Introduction
Breast cancer is one of the most commonly diagnosed
cancers in women worldwide. The incidence rate increases every
year, and breast cancer is becoming more prevalent in younger age
groups (1). The 5- and 10-year
survival rates for metastatic breast cancer are currently estimated
at <25 and 5–10%, respectively (2). Continued investigations into the role
of dysregulated cell cycling in human breast cancer have led to the
identification of several therapeutically attractive targets
(3,4). Recently, the synergistic effects of
autophagy and cyclin-dependent kinase (CDK) inhibitors have been
reported in cancer therapy (5).
Flavopiridol (FP) is a CDK inhibitor that is known for its activity
in arresting cell cycle at the G1-S or G2-M
boundary and in mediating antiproliferative effects by inhibiting
multiple CDKs (6,7). In addition, comprehensive studies
have demonstrated the antitumoral activity of FP against human
tumor cell lines (4,8,9); FP
is considered an important ancillary tumor chemotherapy drug for
its cytotoxicity in resting cells. In human breast cancer cells, FP
may also downregulate the expression of antiapoptosis proteins in
the inhibitor of apoptosis (IAP) family and B cell lymphoma-extra
large (Bcl-XL) and upregulate the expression of
proapoptotic Bcl-2-associated X protein (Bax) to induce cell growth
arrest and apoptosis, and these effects are independent of tumor
estrogen receptor (ER) status (10–12).
However, the potential molecular mechanisms leading to cell
apoptosis are unclear.
Autophagy is a catabolic process that regulates the
degradation and recycling of organelles and proteins to maintain
the general cellular homeostasis (13). Once autophagy is activated, it may
either induce tumor cell death (14) or antiapoptotic survival (15). Numerous scientific reports have
demonstrated the association between autophagy and cancer cell
biologic activities, such as survival, apoptosis and metastasis.
Apoptosis activation serves as a potential anticancer strategy by
repressing various types of tumor cells (16,17).
A previous study demonstrated that significant autophagy flux in
FP-treated chronic lymphocytic leukemia (CLL) cells was initiated
by FP-induced endoplasmic reticulum stress other than the result of
directly inhibition of CDKs. Furthermore, inhibition of autophagy
enhances FP cytotoxicity, which indicated that FP-induced autophagy
may serve a drug resistance role in CLL cells (18).
Autophagy and apoptosis were demonstrated to be
induced by antitumor drugs in human breast cancer cell lines, such
as Paris saponin XA-2 and rapamycin (19). Those two drugs promoted breast
cancer cell apoptosis via the Akt/mammalian target of rapamycin
signaling pathway, which is also independent of cancer cell ER
expression, as evidenced by the caspase activation and cleavage of
Poly (ADP-ribose) polymerase. A recent study demonstrated that
specific inhibition or small interfering siRNA-mediated silencing
of inositol 1,4,5-trisphosphate receptors induced autophagic cell
death through compromised bioenergetics and the generation of
reactive oxygen species in MCF-7 human breast cancer cells
(20). In other human breast tumor
cell lines, such as SKBR-3 and MB-468, FP treatment was revealed to
reduce the expression levels of the antiapoptotic proteins IAP,
Bcl-xL and myeloid cell leukemia 1 to induce cell death
and enhanced the cytotoxicity of epothilone B, which is impeded by
overexpression of Bcl-2 (11).
Bcl-2 is a crucial factor involved in the process turning a normal
cell into a cancerous one. Other than being an antiapoptotic
protein, Bcl-2 also suppresses autophagy by directly targeting
beclin 1, a component of the class III phosphoinositide 3-kinase
(PI3K) pathway that serves a role in autophagosome formation
(21). Although FP-induced
apoptosis and autophagy in different types of cancer cells has been
well studied, the capacity of FP-induced autophagy in human breast
cancer has, to the best of our knowledge, not yet been examined.
The present study investigated whether FP treatment was able to
induce autophagy in MCF-7 cells and determined the role of
autophagy in survival and cell cycle arrest.
Materials and methods
MCF-7 cells in culture
MCF-7 human breast cancer cells were purchased from
Nanjing KeyGen Biotech Co., Ltd. (Nanjing, China) and were grown in
RPMI-1640 medium (GIBCO; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) supplemented with 10% fetal bovine serum (HyClone; GE
Healthcare, Logan, UT, USA), 0.01 mg/ml bovine insulin
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), 100 U/ml
penicillin and 0.1 mg/ml streptomycin (Beyotime Institute of
Biotechnology, Haimen, China) and maintained in an incubator at
37°C with 95% humidity and 5% CO2.
Western blotting
MCF-7 cells were treated with rapamycin (5 µM) or FP
(1 µM) with or without chloroquine (CQ; 500 nM; Sigma-Aldrich;
Merck KGaA) for a total of 48 h at 37°C, based on a previously
published protocol (18). A total
of 1 ×106 MCF-7 cells were harvested by scraping the
cells from culture plates and collected by centrifugation at 40 × g
for 6 min at 4°C. Cells were resuspended in 125 mM Tris buffer (pH
6.8) and sonicated twice for 10 sec each and lysed using an equal
volume of 8% SDS. Cell extracts were boiled for 10 min and chilled
on ice. Protein concentration was measured using the Bicinchoninic
Acid Solution (Sigma-Aldrich; Merck KGaA). The samples (10 µg for
each well) were loaded on 10% SDS-PAGE for separation, and
electrophoretically transferred to a nitrocellulose (NC) membrane.
Subsequently, membranes were blocked at room temperature for 1 h in
western blocking buffer (Beyotime Institute of Biotechnology). Each
membrane was incubated with monoclonal antibodies against
autophagy-related protein LC3B-II, p62/sequestosome 1 (SQSTM1),
autophagy-related 5 (ATG5) and GAPDH (cat. no. 66139-1-Ig,
66184-1-Ig, 60061-1-Ig and 60004-1-Ig, respectively; Wuhan Sanying
Biotechnology, Wuhan, China) at a dilution of 1:1,000, at 4°C
overnight. Blots were washed with PBS with 0.05% Tween-20 and
incubated with horseradish peroxidase-conjugated secondary
antibodies (cat. no. A0216, Beyotime Institute of Biotechnology) at
a dilution of 1:1,000, at room temperature for 1 h. Signal
intensities were measured using a chemiluminescence Detection
System (Pierce; Thermo Fisher Scientific, Inc.). Autoradiograms
were scanned with a Gel Doc 1000 Imaging System (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The bi-dimensional optical
density of proteins on the films was quantified and analyzed with
Molecular Analysis Software (version 1.6.3, Bio-Rad Laboratories,
Inc.). GAPDH was used as the internal control for LC3B-II, SQSTM1
and ATG 5 expression analysis.
siRNA experiments
Exponentially growing untreated MCF-7 cells were
collected and cultured (2×105/well in 2 ml) for 24 h
before transfection. Plated cells were transfected with 80 pmol of
either siRNA against human ATG5 (cat. no. sc-41445; Santa Cruz
Biotechnology, Inc., Dallas, Texas, USA) or a negative control
siRNA (cat. no. sc-37007; Santa Cruz Biotechnology, Inc.) in 100 µl
siRNA transfection media and an X-tremeGENE Transfection Reagent
(Roche Diagnostics, Basel, Switzerland) 8 µl at 37°C for 8 h,
following the manufacturer's protocol. Two days post-siRNA
transfection, cells were treated with FP for 48 h. Following the
final incubation, total protein was harvested and subjected to
western blot analysis.
Cell viability assay
Cell viability was determined using a Cell Counting
kit-8 (CCK-8; Beyotime Institute of Biotechnology). Briefly, cells
(5,000 cells/well) were seeded in culture medium in 96-well plates
and incubated at 37°C for 24 h prior to drug treatments.
Subsequently, the culture medium was replaced with fresh medium
containing either FP (1 µM) alone or FP (1 µM) with CQ (0.5 µM) or
CQ (0.5 µM) alone and cells were incubated for 48 h at 37°C. The
control group was treated with 0.2% DMSO alone. Finally, cells were
incubated with complete medium containing 10 µl CCK-8 solution at
37°C for 2 h and optical density at 450 nm was measured as the
percentage of cells compared with the control group, using a
microplate reader. After ATG5 siRNA (80 pmol/100 µl) was
transfected, cells were divided into three groups: untransfected
group, control siRNA group and siRNA ATG5 group. And cells were
treated with FP or equal volume PBS. The control group was treated
with 0.2% DMSO. The measurement was the same as above.
Cell cycle analysis
For cell cycle analysis, cells (2×104
cells/well) were plated in a 24-well plate and grown to 60%
confluency. The cells were grown in serum free medium for 24 h to
have limited synchronization. Then, normal MCF-7 cells were treated
in complete medium with either FP (1 µM) or FP (1 µM) with CQ (0.5
µM) at 37°C for 48 h. siRNA transfected cells were treated with FP
(1 µM) alone at the same conditions. Following treatments, cells
were harvested by centrifugation at 40 × g for 6 min at 4°C and
fixed in cold ethanol (70% v/v) in PBS for at least 2 h at 4°C.
Fixed cells were washed with PBS and stained with a solution
containing propidium iodide (6 µM), 0.5% Triton X-100 and RNase
(100 µg/ml) for 30 min at room temperature, in the dark. DNA
content was analyzed through flow cytometry. The percentage of
cells in G0-G1, S and G2-M phases
was calculated using MultiCycle Software (version 3.0; Phoenix Flow
Systems, San Diego, CA).
Statistical analysis
Data are presented as the mean ± standard deviation
from three experiments. Two-way t-tests were used to analyze the
variance in two groups for possible significance. One-way analysis
of variance followed Tukey's method was used for multiple
comparisons. Statistical analysis was performed using GraphPad
Prism Software version 7.00 (GraphPad Software, Inc., La Jolla, CA,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
MCF-7 cells are susceptible to
autophagy induced by FP
As induction of autophagy in human breast cancer
cell lines has been fully confirmed including MCF-7 cells (20), it was determined whether MCF-7
cells undergo autophagy with FP during this process (19). MCF-7 cells were incubated for 48 h
with 1 µM FP or with 5 µM rapamycin (a well-characterized initiator
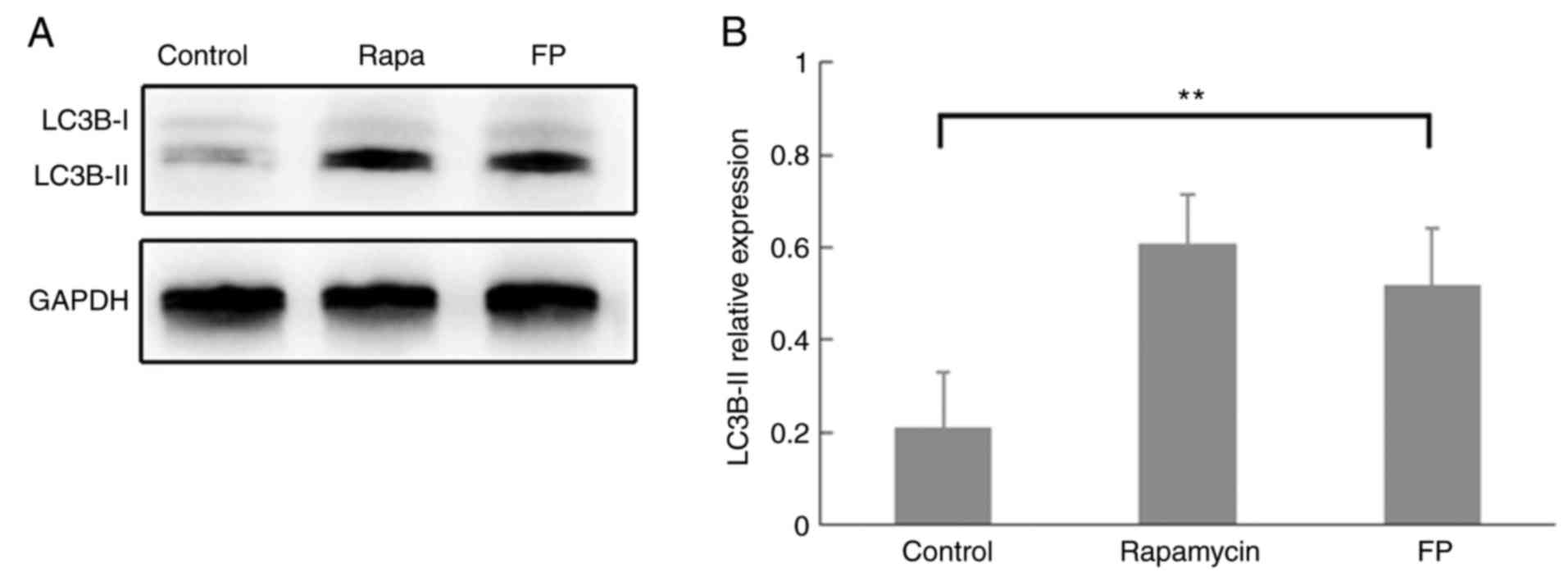
of autophagy). Western blot analysis demonstrated classic autophagy
performance of LC3B protein expression in the rapamycin-treated
MCF-7 cells, although this was not quantified (Fig. 1A). Western blot analysis also
demonstrated that LC3B-II expression was significantly increased in
FP-treated cells compared with the control (Fig. 1B; P<0.01), which indicated the
induction of autophagosome formation by FP incubation.
Verification of autophagy induced by
FP
LC3 protein was previously reported to be localized
in structures other than autophagosomes in some tissues (22,23);
however, immunodetection of LC3 in cytoplasmic granules is not
sufficient as an absolute marker to monitor autophagy. Recent
guidelines for the use and interpretation of assays for monitoring
autophagy recommended using p62/SQSTM1 and LC3B-II turnover to
monitor flux (24). CQ is a
proteasome inhibitor; it interrupts the autophagy flux by
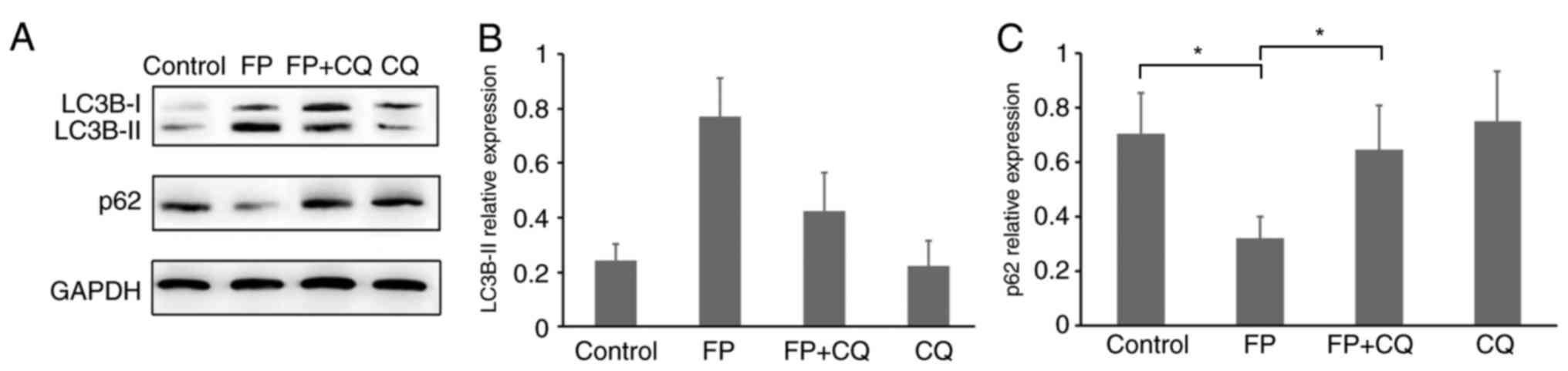
inhibiting lysosome-mediated proteolysis (25). Therefore, the present study
analyzed the protein expression levels of both LC3B-II and
p62/SQSTM1 (Fig. 2A-C). Western
blotting results revealed the expected decrease of p62/SQSTM1
protein expression in FP-treated MCF-7 cells and restored by FP and
CQ co-treatment (Fig. 2A and C,
P<0.05). This could imply that the change in LC3B and p62/SQSTM1
expression levels could be the result of FP-induced autophagy in
MCF-7 cells and it was successfully blocked by co-treatment with
CQ.
Inhibition of autophagy antagonizes FP
cytotoxicity
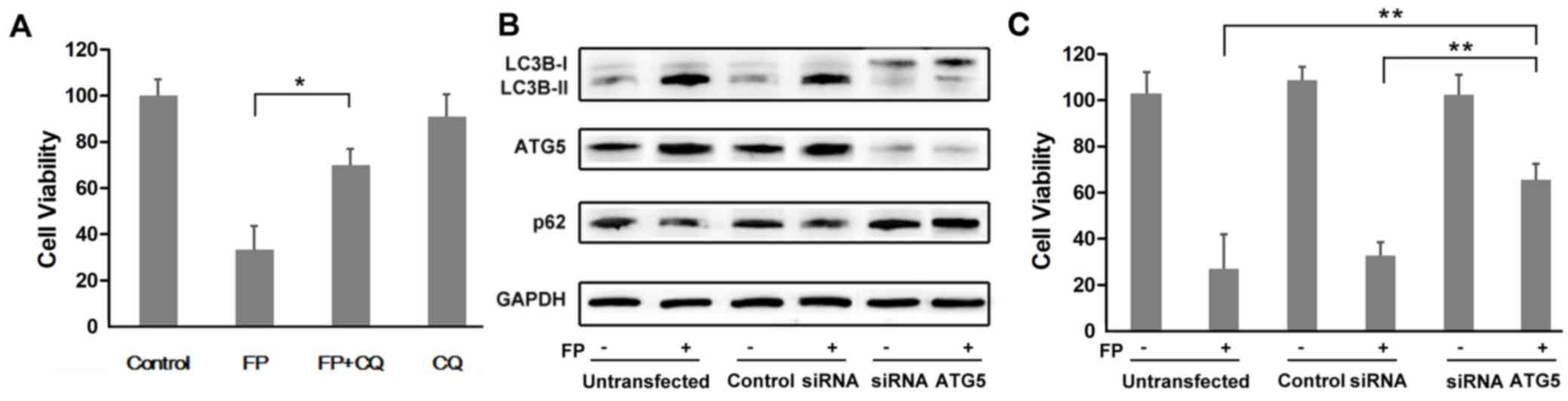
Co-treatment with FP and CQ significantly
antagonizes the cytotoxicity of FP in MCF-7 cells (P<0.05;
Fig. 3A). As CQ is not a specific
autophagy inhibitor, it may affect other cellular processes in
addition to autophagy. After transfected with siRNA ATG5, the
expression of ATG5 was knocked down (Fig. 3B). The expression trend of both
LC3B and p62/SQSTM1 validated that autophagy was blocked after
transfection compared with the untransfected cells and control
siRNA-transfected cells (Fig. 3B).
There was a significantly higher rate in cell survival in
ATG5-silenced MCF-7 cells compared with the untransfected and the
control siRNA-transfected cells treated with FP (Fig. 3C; P<0.01).
FP-induced cell cycle arrest is not
impaired by autophagy inhibition
Autophagy is generally associated with metabolic
activity, including proliferation and cell cycle. It has been
proved that FP blocked MCF-7 cells at G1 stage, inhibition of
resveratrol-induced autophagy abolished cell cycle arrest by
restoring the levels of cyclin A and B to normal levels in glioma
cells (26). In the present study,
autophagy inhibition by CQ or ATG5 siRNA treatment did not notably
impair the G0-G1 phase of the cell cycle
(P>0.05; Table I). These data
suggested that FP may have a cell cycle regulation role but this
needs to be determined.
 | Table I.Cell cycle effects of autophagy
inhibitiona on MCF-7
cells. |
Table I.
Cell cycle effects of autophagy
inhibitiona on MCF-7
cells.
| Treatment |
G0-G1 (%) | S (%) | G2-M
(%) |
|---|
| Control | 45±2 | 26±3 | 29±7 |
| FP | 61±5 | 19±4 | 20±6 |
| FP + CQ | 63±9b | 22±8 | 15±2 |
| FP + ATG5 siRNA | 64±3b | 20±3 | 16±5 |
Discussion
CDK inhibition is an important target for breast
cancer treatments, which is resistant to endocrine therapy
(4). Flavopiridol is a
semisynthetic flavonoid that is derived from rohitukine.
Significant effort has been made to understand the detailed
pathways of FP action (27).
Briefly, the mechanisms of antitumoral effects may lie in direct
inhibition of CDK expression or activation by binding to the
ATP-binding site or by the prevention of CDK phosphorylation, which
is overexpressed in many human neoplasias (28,29).
However, this does not offer a sufficient explanation as to how FP
induces cell death in growth-arrested cancer cells.
An important biochemical mechanism that is involved
in FP-induced proapoptotic cell death is autophagy (10). The present study demonstrated that
MCF-7 cells expressed crucial components of the autophagy
machinery, which may be robustly activated by FP. FP induced
aggregation of LC3B-II and decreased p62/SQSTM1 expression levels
which was partially abolished by CQ co-treatment and siRNA ATG5
transfection. Autophagy inhibition by CQ resulted in notably higher
cell survival rate compared with cells treated with FP alone.
Furthermore, this was also confirmed by ATG5 silencing. Autophagy
inhibition did not impair the effects on cell cycle arrest in
FP-treated MCF-7 cells.
As autophagy has been considered to be paradoxical
in character when MCF-7 cells were treated with different drugs:
One involves augmenting proapoptotic autophagy to induce cell death
(30), and the other involves the
upregulation of pro-survival drug resistance (31). These inconsistent results from
previously published work may attribute to the complex role of
autophagy in cancer and it is likely dependent on tumor type,
stage, and genetic context (32).
Further clarification of autophagy may result in new strategies for
cancer therapy. Antiapoptotic Bcl-2 protein expression leads to
oncogenic transformation and drug resistance in many human cancers,
including breast cancer (33,34).
Bcl-2 suppresses autophagy by directly binding the BH3 domain in
beclin 1, which is part of a class III PI3K complex that
participates in autophagosome formation (35). A previous study using MCF-7 cells
demonstrated that the Bcl-2/beclin 1 complex may function as a
rheostat that ensures that the levels of autophagy remain within a
homeostatic rather than a nonphysiological range that triggers cell
death (36). Chemotherapeutics may
alter Bcl-2/beclin 1 binding activity or Bcl-2 expression to
regulate autophagy and apoptosis in the cancer cells. These two
Bcl-2 regulations may cause different results of cancer cell
survival. Inhibition of Bcl-2/beclin 1 binding activity may not
affect the antiapoptotic function of Bcl-2. Such as Pseudolaric
acid B (PAB), which has been demonstrated to exert an antitumor
effect in MCF-7 cells. PAB treatment inhibits the formation of the
Bcl-2/beclin 1 complex, leaving beclin 1 free to participate in
autophagy. However, PAB may also increase Bcl-2 expression and it
was concluded that PAB-activated autophagy in MCF-7 cells may
contribute to resistance to cell death (37). On the other hand, if Bcl-2
expression was decreased; both autophagy and apoptosis were
enhanced. FP has been reported to downregulate the Bcl-2 expression
in several types of human breast cancer cell lines (4), which may explain why FP is useful as
a medication for cancer therapy. Altogether, the present results
demonstrated that FP exposure induced autophagy in MCF-7 human
breast cancer cells and autophagy, considering FP as a potential
autophagy regulatory molecule for cancer therapy, but further
experimentation is required.
Acknowledgements
This work was supported by The National Natural
Science Foundation of China (grant no. 81601535).
Glossary
Abbreviations
Abbreviations:
|
CDK
|
cyclin-dependent kinase
|
|
CQ
|
chloroquine
|
|
FP
|
flavopiridol
|
References
|
1
|
Howell A, Anderson AS, Clarke RB, Duffy
SW, Evans DG, Garcia-Closas M, Gescher AJ, Key TJ, Saxton JM and
Harvie MN: Risk determination and prevention of breast cancer.
Breast Cancer Res. 16:4462014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Austreid E, Lonning PE and Eikesdal HP:
The emergence of targeted drugs in breast cancer to prevent
resistance to endocrine treatment and chemotherapy. Expert Opin
Pharmacother. 15:681–700. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Asghar U, Witkiewicz AK, Turner NC and
Knudsen ES: The history and future of targeting cyclin-dependent
kinases in cancer therapy. Nat Rev Drug Discov. 14:130–146. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
DiPippo AJ, Patel NK and Barnett CM:
Cyclin-dependent kinase inhibitors for the treatment of breast
cancer: Past, Present, and Future. Pharmacotherapy. 36:652–667.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Senderowicz AM and Sausville EA:
Preclinical and clinical development of cyclin-dependent kinase
modulators. J Natl Cancer Inst. 92:376–387. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sedlacek HH: Mechanisms of action of
flavopiridol. Crit Rev Oncol Hematol. 38:139–170. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ravishankar D, Rajora AK, Greco F and
Osborn HM: Flavonoids as prospective compounds for anti-cancer
therapy. Int J Biochem Cell Biol. 45:2821–2831. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Iyer SP, Mejia J, Rosato AE, Grant S and
Rosato RR: Flavopiridol-mediated multiple modulatory effects
synergistically increase sensitivity to TRAIL-induced cell death in
human leukemia cells. Cancer Res. 73(8 Suppl): S29602013.
View Article : Google Scholar
|
|
9
|
Ali S, El-Rayes BF, Aranha O, Sarkar FH
and Philip PA: Sequence dependent potentiation of gemcitabine by
flavopiridol in human breast cancer cells. Breast Cancer Res Treat.
90:25–31. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wittmann S, Bali P, Donapaty S,
Nimmanapalli R, Guo F, Yamaguchi H, Huang M, Jove R, Wang HG and
Bhalla K: Flavopiridol down-regulates antiapoptotic proteins and
sensitizes human breast cancer cells to epothilone B-induced
apoptosis. Cancer Res. 63:93–99. 2003.PubMed/NCBI
|
|
11
|
Li Y, Bhuiyan M, Alhasan S, Senderowicz AM
and Sarkar FH: Induction of apoptosis and inhibition of c-erbB-2 in
breast cancer cells by flavopiridol. Clin Cancer Res. 6:223–229.
2000.PubMed/NCBI
|
|
12
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Weiner LM and Lotze MT: Tumor-cell death,
autophagy, and immunity. N Engl J Med. 366:1156–1158. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mahoney E, Byrd JC and Johnson AJ:
Autophagy and ER stress play an essential role in the mechanism of
action and drug resistance of the cyclin-dependent kinase inhibitor
flavopiridol. Autophagy. 9:434–435. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Galluzzi L, Pietrocola F, Bravo-San Pedro
JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J,
Gewirtz DA, Karantza V, et al: Autophagy in malignant
transformation and cancer progression. EMBO J. 34:856–880. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rosenfeldt MT and Ryan KM: The multiple
roles of autophagy in cancer. Carcinogenesis. 32:955–963. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mahoney E, Lucas DM, Gupta SV, Wagner AJ,
Herman SE, Smith LL, Yeh YY, Andritsos L, Jones JA, Flynn JM, et
al: ER stress and autophagy: New discoveries in the mechanism of
action and drug resistance of the cyclin-dependent kinase inhibitor
flavopiridol. Blood. 120:1262–1273. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xie ZZ, Li MM, Deng PF, Wang S, Wang L, Lu
XP, Hu LB, Chen Z, Jie HY, Wang YF, et al: Paris saponin-induced
autophagy promotes breast cancer cell apoptosis via the Akt/mTOR
signaling pathway. Chem Biol Interact. 264:1–9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Singh A, Chagtoo M, Tiwari S, George N,
Chakrabarti B, Khan S, Lakshmi S and Godbole MM: Inhibition of
inositol 1,4,5-trisphosphate receptor induce breast cancer cell
death through deregulated autophagy and cellular bioenergetics. J
Cell Biochem. 118:2333–2346. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shibata M, Yoshimura K, Furuya N, Koike M,
Ueno T, Komatsu M, Arai H, Tanaka K, Kominami E and Uchiyama Y: The
MAP1-LC3 conjugation system is involved in lipid droplet formation.
Biochem Biophys Res Commun. 382:419–423. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shibata M, Yoshimura K, Tamura H, Ueno T,
Nishimura T, Inoue T, Sasaki M, Koike M, Arai H, Kominami E and
Uchiyama Y: LC3, a microtubule-associated protein1A/B light chain3,
is involved in cytoplasmic lipid droplet formation. Biochem Biophys
Res Commun. 393:274–279. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Arozena A Acevedo, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the Use and Interpretation of Assays
for Monitoring Autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ye H, Chen M, Cao F, Huang H, Zhan R and
Zheng X: Chloroquine, an autophagy inhibitor, potentiates the
radiosensitivity of glioma initiating cells by inhibiting autophagy
and activating apoptosis. BMC Neurol. 16:1782016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shao X, Gao D, Wang Y, Jin F, Wu Q and Liu
H: Application of metabolomics to investigate the antitumor
mechanism of flavopiridol in MCF-7 breast cancer cells. J
Chromatogr B Analyt Technol Biomed Life Sci. 1025:40–47. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Filippi-Chiela EC, Villodre ES, Zamin LL
and Lenz G: Autophagy interplay with apoptosis and cell cycle
regulation in the growth inhibiting effect of resveratrol in glioma
cells. PLoS One. 6:e208492011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Asghar U, Witkiewicz AK, Turner NC and
Knudsen ES: The history and future of targeting cyclin-dependent
kinases in cancer therapy. Nat Rev Drug Discov. 14:130–146. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Carlson B, Lahusen T, Singh S,
Loaiza-Perez A, Worland PJ, Pestell R, Albanese C, Sausville EA and
Senderowicz AM: Down-regulation of cyclin D1 by transcriptional
repression in MCF-7 human breast carcinoma cells induced by
flavopiridol. Cancer Res. 59:4634–4641. 1999.PubMed/NCBI
|
|
28
|
Carlson B, Pearlstein R, Naik R, Sedlacek
H, Sausville E and Worland P: Inhibition of CDK2, CDK4 and CDK7 by
flavopiridol and structural analogs. Proc Am Assoc Cancer Res.
37:pp. 4241996;
|
|
29
|
Worland PJ, Kaur G, Stetler-Stevenson M,
Sebers S, Sartor O and Sausville EA: Alteration of the
phosphorylation state of p34cdc2 kinase by the flavone L86-8275 in
breast carcinoma cells: Correlation with decreased H1 kinase
activity. Biochem Pharmacol. 46:1831–1840. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim DE, Kim Y, Cho DH, Jeong SY, Kim SB,
Suh N, Lee JS, Choi EK, Koh JY, Hwang JJ and Kim CS: Raloxifene
induces autophagy-dependent cell death in breast cancer cells via
the activation of AMP-activated protein kinase. Mol Cells.
38:138–144. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sun WL, Chen J, Wang YP and Zheng H:
Autophagy protects breast cancer cells from epirubicin-induced
apoptosis and facilitates epirubicin-resistance development.
Autophagy. 7:1035–1044. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kimmelman AC: The dynamic nature of
autophagy in cancer. Genes Dev. 25:1999–2010. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Akkoç Y, Berrak Ö, Arısan ED, Obakan
PÇoker-Gürkan A and Palavan-Ünsal N: Inhiition of PI3K signaling
triggered apoptotic potential of curcumin which is hindered by
Bcl-2 through activation of autophagy in MCF-7 cells. Biomed
Pharmacother. 71:161–171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kelly PN and Strasser A: The role of Bcl-2
and its pro-survival relatives in tumourigenesis and cancer
therapy. Cell Death Differ. 18:1414–1424. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liang XH, Kleeman LK, Jiang HH, Gordon G,
Goldman JE, Berry G, Herman B and Levine B: Protection against
fatal Sindbis virus encephalitis by beclin, a novel
Bcl-2-interacting protein. J Virol. 72:8586–8596. 1998.PubMed/NCBI
|
|
36
|
Pattingre S, Tassa A, Qu X, Garuti R,
Liang XH, Mizushima N, Packer M, Schneider MD and Levine B: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yu J, Chen C, Xu T, Yan M, Xue B, Wang Y,
Liu C, Zhong T, Wang Z, Meng X, et al: Pseudolaric acid B activates
autophagy in MCF 7 human breast cancer cells to prevent cell death.
Oncol Lett. 11:1731–1737. 2016.PubMed/NCBI
|