to development of ALI and inhibition of IL-17 using neutralizing
antibody resulted in attenuating LPS-induced lung inflammation,
which was consistent with previous studies. However, the
anti-inflammatory mechanisms of the IL-17 neutralizing antibody was
elucidated, which was associated with repressing activation of the
ERK1/2 and NF-κB signaling pathway. These findings increased the
understanding of how IL-17 affects lung inflammation in ALI,
providing promising potential therapeutic targets for clinical
ALI.
Acknowledgements
The authors would like to thank Professor Lian-li
Zhao for critical readings of the manuscript. The present study was
supported by a grant from the Natural Science Foundation of Hebei,
China (grant no. H2015209309) and the Science and Technology
Development Project (grant no. 12140209A-31).
Glossary
Abbreviations
Abbreviations:
|
ALI
|
acute lung injury
|
|
LPS
|
lipopolysaccharide
|
|
IL-17
|
Interleukin-17
|
|
TNF-α
|
tumor necrosis factor-α
|
|
IL-1β
|
interleukin-1β
|
|
IL-6
|
interleukin-6
|
|
NF-κB
|
nuclear factor-κB
|
|
ERK1/2
|
extracellular signal-regulated
kinase1/2
|
|
BALF
|
bronchoalveolar lavage fluid
|
References
|
1
|
Sadowitz B, Jain S, Kollisch-Singule M,
Satalin J, Andrews P, Habashi N, Gatto LA and Nieman G: Preemptive
mechanical ventilation can block progressive acute lung injury.
World J Crit Care Med. 5:74–82. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li C, Bo L, Liu W, Lu X and Jin F: Enteral
immunomodulatory diet (Omega-3 Fatty, Acid, γ-Linolenic Acid and
Antioxidant Supplementation) for acute lung injury and acute
respiratory distress syndrome: An updated systematic review and
meta-analysis. Nutrients. 7:5572–5585. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Imam F, Al-Harbi NO, Al-Harbi MM, Ansari
MA, Zoheir KM, Iqbal M, Anwer MK, Al Hoshani AR, Attia SM and Ahmad
SF: Diosmin downregulates the expression of T cell receptors,
pro-inflammatory cytokines and NF-κB activation against LPS-induced
acute lung injury in mice. Pharmacol Res. 102:1–11. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fu J, Wang Y, Zhang J, Wu W, Chen X and
Yang Y: Anti-inflammatory and anti-apoptotic effects of
oxysophoridine on lipopolysaccharide-induced acute lung injury in
mice. Am J Transl Res. 7:2672–2682. 2015.PubMed/NCBI
|
|
5
|
Wang YY, Qiu XG and Ren HL: Inhibition of
acute lung injury by rubriflordilactone in LPS-induced rat model
through suppression of inflammatory factor expression. Int J Clin
Exp Pathol. 8:15954–15959. 2015.PubMed/NCBI
|
|
6
|
Yan Z, Xiaoyu Z, Zhixin S, Di Q, Xinyu D,
Jing X, Jing H, Wang D, Xi Z, Chunrong Z and Daoxin W: Rapamycin
attenuates acute lung injury induced by LPS through inhibition of
Th17 cell proliferation in mice. Sci Rep. 6:201562016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gross CM, Rafikov R, Kumar S, Aggarwal S,
Ham PB III, Meadows ML, Cherian-Shaw M, Kangath A, Sridhar S, Lucas
R and Black SM: Endothelial nitric oxide synthase deficient mice
are protected from lipopolysaccharide induced acute lung injury.
PLoS One. 10:e01199182015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xu C, Chen G, Yang W, Xu Y, Xu Y, Huang X,
Liu J, Feng Y, Xu Y and Liu B: Hyaluronan ameliorates LPS-induced
acute lung injury in mice via Toll-like receptor (TLR) 4-dependent
signaling pathways. Int Immunopharmacol. 28:1050–1058. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Feng G, Jiang ZY, Sun B, Fu J and Li TZ:
Fisetin alleviates lipopolysaccharide-induced acute lung injury via
TLR4-Mediated NF-κB signaling pathway in rats. Inflammation.
39:148–157. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Aggarwal S, Dimitropoulou C, Lu Q, Black
SM and Sharma S: Glutathione supplementation attenuates
lipopolysaccharide-induced mitochondrial dysfunction and apoptosis
in a mouse model of acute lung injury. Front Physiol. 3:1612012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
You QH, Zhang D, Niu CC, Zhu ZM, Wang N,
Yue Y and Sun GY: Expression of IL-17A and IL-17F in
lipopolysaccharide-induced acute lung injury and the counteraction
of anisodamine or methylprednisolone. Cytokine. 66:78–86. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gong F, Liu Z, Liu J, Zhou P, Liu Y and Lu
X: The paradoxical role of IL-17 in atherosclerosis. Cell Immunol.
297:33–39. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Khan D and Ahmed S Ansar: Regulation of
IL-17 in autoimmune diseases by transcriptional factors and
microRNAs. Front Genet. 6:2362015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kugyelka R, Kohl Z, Olasz K, Mikecz K,
Rauch TA, Glant TT and Boldizsar F: Enigma of IL-17 and Th17 cells
in rheumatoid arthritis and in autoimmune animal models of
arthritis. Mediators Inflamm. 2016:61458102016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qian X, Chen H, Wu X, Hu L, Huang Q and
Jin Y: Interleukin-17 acts as double-edged sword in anti-tumor
immunity and tumorigenesis. Cytokine. 89:34–44. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Beringer A, Noack M and Miossec P: IL-17
in chronic inflammation: From discovery to targeting. Trends Mol
Med. 22:230–241. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li Q, Gu Y, Tu Q, Wang K, Gu X and Ren T:
Blockade of Interleukin-17 restrains the development of acute lung
injury. Scand J Immunol. 83:203–211. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Onishi RM and Gaffen SL: Interleukin-17
and its target genes: Mechanisms of interleukin-17 function in
disease. Immunology. 129:311–321. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Matute-Bello G, Frevert CW and Martin TR:
Animal models of acute lung injury. Am J Physiol Lung Cell Mol
Physiol. 295:L379–L399. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xie K, Yu Y, Pei Y, Hou L, Chen S, Xiong L
and Wang G: Protective effects of hydrogen gas on murine
polymicrobial sepsis via reducing oxidative stress and HMGB1
release. Shock. 34:90–97. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shabgah AG, Fattahi E and Shahneh FZ:
Interleukin-17 in human inflammatory diseases. Postepy Dermatol
Alergol. 31:256–261. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ferretti S, Bonneau O, Dubois GR, Jones CE
and Trifilieff A: IL-17, produced by lymphocytes and neutrophils,
is necessary for lipopolysaccharide-induced airway neutrophilia:
IL-15 as a possible trigger. J Immunol. 170:2106–2112. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shih RH, Wang CY and Yang CM: NF-kappaB
signaling pathways in neurological inflammation: A mini review.
Front Mol Neurosci. 8:772015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang H and Sun SC: NF-κB in inflammation
and renal diseases. Cell Biosci. 5:632015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schuliga M: NF-kappaB signaling in chronic
inflammatory airway disease. Biomolecules. 5:1266–1283. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hata K, Andoh A, Shimada M, Fujino S,
Bamba S, Araki Y, Okuno T, Fujiyama Y and Bamba T: IL-17 stimulates
inflammatory responses via NF-kappaB and MAP kinase pathways in
human colonic myofibroblasts. Am J Physiol Gastrointest Liver
Physiol. 282:G1035–G1044. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bulek K, Liu C, Swaidani S, Wang L, Page
RC, Gulen MF, Herjan T, Abbadi A, Qian W, Sun D, et al: The
inducible kinase IKKi is required for IL-17-dependent signaling
associated with neutrophilia and pulmonary inflammation. Nat
Immunol. 12:844–852. 2011. View
Article : Google Scholar : PubMed/NCBI
|