Introduction
Autophagy, an evolutionarily conserved cellular
degradation pathway, has been shown to be adaptable to starvation
and some other stress conditions (1). Autophagy plays a critical role in
biological processes, including cell metabolism, survival, death
and degradation, and recycling of cellular components.
Additionally, autophagy is involved in the pathogenesis of
essential diseases including neurodegenerative disease, metabolic
disorders, and noteworthy cancer (2–5). The
role of autophagy in cancer is one of a double-edged sword: While
it renders tumor cells the ability to surmount metabolic stress,
including hypoxia and insufficient nutrients, it can also inhibit
cancer cells proliferation through oncogenic proteins degradation
(3). In addition, autophagy in
tumor cells is elicited during tumor progression to accommodate the
metabolic stress (6). However, if
the cancer cells cannot sustain such metabolic stress, autophagic
cell death might occur, suggesting a new target for cancer therapy
(7,8); Many drugs, including mammalian target
of rapamycin (mTOR) inhibitors, proteasome, and histone
deacetylases, can induce autophagy, and are thus considered
efficacious tools (3,4).
Histone deacetylase inhibitors (HDACIs) are a class
of promising target molecules for cancer treatment, which act
through the regulation of the acetylation states of histone
proteins and other non-histone protein targets. HDACIs induces cell
cycle arrest, differentiation, apoptosis, and autophagy (9,10).
Generally, induction of apoptosis is essential for the antitumor
activity of HADCIs (11). There is
numerous evidence indicating that HDACIs can cause morphological
alteration and cell cycle arrest in oncogene-transformed or tumor
cells (12). Moreover, recent
studies have exploited the antitumor effect of HDACIs including
suberoylanilide hydroxamic acid (SAHA) and trichostatin A (TSA)
through inducing autophagy (13–15).
The forkhead box proteins (FOXOs) including FOXO1,
FOXO3, FOXO4 and FOXO6 are essential for genes regulation (16). Among these proteins, the functions
of FOXO1 like cell cycle arrest, apoptosis, defense against
oxidative stress and DNA repair are the most clearly elucidated
(16,17). Recently, it has been reported that
FOXO1 acetylation is involved in HDACIs-mediated autophagy
(10). Nonetheless, the
involvement of FOXO1 in HDACIs-caused oncogene-transformed
mammalian cells death remains unclear and requires further
investigation.
In the present study, we investigate the antitumor
effect of TSA in H-ras-transformed human breast epithelial cells
(MCF10A-ras cells) through a FOXO1-dependent pathway. In addition,
we found that TSA could induce autophagy in MCF10A-ras cells
through blocking mTOR pathway; such autophagy served as a
pro-survival mechanism in TSA-mediated cell death. Finally, we
found that combination of TSA and autophagy inhibitor chloroquine
(CQ) exerted a synergistic antitumor effect.
Materials and methods
Cell cultures
MCF10A and MCF10A-ras cells were provided by Prof.
Shen Hanming (National University of Singapore). All cells were
maintained in DMEM (D1152; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) supplemented with 5% horse serum, 0.5 µg/ml
hydrocortisone, 10 µg/ml insulin, 20 ng/ml epidermal growth factor
(EGF), 0.1 µg/ml cholera enterotoxin, 100 U/ml
penicillin-streptomycin, 2.5 mM L-glutamine and 0.5 µg/ml
fungizone, in a humidified atmosphere containing 5%
CO2/95% air at 37°C. The culture medium was replaced
every 2 days.
Reagents and antibodies
The chemicals and reagents used in our experiments
were purchased as follows: TSA (T8552; Sigma-Aldrich; Merck KGaA);
CQ (C6628; Sigma-Aldrich; Merck KGaA); FOXO1 (2880; Cell Signaling
Technology, Inc., Danvers, MA, USA); microtubule-associated
protein1 light chain 3/LC3 (L7543; Sigma-Aldrich; Merck KGaA);
poly-ADP-ribose polymerase-1 (PARP1; 9542; Cell Signaling
Technology, Inc.); CDKN1A/P21 (2947; Cell Signaling Technology,
Inc.); P62/SQSTM1 (P0067; Sigma-Aldrich; Merck KGaA); phospho-S6
(S235/236; 2211; Cell Signaling Technology, Inc.); phosphor-AKT
(ser473; 4060; Cell Signaling Technology, Inc.); HSP70 (4872; Cell
Signaling Technology, Inc.); PARP1 (9542; Cell Signaling
Technology, Inc.); caspase-3 (9662; Cell Signaling Technology,
Inc.); Cathepsin D (2284; Cell Signaling Technology, Inc.); and
β-actin (A5441; Sigma-Aldrich; Merck KGaA).
Reverse transcription-quantitative
PCR
RNA was extracted using RNeasy kit (217004; Qiagen
GmbH, Hilden, Germany). A reverse transcription reaction was
performed using 1 µg of total RNA with High Capacity cDNA Reverse
Transcription kit (4368814; Applied Biosystems; Thermo Fisher
Scientific, Inc.), following the manufacturer's instructions. The
mRNA expression levels were determined by qPCR using
SsoFast™ EvaGreen Supermix (172–5201; Bio-Rad
Laboratories, Inc., Hercules, CA, USA) and CFX96 Touch™
Real-Time PCR Detection System (Bio-Rad Laboratories, Inc.).
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an
internal control of RNA integrity. qPCR was performed in
triplicate.
Small interfering RNA (siRNA) and
transient transfection
DharmaFECT 4 Transfection Reagent (T-2001-02; GE
Healthcare Dharmacon, Inc., Lafayette, CO, USA) was used to
transfected the scramble RNAi oligonucleotides and siRNAs targeting
FOXO1 (6242; Cell Signaling Technology, Inc.) into MCF10A-ras cells
according to manufacturer's instructions. After transfection, cell
lysates were detected by western blotting.
Western blotting
The treated cells were lysed in Laemmli SDS buffer
(62.5 mM Tris at pH 6.8) 25% glycerol, 2% SDS, phosphatase
inhibitor (78428; Pierce; Thermo Fisher Scientific, Inc.) and
proteinase inhibitor cocktail (11836153001; Roche Applied Science,
Madison, WI, USA). The cell lysates were boiled and then prepared
for western blotting after lysis. Equal amount of proteins was
resolved by SDS-PAGE and then transferred onto PVDF membrane. The
membranes were probed with selected primary and secondary
antibodies after blocking with 5% nonfat milk, and then were
developed using enhanced chemiluminescence method. Finally, the
membranes were visualized using the Kodak Image Station 4000R
(Kodak, Rochester, NY, USA).
Detection of viable and dead
cells
Morphological changes under phase-contrast
microscopy and propidium iodide (PI) live cell uptake assay,
coupled with flow cytometry were used to quantitatively and
qualitatively examine cell death. For PI staining, the medium in
each plastic well was collected and cells were harvested with
trypsin after treatments. Then, cells were resuspended in 1×
phosphate-buffered saline containing PI at a final concentration of
5 µg/ml and incubated at 37°C for 10 min. 10,000 cells from each
sample were analyzed through FACSCalibur flow cytometry (BD
Biosciences, San Jose, CA, USA) using CellQuest software.
Statistical analysis
All western blotting and image data presented in
this study are representatives from at least 3 independent
experiments. All data are illustrated as the mean ± standard
deviation from triplicate independent experiments performed in a
parallel manner and analyzed through the ANOVA followed by
Dunnett's post hoc test. The statistical significance is indicated
as P<0.05, P<0.01 and P<0.001.
Results
Morphological changes and cell
viability by TSA
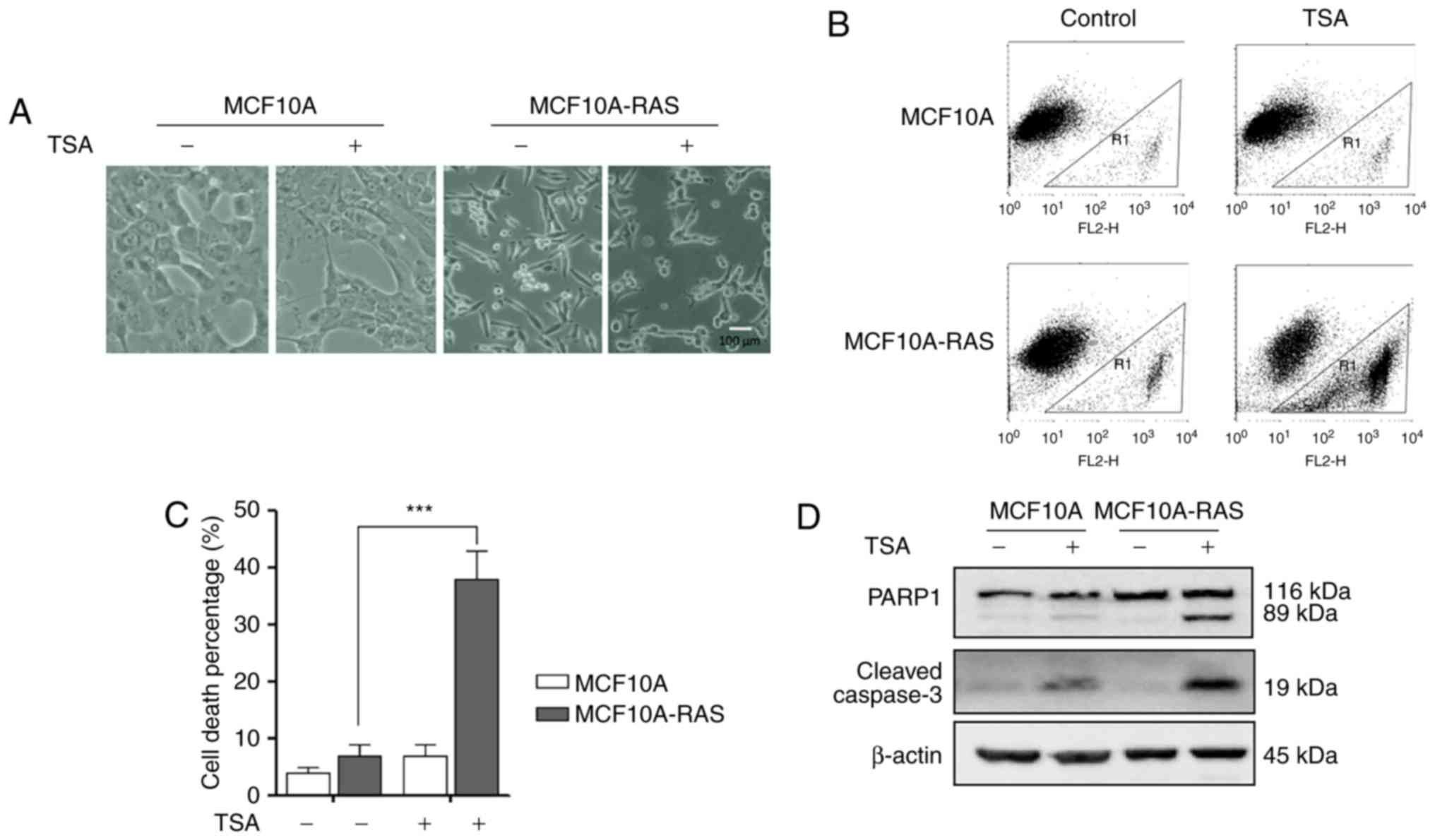
As shown in Fig.
1A, MCF10A-ras cells changed their shape from rounded to
spindle, indicating that an oncogenic transformation caused
significant morphological alterations in these cells. After
treatment with 0.5 µM TSA for 24 h, the morphology of
MCF10A-ras cells dramatically shifted to an elongated shape
with filamentous protrusions; while, no discernable changes were
found in MCF10A cells (Fig. 1A).
Furthermore, after treatment with 0.5 µM TSA for 24 h,
significantly higher cell death percentage was observed in
MCF10A-ras cells (Fig. 1B and
C).
TSA treatment causes MCF10A-ras cell
apoptosis
The effects of TSA on the cleavage of PARP1 and
Caspase-3 were examined to determine the underlying molecular
mechanism of the TSA-induced cell death. As shown in Fig. 1D, TSA significantly elevated the
levels of cleaved Caspase-3 and PARP1 in MCF10A-ras cells compared
to MCF10A cells. These results demonstrated that TSA could induce
MCF10A-ras cell apoptosis.
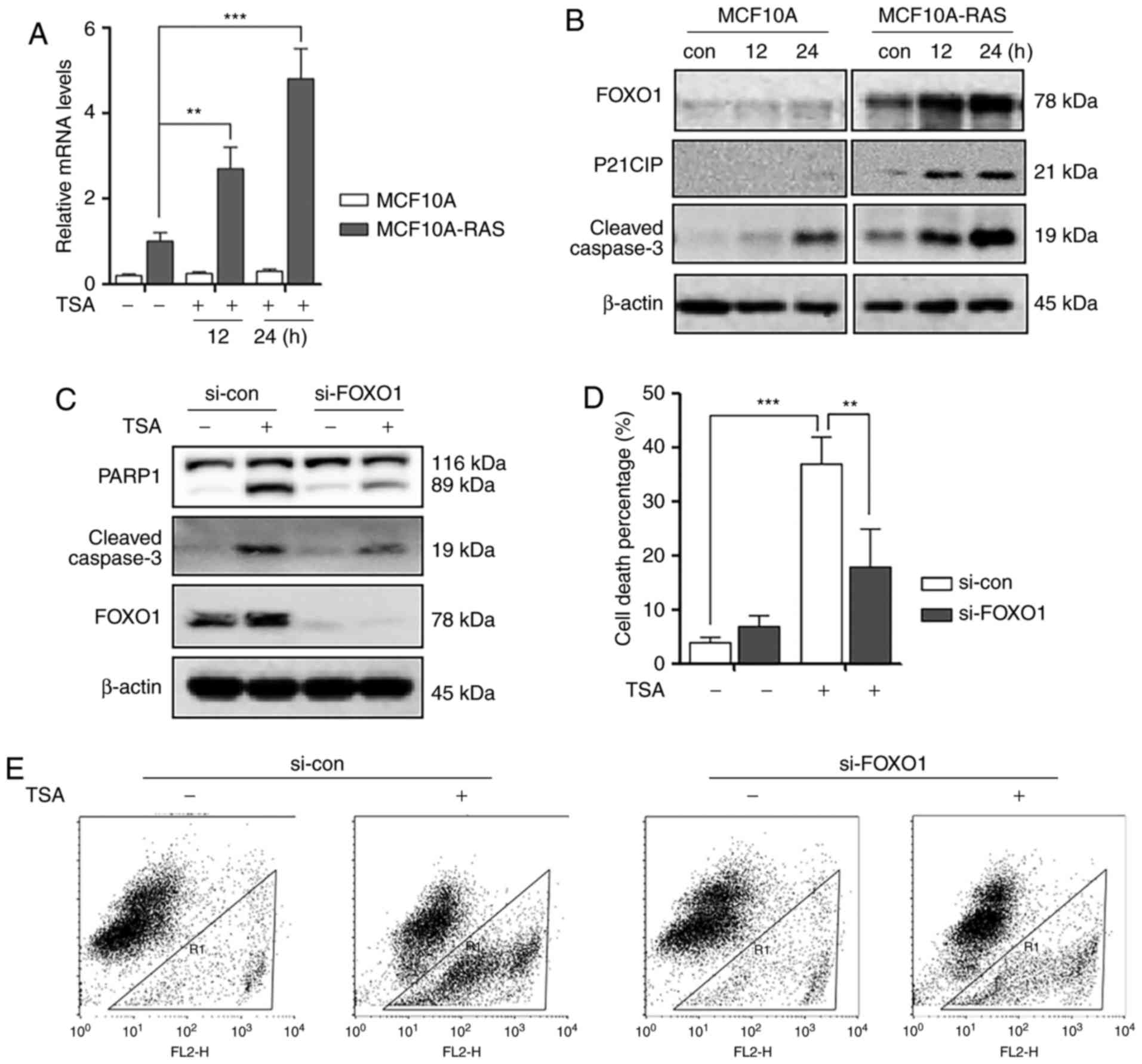
TSA treatment increases the activity
of FOXO1
It was reported that FOXO1 is essential in
regulating apoptosis and autophagy (16). Therefore, the possibility of
involvement of FOXO1 in TSA-induced apoptosis was investigated.
Firstly, we investigated the transcriptional level changes of FOXO1
in MCF10A and MCF10A-ras cells. As shown in Fig. 2A, we performed qPCR to measure the
mRNA levels of FOXO1, and found that TSA treatment induced
significant increase of FOXO1 mRNA level in MCF10A-ras cells
compared to MCF10A cells. Secondly, TSA induced an increase in
FOXO1, P21 and cleaved Caspase-3 expression n MCF10A-ras cell lines
compared to MCF10A cells (Fig.
2B).
Furthermore, to confirm the role of FOXO1 in HDACIs
TSA-mediated MCF10A-ras cell death, FOXO1 was silenced by siRNA. As
expected, knockdown of FOXO1 markedly reduced the expression level
of cleaved Caspase-3 and reduced cell death percentage in
MCF10A-ras cells (Fig. 2C-E).
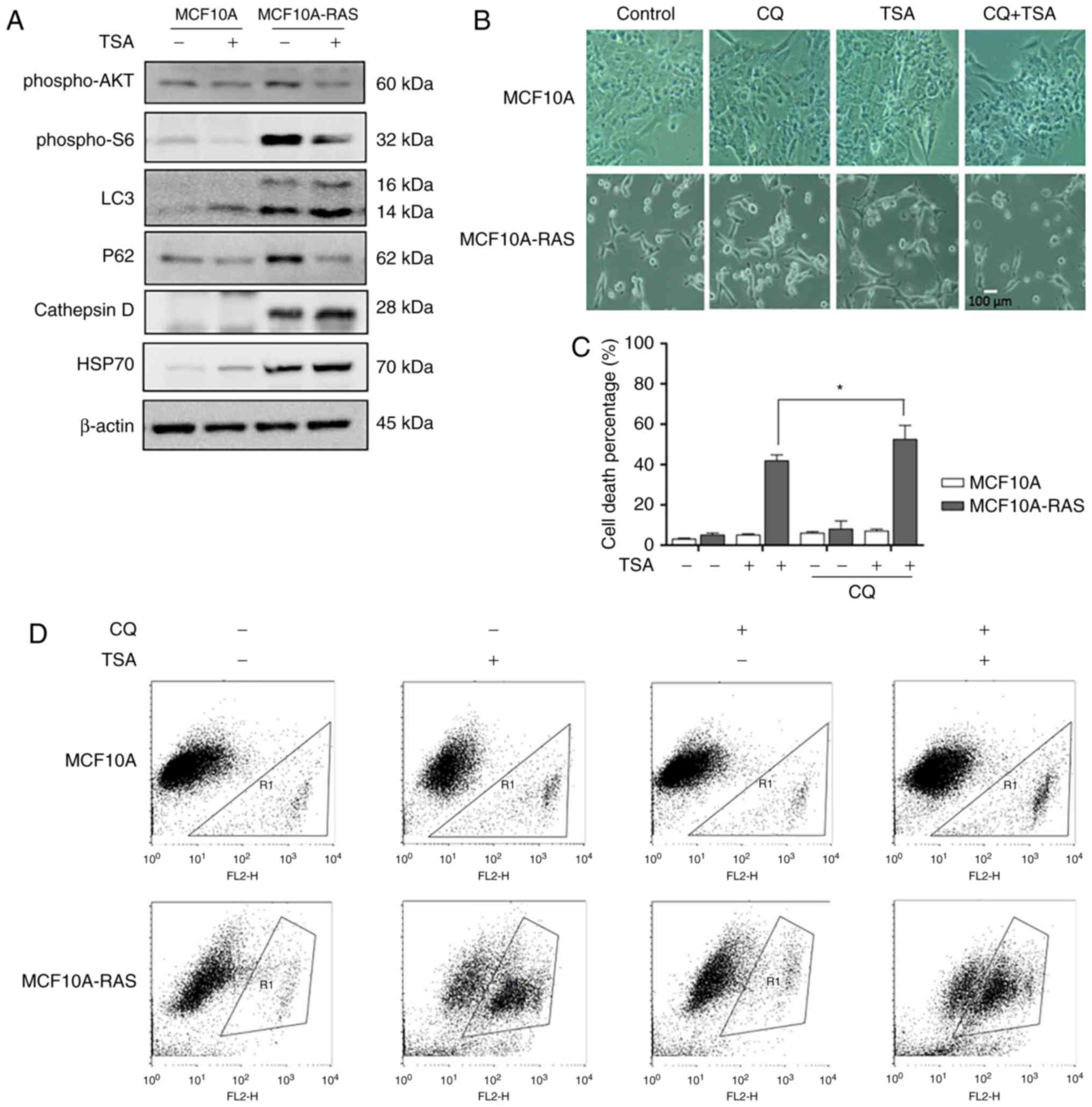
TSA treatment induces autophagy via
blocking mTOR pathway
TSA can suppress cell proliferation and induce cell
death through effective inhibition of HDAC enzyme activity at
nanomolar concentrations (18). To
investigate TSA's effect on autophagy, we treated MCF10A-ras cells
with TSA. Briefly, our results indicated that TSA enhanced
autophagy through increasing LC3, Cathepsin D and HSP70, and
decreasing P62 in both MCF10A and MCF10A-ras cells. It has been
reported that HDACIs can induce autophagy via downregulation of
AKT-mTOR signaling (13).
Therefore, we determined the role of mTOR pathway in autophagy
induction by TSA. As shown in Fig.
3A, TSA reduced phospho-AKT and phosphor-S6 expression levels
in both MCF10A and MCF10A-ras cells, suggesting the suppression of
mTOR activity.
Suppression of autophagy sensitizes
TSA-caused cell death
Previous results showed that TSA could induce
autophagy in MCF10A-ras cells, hence we investigated whether the
inhibition of autophagy would sensitize TSA-caused cell death.
Briefly, we treated the tumor cells with a pharmacological
inhibitor CQ, an inhibitor of the lysosomal pH gradient. Fig. 3B revealed the morphological changes
with the treatment of combination of CQ and TSA. TSA treatment
triggered more MCF10A-ras cell deaths in the presence of CQ
(Fig. 3C, D), supporting the
notion that TSA-induced autophagy served as a cell survival
mechanism. This result was consistent with our previous studies
(10).
Discussion
HDACIs are new antitumor agents, which exert a great
influence on cancer cells promoting cell death, apoptosis, cell
cycle arrest and autophagy (9–11).
In our study, TSA was found to induce apoptosis in MCF10A-ras cells
through activation of FOXO1. In addition, siRNA knockdown of FOXO1
in MCF10A-ras cells markedly reduced TSA-induced apoptosis and
dramatically attenuated the antitumor effect of TSA.
Deacetylase and acetyltransferase, mediating
post-transcriptional regulation, are involved in the regulation of
apoptosis (19). It has been shown
that the HACIs can facilitate apoptosis though upregulation of
pro-apoptotic gens and downregulation of anti-apoptotic genes
(9). TSA, one of HDACIs, has been
shown to significantly induce the apoptosis of MCF10A-ras cells by
promoting expression of Bax (20).
Our data were generally concordant with earlier reports suggesting
that TSA was capable of inducing apoptosis in MCF10A-ras cells
(20). Furthermore, our study
clearly showed that the FOXO1 played a critical role in TSA-induced
apoptosis. A previous study has proved that HDACIs could induce
apoptosis through the FOXO1-Bim pathway and that FOXO1 knockdown
could protect from TSA-caused cell death (10,21).
In our study, TSA, one of HDACIs, concurrently increased the
expression levels of FOXO1 in MCF10A-ras cells with the expression
of cleavage of Caspase-3. Conversely, in the context of the
knockdown of FOXO1, the expression of cleaved Caspase3 was
attenuated indicating that the apoptosis of MCF10A-ras cells
induced by TSA was inhibited. As a result, the cell death was
reduced in the MCF10A-ras cell. Overall, to the best of our
knowledge this was the first study showing that TSA-induced
apoptosis observed in MCF10A-ras cells was dependent on FOXO1
activity.
Previous studies have shown that HDACIs facilitated
autophagy in cancer cells (14,22).
Nonetheless, the role of autophagy in HDACIs-mediated cancer cell
death is still controversial. Some studies have shown that
autophagy served as a pro-death role through autophagy inhibition
and reduction of HDACIs cytotoxicity through Atgs (autophagy
associated gene) knockdown (23,24).
On the contrary, some other studies have revealed that autophagy
served as a pro-survival mechanism (10,25,26).
We found that TSA reduced the expression of phospho-AKT and
phospho-S6 and increased the levels of expression of LC3. These
results clearly showed that TSA treatment led to autophagy in
MCF10A-ras cells, which was consistent with earlier reports on
HDACI-induced autophagy in human cancer cells (14). Moreover, it revealed that
inhibition of autophagy by CQ significantly enhanced TSA-caused
cell death, suggesting that autophagy served as a cell survival
mechanism in TSA-treated MCF10A-ras cells. Therefore, our results
supported the notion that autophagy serves as a pro-survival
mechanism. Based upon our findings, we concluded that with
treatment of HDACIs in cancer cells autophagy could delay the onset
of apoptosis through various mechanism including elimination of
reactive oxygen species (ROS) (27). Our data support the hypothesis that
combinations of HDCAIs and autophagy inhibitors might be a
promising therapeutic strategy for cancer patients.
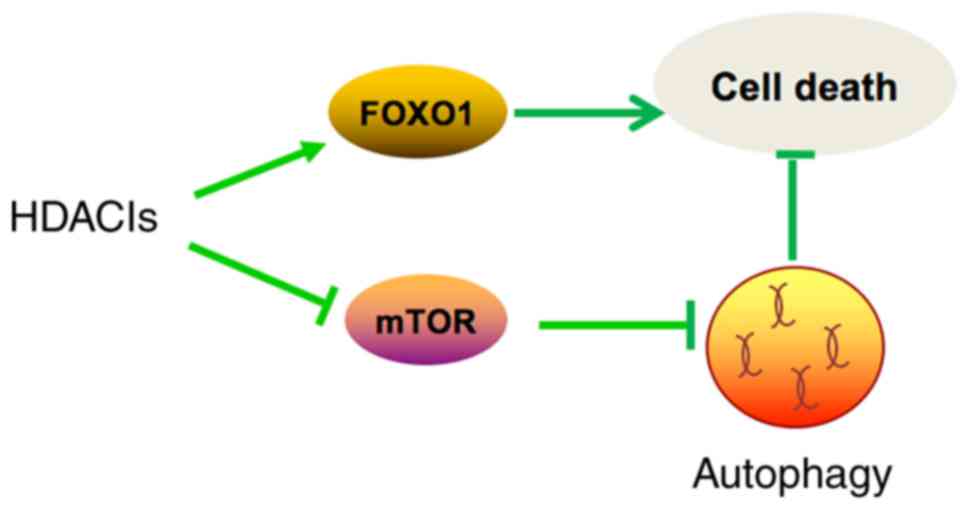
In summary (Fig.
4), TSA caused morphological changes and induced apoptosis in
MCF10A-ras cells through activation of FOXO1. In addition, TSA also
induced autophagy through inhibition of mTOR pathway. Moreover,
inhibition of autophagy synergistically enhances the antitumor
effect of TSA. Our results shed some light on developing more
effective cancer therapeutic strategies by combining HDACIs and
autophagy inhibitors.
Acknowledgements
This study was financially supported by the Science
Technology Department of Zhejiang Province, China (grant no.
2015C33173), the Traditional Chinese Medicine Fund of Zhejiang
Province, China (grant no. 2011ZA010) and the Zhejiang Provincial
Natural Science Foundation of China (grant no. LQ18H280006).
Glossary
Abbreviations
Abbreviations:
|
CQ
|
chloroquine
|
|
FOXO1
|
forkhead box O1
|
|
HDACIs
|
histone deacetylase inhibitors
|
|
mTOR
|
mammailian target of rapamycin
|
|
PARP1
|
poly-ADP-ribose polymerase-1
|
|
TSA
|
trichostatin A
|
References
|
1
|
Jiang P and Mizushima N: Autophagy and
human diseases. Cell Res. 24:69–79. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:651–662. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Janku F, McConkey DJ, Hong DS and Kurzrock
R: Autophagy as a target for anticancer therapy. Nat Rev Clin
Oncol. 8:528–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Santana-Codina N, Mancias JD and Kimmelman
AC: The Role of Autophagy in Cancer. Ann Rev Cancer Biol. 1:19–39.
2017. View Article : Google Scholar
|
|
5
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Degenhardt K, Mathew R, Beaudoin B, Bray
K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lum JJ, Bauer DE, Kong M, Harris MH, Li C,
Lindsten T and Thompson CB: Growth factor regulation of autophagy
and cell survival in the absence of apoptosis. Cell. 120:237–248.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jin S and White E: Role of autophagy in
cancer: Management of metabolic stress. Autophagy. 3:28–31. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Emanuele S, Lauricella M and Tesoriere G:
Histone deacetylase inhibitors: Apoptotic effects and clinical
implications (Review). Int J Oncol. 33:637–646. 2008.PubMed/NCBI
|
|
10
|
Zhang J, Ng S, Wang J, Zhou J, Tan SH,
Yang N, Lin Q, Xia D and Shen HM: Histone deacetylase inhibitors
induce autophagy through FOXO1-dependent pathways. Autophagy.
11:629–642. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim HJ and Bae SC: Histone deacetylase
inhibitors: Molecular mechanisms of action and clinical trials as
anti-cancer drugs. Am J Transl Res. 3:166–179. 2011.PubMed/NCBI
|
|
12
|
Hoshikawa Y, Kwon HJ, Yoshida M,
Horinouchi S and Beppu T: Trichostatin A induces morphological
changes and gelsolin expression by inhibiting histone deacetylase
in human carcinoma cell lines. Exp Cell Res. 214:189–197. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu YL, Yang PM, Shun CT, Wu MS, Weng JR
and Chen CC: Autophagy potentiates the anti-cancer effects of the
histone deacetylase inhibitors in hepatocellular carcinoma.
Autophagy. 6:1057–1065. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gammoh N, Lam D, Puente C, Ganley I, Marks
PA and Jiang X: Role of autophagy in histone deacetylase
inhibitor-induced apoptotic and nonapoptotic cell death. Proc Natl
Acad Sci USA. 109:pp. 6561–6565. 2012; View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chiao MT, Cheng WY, Yang YC, Shen CC and
Ko JL: Suberoylanilide hydroxamic acid (SAHA) causes tumor growth
slowdown and triggers autophagy in glioblastoma stem cells.
Autophagy. 9:1509–1526. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Eijkelenboom A and Burgering BM: FOXOs:
Signalling integrators for homeostasis maintenance. Nat Rev Mol
Cell Biol. 14:83–97. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Burgering BM: A brief introduction to
FOXOlogy. Oncogene. 27:2258–2262. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Michaelis M, Suhan T, Michaelis UR, Beek
K, Rothweiler F, Tausch L, Werz O, Eikel D, Zörnig M, Nau H, et al:
Valproic acid induces extracellular signal-regulated kinase 1/2
activation and inhibits apoptosis in endothelial cells. Cell Death
Differ. 13:446–453. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jazirehi AR: Regulation of
apoptosis-associated genes by histone deacetylase inhibitors:
Implications in cancer therapy. Anticancer Drugs. 21:805–813. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Park H, Lee YJ, Kim TH, Lee J, Yoon S,
Choi WS, Myung CS and Kim HS: Effects of trichostatin A, a histone
deacetylase inhibitor, on the regulation of apoptosis in
H-ras-transformed breast epithelial cells. Int J Mol Med.
22:605–611. 2008.PubMed/NCBI
|
|
21
|
Yang Y, Zhao Y, Liao W, Yang J, Wu L,
Zheng Z, Yu Y, Zhou W, Li L, Feng J, et al: Acetylation of FoxO1
activates Bim expression to induce apoptosis in response to histone
deacetylase inhibitor depsipeptide treatment. Neoplasia.
11:313–324. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Del Bufalo D, Desideri M, De Luca T, Di
Martile M, Gabellini C, Monica V, Busso S, Eramo A, De Maria R,
Milella M and Trisciuoglio D: Histone deacetylase inhibition
synergistically enhances pemetrexed cytotoxicity through induction
of apoptosis and autophagy in non-small cell lung cancer. Mol
Cancer. 13:2302014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang PM and Chen CC: Life or death?
Autophagy in anticancer therapies with statins and histone
deacetylase inhibitors. Autophagy. 7:107–108. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hrzenjak A, Kremser ML, Strohmeier B,
Moinfar F, Zatloukal K and Denk H: SAHA induces
caspase-independent, autophagic cell death of endometrial stromal
sarcoma cells by influencing the mTOR pathway. J Pathol.
216:495–504. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Carew JS, Medina EC, Esquivel JA II,
Mahalingam D, Swords R, Kelly K, Zhang H, Huang P, Mita AC, Mita
MM, et al: Autophagy inhibition enhances vorinostat-induced
apoptosis via ubiquitinated protein accumulation. J Cell Mol Med.
14:2448–2459. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lopez G, Torres K, Liu J, Hernandez B,
Young E, Belousov R, Bolshakov S, Lazar AJ, Slopis JM, McCutcheon
IE, et al: Autophagic survival in resistance to histone deacetylase
inhibitors: Novel strategies to treat malignant peripheral nerve
sheath tumors. Cancer Res. 71:185–196. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ungerstedt JS, Sowa Y, Xu WS, Shao Y,
Dokmanovic M, Perez G, Ngo L, Holmgren A, Jiang X and Marks PA:
Role of thioredoxin in the response of normal and transformed cells
to histone deacetylase inhibitors. Proc Natl Acad Sci USA. 102:pp.
673–678. 2005; View Article : Google Scholar : PubMed/NCBI
|