Introduction
Proliferative vitreoretinopathy (PVR) is a clinical
syndrome that occurs following rhegmatogenous retinal detachment
and is caused by breaks in the retina, including retinal tears,
retinal holes or surgical repair. The basic pathophysiological
process is damage to the blood-retinal barrier. A variety of cells
participate in PVR, including retinal pigment epithelium (RPE)
cells, glial cells, fibroblasts and inflammatory cells (1). RPE cells are the primary component of
the cell proliferation membrane and are the dominant cell in the
pathogenesis of PVR (2). The main
pathophysiological mechanism of PVR is considered to be the
conversion of RPE cells into mesenchymal cells via
epithelial-mesenchymal transition (EMT) (3). A number of studies have observed the
prevention of PVR by inhibiting EMT (4,5).
However, the molecular mechanism underlying transforming growth
factor (TGF)-β1-induced EMT is poorly understood.
EMT may be induced and regulated by various factors,
including TGF-β, thrombin, platelet-derived growth factor and bone
morphogenetic proteins (6).
TGF-β-induced EMT has been investigated in the majority of
epithelial cell types (7).
Expression of the TGF family is upregulated in the vitreous or
epiretinal proliferative membranes in patients with PVR (8,9). In
addition, TGF-β expression levels are positively associated with
the severity of PVR in vitreous or PVR experimental models
(10). Blocking the TGF-β
signaling pathway may inhibit the conversion of RPE cells via EMT
in vivo and in vitro (11,12).
Therefore, TGF-β serves an important role in the pathogenesis of
PVR. TGF-β1 resulted in EMT of RPEs and the development of PVR,
which has become a classical model of EMT (13,14).
Studies have reported that chronic inflammation has
a role in the pathogenesis of PVR (15) and is an important pathological
factor for promoting the development of proliferative retinopathy.
The complement and blood coagulation cascades are the most
important Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways
in the pathological process of PVR (16). The cascade consists of the
kinin-kallikrein system (KKS), the blood coagulation system and the
complement system. As the intermediate process connecting the
complement pathway and thrombin, the KKS serves a key role in
regulation. The KKS is primarily composed of kallikrein, kininogen,
kinin, bradykinin 1 receptor (B1R), bradykinin 2 receptor (B2R) and
kininase. Kinin includes bradykinin (BK) and kallidin (KD), and KD
may be converted into BK under the action of enzymes. Therefore,
BK, as the final effector molecule, is the primary kinin under
physiological conditions. The expression of BK is significantly
increased in experimental animal models with severe PVR (17). Therefore, it was hypothesized in
the present study that BK may regulate KKS, the blood coagulation
system and the complement system, and ultimately act via KEGG
pathways to induce PVR. To elucidate the underlying mechanisms
behind PVR and enhance its treatment in a clinical setting, the
role of BK in its pathophysiology requires further
investigation.
Materials and methods
Cell culture
Human retinal pigment epithelial cells (ARPE-19)
were obtained from Cell Biosciences Pty, Ltd. (Heidelberg,
Australia). ARPE-19 cells were cultured in Dulbecco's modified
Eagle's medium (DMEM)/H (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS;
Gibco; Thermo Fisher Scientific, Inc.). Cells were incubated at
37°C in a humidified incubator containing 5% CO2. The
cell culture medium was changed every 3–4 days. At 50–70%
confluence, the cells were growth-arrested in serum-free medium for
12 h at 37°C prior to the addition of 10 nM BK for 0.5 h.
Subsequently, 10 ng/ml TGF-β1 (PeproTech, Inc., Rocky Hill, NJ,
USA) was added for 48 h at 37°C. The TGF-β1 concentration and
incubation time were determined from dose response experiments (0
to 12.5 ng/ml) and time course experiments (24 and 48 h) to induce
EMT and provide the optimal balance between cytotoxicity and cell
viability (Fig. 1A). B2R activity
was blocked by pre-incubating the cells for 1 h at 37°C with 100 uM
of the B2R antagonist, HOE-140 (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany).
Cell Counting Kit-8 (CCK-8) assay. ARPE-19 cells
were seeded into 96-well plates at 5×103 cells/well and
cultured in DMEM. At 50% confluence, the medium was replaced with
FBS-free medium, and culture was continued for 12 h. TGF-β1 (0,
0.1, 0.5, 2.5, 10 and 12.5 ng/ml) was added to the medium as
aforementioned. Then ARPE-19 cells were incubated for 24 and 48 h
respectively at 37°C in a humidified incubator containing 5%
CO2. CCK-8 reagents (Nanjing AnboRuila Biotechnology
Co., Ltd., Nanjing, China) were added to each well and incubated at
37°C for 4 h. The absorbance was measured at a wavelength of 450 nm
using an ELISA plate reader (BioTek China, Beijing, China).
Cell morphology
ARPE-19 cells were seeded into 6-well plates at
5×105 cells/well containing DMEM with 10% FBS. After
reaching 70% confluence, the medium was replaced with FBS-free
medium, and culture was continued for 12 h at 37°C. A total of 100
nM BK, 10 ng/ml TGF-β1, 100 nM BK plus 10 ng/ml TGF-β1, 10 ng/ml
TGF-β1 plus 10 uM HOE140, 100 nM BK plus 10 ng/ml TGF-β1 plus 10 uM
HOE140, or DMEM/H vehicle, was added to the medium. The cells were
growth-arrested in serum-free medium for 12 h at 37°C prior to the
addition of BK for 0.5 h. Subsequently, TGF-β1 was added for 48 h
at 37°C. B2R activity was blocked by pre-incubating the cells for 1
h at 37°C with of the B2R antagonist, HOE-140. The morphological
alterations were observed with an inverted phase-contrast
microscope (Shanxi AntaiGroup Co., Ltd., Shanxi, China,
magnification, ×100). A total of five fields were analyzed and
experiments were repeated three times in duplicate.
Western blot analysis
ARPE-19 cells were homogenized using lysis buffer
containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1%
sodium deoxycholate, 0.1% SDS, 5 mM sodium orthovanadate, 5 mM
EDTA, 1 mM phenylmethanesulfonyl fluoride and 1 mM NaF. Total
protein was quantified by bicinchoninic acid assay (Beyotime
Institute of Biotechnology, Haimen, China). A total of 25 µg
protein was analyzed for EMT-associated protein expression,
including the epithelial marker E-cadherin, the mesenchymal markers
α-smooth muscle actin (SMA) and vimentin, and phosphorylated (p)
mothers against decapentaplegic homolog (Smad)3 and Smad7. Protein
samples were subjected to SDS-PAGE (6–12% polyacrylamide gels) and
transferred onto a polyvinylidene diflouride membrane (EMD
Millipore, Billerica, MA, USA). Membranes were blocked with 5%
non-fat milk for 1 h at 37°C and probed with antibodies overnight
at 4°C. The antibodies and dilution ratios were as follows:
Anti-E-cadherin (1:500; cat. no. GTX100443, GeneTex International
Corporation, Hsinchu, Taiwan), mouse anti-α-SMA (1:500; cat. no.
BS70000, Bioworld Technology, Inc., St. Louis Park, MN, USA),
vimentin (cat. no. BS1491, 1:500; Bioworld Technology, Inc.),
anti-pSmad3 (1:500; cat. no. BS4173, Bioworld Technology, Inc.),
anti-Smad7 (1:500; cat. no. BS60366, Bioworld Technology, Inc.) and
anti-GAPDH (1:500; cat. no. G9545, Sigma-Aldrich; Merck KGaA).
Following washing with 0.1% TBS-Tween 20 containing 5% skim milk at
room temperature, membranes were incubated with horseradish
peroxidase-conjugated sheep anti-mouse secondary antibodies
(1:2,000; cat. no. A5906, Sigma-Aldrich; Merck KGaA) or
peroxidase-conjugated goat anti-rabbit secondary antibodies
(1:2,000; cat. no. A6154, Sigma-Aldrich; Merck KGaA) for 1 h at
room temperature. Image Quant LAS 4000 (GE Healthcare, Chicago, IL,
USA) with ImageJ software (version 1.38e, National Institutes of
Health, Bethesda, MD, USA) were used to quantify band
intensities.
Cell immunofluorescence analysis
ARPE-19 cells were seeded into 24-well plates at
4×104 cells/well containing DMEM with 10% FBS. After
reaching 70% confluence, the medium was replaced with FBS-free
medium, and culture was continued for 12 h at 37°C. Following
treatment with 100 nM BK, 10 ng/ml TGF-β1, 100 nM BK plus 10 ng/ml
TGF-β1, 10 ng/ml TGF-β1 plus 10 uM HOE140, or 100 nM BK plus 10
ng/ml TGF-β1 plus 10 uM HOE140, or DMEM/H vehicle, was added to the
medium. The cells were growth-arrested in serum-free medium for 12
h at 37°C prior to the addition of BK for 0.5 h. Subsequently,
TGF-β1 was added for 48 h at 37°C. B2R activity was blocked by
pre-incubating the cells for 1 h at 37°C with of the B2R
antagonist, HOE-140. Then cells of different groups were washed
three times with PBS and fixed with 4% paraformaldehyde for 20 min
at room temperature. Following three washings with PBS, cells were
blocked with 10% bovine serum albumin (1:2,000; cat. no. B2064,
Sigma-Aldrich; Merck KGaA) for 30 min at 37°C and incubated
overnight at 4°C with the primary antibodies described above. The
dilutions were as follows: Rabbit anti-E-cadherin (1:100), mouse
anti-a-SMA (1:100), vimentin (1:100), anti-p Smad3 (1:100) and
anti-Smad7 (1:100). Cells were stained with FITC-labeled goat
anti-rabbit immunoglobulin G (1:500; cat. no. F0382, Sigma-Aldrich;
Merck KGaA) for 2 h at 37°C andDAPI (1:5,000; cat. no. D9542,
Sigma-Aldrich; Merck KGaA) for 5 min at 37°C to visualize the
nuclei. Images were captured using a confocal microscope (Zeiss
GmbH, Jena, Germany, magnification, ×200).
Wound healing test
ARPE-19 cells were seeded in six-well plates at a
density of 5×105 cells/well and cultured with DMEM/H
containing 10% FBS. At 70–80% confluence, the cells were
serum-starved for 12 h at 37°C. A total of 100 nM BK, 10 ng/ml
TGF-β1, 100 nM BK plus 10 ng/ml TGF-β1, 10 ng/ml TGF-β1 plus 10 uM
HOE140, 100 nM BK plus 10 ng/ml TGF-β1 plus 10 uM HOE140, or DMEM/H
vehicle, was added to the medium. The cells were growth-arrested in
serum-free medium for 12 h at 37°C prior to the addition of BK for
0.5 h. Subsequently, TGF-β1 was added for 48 h at 37°C. B2R
activity was blocked by pre-incubating the cells for 1 h at 37°C
with of the B2R antagonist, HOE-140. A sterile 200 µl pipette tip
was used to create a wound in the monolayer by scraping. The cells
were washed with PBS and cultured in FBS-free medium for 24 h at
37°C in a humidified incubator containing 5% CO2.
Micrographs were captured with an inverted phase contrast
microscope (magnification, ×50). Experiments were repeated three
times in duplicate.
Transwell migration assay
ARPE-19 cells were seeded in six-well plates at a
density of 5×105 cells/well and cultured with DMEM/H
containing 10% FBS. At 50–70% confluence, the cells were starved
for 12 h at 37°C. Cells were treated as aforementioned. A total of
1×106 cells were added to the top chamber of 24-well
Transwell plates (8 µm pore size; Corning Incorporated, Corning,
NY, USA) in 200 µl medium. The bottom chambers were filled with 500
µl medium with 5% FBS. Following 18 h of incubation at 37°C in the
5% CO2 atmosphere, and the chambers were then washed
with PBS. Cells that did not invade through the membrane were
removed, while the invading cells on the lower surface of the
membrane, were fixed with 4% paraformaldehyde for 15 min at room
temperature and then stained with 0.1% crystal violet for 20 min at
room temperature. The number of migratory cells in each chamber was
quantified by counting five fields under a light microscope
(OLYMPUS CKX41; Olympus Corporation, Tokyo, Japan, magnification,
×50). Another counting method was used, where cells were bleached
with 33% acetic acid for 15 min at room temperature following
staining with 0.1% crystal violet, prior to adding CCK-8 reagent
(Nanjing AnboRuila Biotechnology Co., Ltd.) to the eluent. The
optical density value was measured at a wavelength of 570 nm using
an ELISA plate reader (BioTek China). All experiments were repeated
at least three times.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA of cells was extracted using TRIzol
reagent, according to the manufacturer's protocol (Invitrogen;
Thermo Fisher Scientific, Inc.). The concentration of RNA was
measured with a Nanodrop 2000 spectrophotometer (Thermo Fisher
Scientific, Inc., Wilmington, DE, USA), and cDNA was synthesized
from 1 µg total RNA using a PrimeScript™ RT-PCR kit
(cat. no. RR037A; Takara Biotechnology Co., Ltd., Dalian, China).
Specific primers (Bioworld Technology, Inc.) were used to detect
the expression of EMT-associated genes, using GAPDH as an internal
control. miRNA quantification was performed using the 7500 Fast
Real-time PCR system (Applied Biosystems; Thermo Fisher Scientific,
Inc.) using a SYBR®PrimeScript™ RT-qPCR kit (cat. no.
RR081A; Takara Bio, Inc.). The 20 µl reaction system contained the
following: 10 µl SYBR Premix Ex Taq (2X), 0.4 µl ROX Dye II, 6 µl
dH2O, 0.8 µl PCR forward primer, 0.8 µl PCR reverse primer and 2 µl
cDNA. qPCR thermal cycling was performed as follows: One cycle at
95°C for 30 sec, followed by 40 cycles of 95°C for 5 sec and 60°C
for 34 sec, and one cycle of 95°C for 15 sec, 60°C for 30 sec and
95°C for 15 sec for fluorescence signal acquisition. The relative
quantity of each gene was measured using the 2−ΔΔCq
method (18). All RT-qPCR
experiments were performed in triplicate. Primer sequences are
listed in Table I.
 | Table I.Primers used for reverse
transcription-quantitative polymerase chain reaction
amplification. |
Table I.
Primers used for reverse
transcription-quantitative polymerase chain reaction
amplification.
| Gene | Forward 5′→3′ | Reverse 5′→3′ |
|---|
| pSmad3 |
TGGACGCAGGTTCTCCAAAC |
CCGGCTCGCAGTAGGTAAC |
| Smad7 |
GGACAGCTCAATTCGGACAAC |
GTACACCCACACACCATCCAC |
| GAPDH |
AGAAGGCTGGGGCTCATTTG |
AGGGGCCATCCACAGTCTTC |
Statistical analysis
Independent sample t-test was used for two-group
comparisons. One-way analysis of variance followed by a
Student-Newman-Keuls test, was used for multiple sample analysis.
P<0.05 was considered to indicate a statistically significant
difference. The data were analyzed with SPSS version 19.0 software
(IBM Corp., Armonk, NY, USA). The data were expressed as the mean ±
standard deviation.
Results
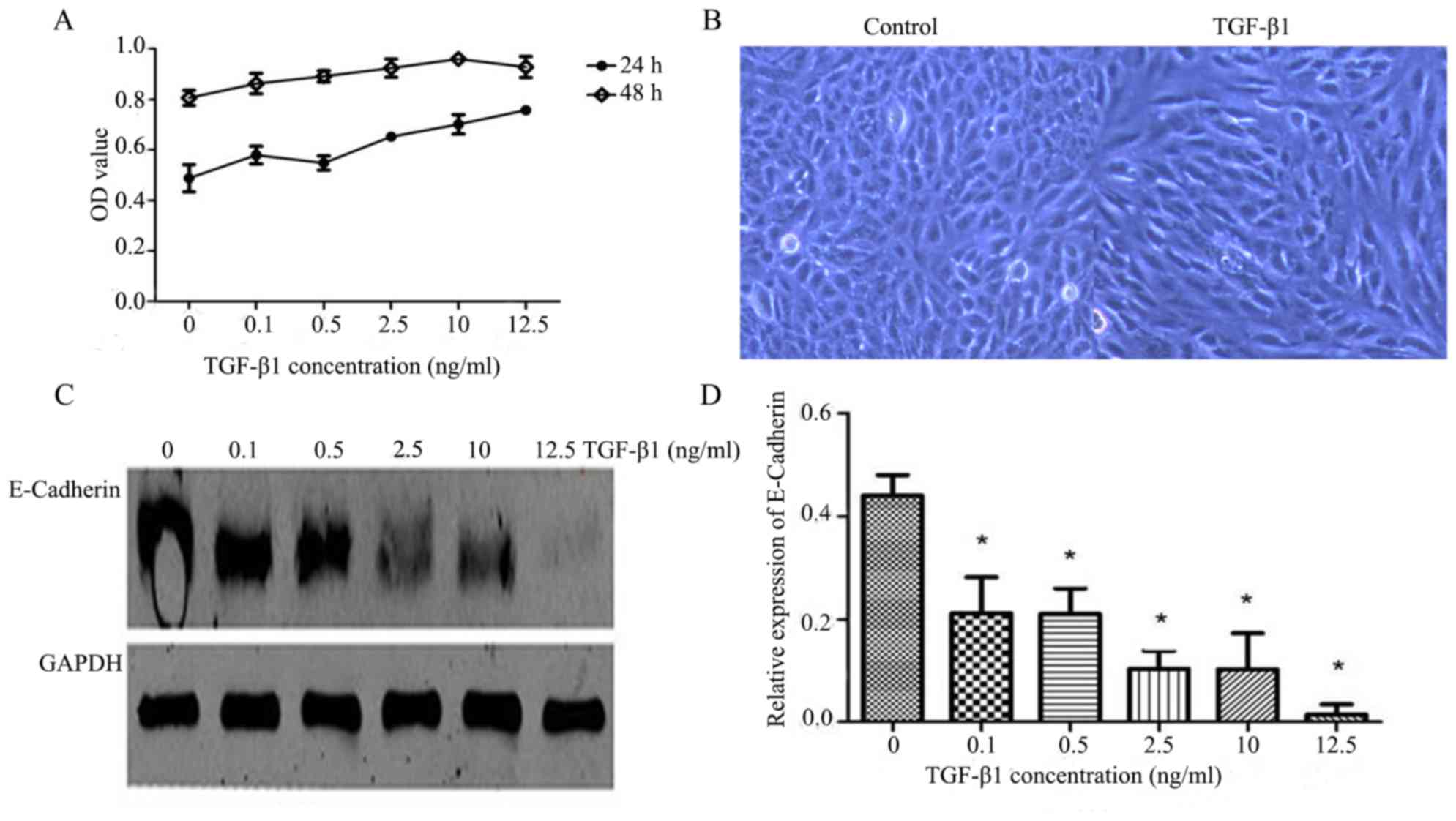
Treatment with 10 ng/ml TGF-β1 induced
EMT in ARPE-19 cells
To assess the optimal concentration and time of
treatment with TGF-β1 that resulted in the success of the
experimental EMT model in ARPE-19 cells, a CCK-8 assay was used to
investigate cell proliferation. TGF-β1-treated groups had elevated
optical density values compared with the control group (Fig. 1A). ARPE-19 cell proliferation was
more obvious following 48 h TGF-β1 treatment compared with 24 h,
and cell proliferation was most marked when the concentration of
TGF-β1 was 10 ng/ml at 48 h (Table
II and Fig. 1A). An inverted
phase-contrast microscope was used to observe the cell morphology.
ARPE-19 cell morphology was altered compared with the control group
at 10 ng/ml TGF-β1 and 48 h. Cell morphology gradually changed from
the typical cobblestone-like to the long spindle-shaped
mesenchymal-like cells (Fig. 1B).
In addition, the expression levels of E-cadherin were detected by
western blot analysis. When ARPE-19 cells were treated with
different concentrations of TGF-β1 for 48 h, E-cadherin expression
was reduced compared with the control group (P<0.05; Fig. 1C and D). E-cadherin expression in
ARPE-19 cells was negatively associated with TGF-β1 concentration
(Fig. 1C and D). Therefore, the
EMT experimental model was successfully established when ARPE-19
cells were treated with TGF-β1 (10 ng/ml) for 48 h. This time point
was chosen to examine the function of BK in the following
experiments.
| Table II.Different concentrations of TGF-β1
affect the proliferation of ARPE-19 cells. |
Table II.
Different concentrations of TGF-β1
affect the proliferation of ARPE-19 cells.
|
| Optical density
value |
|---|
|
|
|
|---|
| Concentration of
TGF-β1 (ng/ml) | 24 h | 48 h |
|---|
| 0 | 0.49±0.05 | 0.81±0.02 |
| 0.1 |
0.58±0.04a |
0.86±0.02a |
| 0.5 |
0.55±0.03a |
0.89±0.01a |
| 2.5 |
0.65±0.01a |
0.92±0.01a |
| 10 |
0.70±0.04a |
0.96±0.02a |
| 12.5 |
0.76±0.01a |
0.93±0.01a |
BK inhibits TGF-β1-induced EMT in
ARPE-19 cells, and the B2R inhibitor attenuates the role of BK
To determine whether BK serves an important role in
TGF-β1-induced EMT, the alterations in cell morphology were
observed and the expression levels of E-cadherin, α-SMA and
vimentin in each group were determined. Fig. 2A demonstrated that BK+TGF-β1 group
may prevent TGF-β1-induced EMT; the number of spindle-shaped
mesenchymal cells decreased, whereas BK+TGF-β1+HOE-140 weakened the
role of BK and the mesenchyme transformation of the cells increased
significantly. Western blot analysis revealed that BK+TGF-β1 group
increased E-cadherin protein expression levels, and decreased the
expression levels of α-SMA and vimentin compared with
TGF-β1-treated cells (P<0.05). The B2R inhibitor HOE-140 in
BK+TGF-β1+HOE-140 group blocked BK, weakening its effect compared
with BK+TGF-β1 group (P<0.05, Fig.
2B). The immunofluorescence results revealed that BK in
BK+TGF-β1 group significantly attenuated the downregulation of
E-cadherin, and the upregulation of α-SMA and vimentin compared
with TGF-β1-treated cells, whereas B2R antagonism in
BK+TGF-β1+HOE-140 group reversed this effect compared with
BK+TGF-β1group (Fig. 2C). In
addition, BK in BK+TGF-β1 group inhibited the ARPE-19 cell
migration enhanced by TGF-β1. With HOE-140 in BK+TGF-β1+HOE-140
group, cell migration ability was enhanced compared with BK+TGF-β1
group (Table III; Fig. 2D-F). These results suggested that
BK may inhibit TGF-β1-induced EMT in ARPE-19 cells.
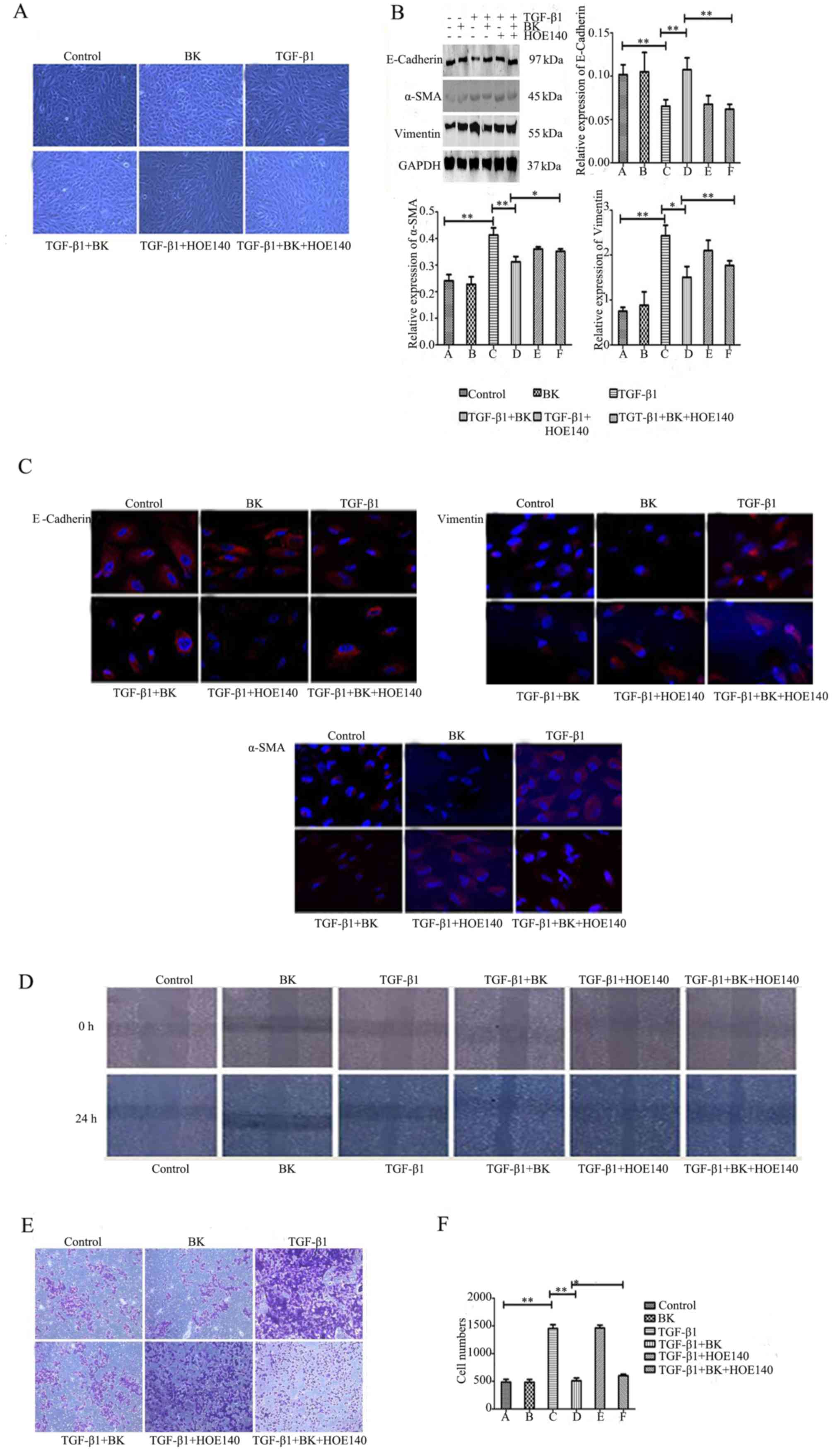 | Figure 2.BK inhibits TGF-β1-induced
epithelial-mesenchymal transitionin ARPE-19 cells, and a B2R
inhibitor attenuates the role of BK. ARPE-19 cells were treated
with BK, TGF-β1, BK plus TGF-β1, TGF-β1 plus HOE140, BK plus TGF-β1
plus HOE140, and Dulbecco's modified Eagle's medium for 48 h prior
todetection. (A) Morphological alterations were detected using an
inverted phase-contrast microscope (magnification, ×100). (B)
Western blot analysis detected the expression levels of E-cadherin,
α-SMA and vimentin in each group. (C) The protein expression of
E-cadherin, α-SMA and vimentin was identified by immunofluorescence
staining (magnification, ×200). (D) A wound healing assay tested
the cell migration of a scratched edge. (E) Cells that had migrated
through the Transwell chamber filter over 18 h were stained with
crystal violet, and imaged at ×50 magnification. (F) The number of
migrated ARPE-19 cells in each group. **P<0.001 and *P<0.05.
TGF-β1, transforming growth factor-β1; BK, bradykinin; α-SMA,
α-smooth muscle actin. |
| Table III.The effect of TGF-β1 on migration of
ARPE-19 cells. |
Table III.
The effect of TGF-β1 on migration of
ARPE-19 cells.
| A, Migration
distance of ARPE-19 cells |
|---|
| Group | Migration distance,
µm (mean ± standard deviation) |
|---|
| DMEM (control) | 3.60±0.62 |
| BK | 3.53±0.50 |
| TGF-β1 | 3.98±0.36 |
| TGF-β1+BK | 3.55±0.47 |
| TGF-β1+HOE-140 | 3.86±0.22 |
|
TGF-β1+BK+HOE-140 | 3.77±0.32 |
|
| B, Optical
density value of cell migration using the indirect enumeration
method |
|
| Group | Migration
distance, µm (mean ± standard deviation) |
|
| DMEM (control) | 0.431±0.059 |
| BK | 0.430±0.054 |
| TGF-β1 |
1.272±0.059a |
| TGF-β1+BK |
0.562±0.018b |
| TGF-β1+HOE-140 | 1.208±0.038 |
|
TGF-β1+BK+HOE-140 |
0.666±0.048c |
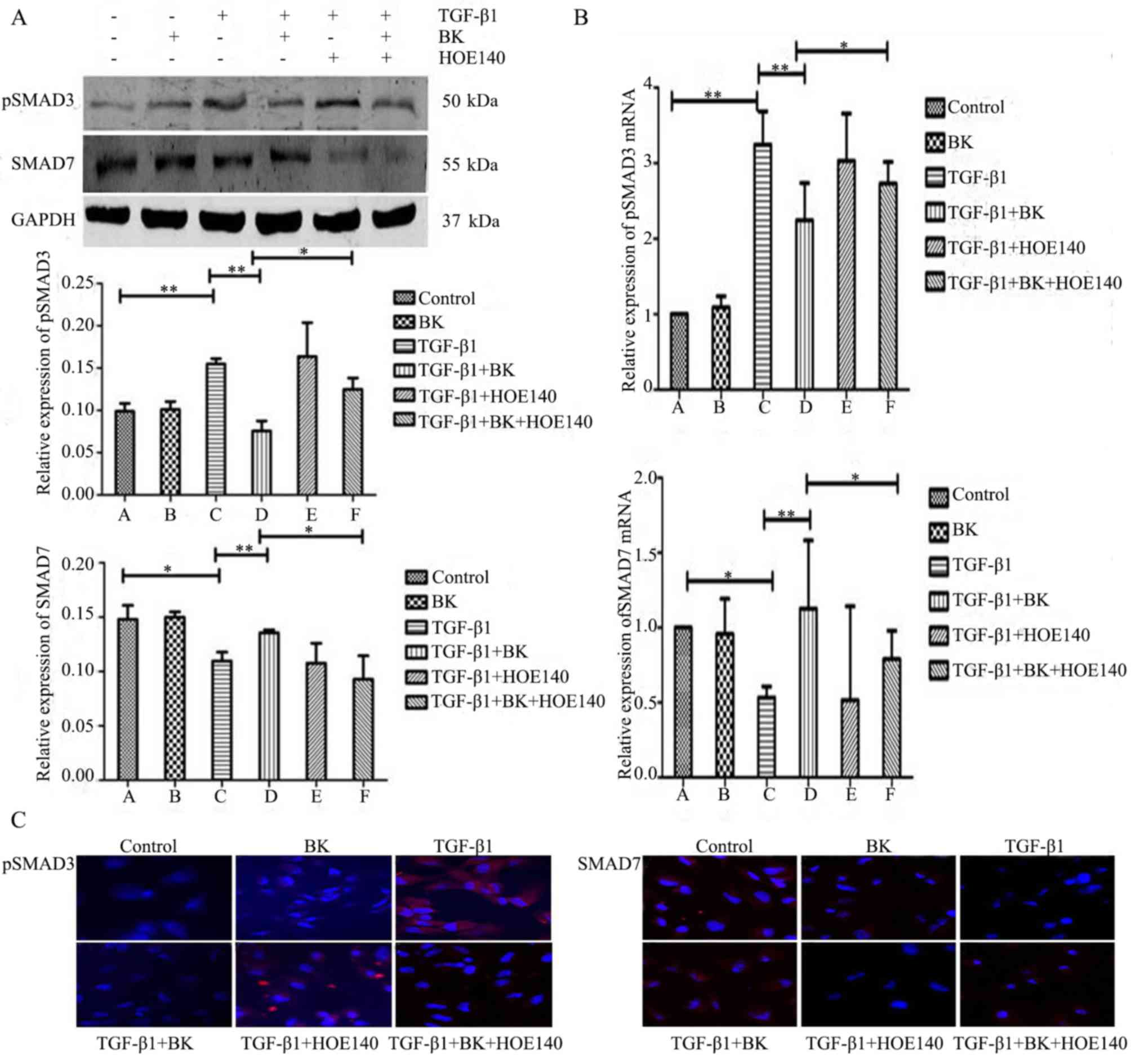
BK inhibits TGF-β1-induced EMT in
ARPE-19 cells via the TGF/Smad signaling pathway
To determine whether BK inhibits TGF-β1-induced EMT
in ARPE-19 cells via the TGF/Smad signaling pathway, pSmad3 and
Smad7 expression levels were measured. As determined by western
blot analysis (Fig. 3A), BK+TGF-β1
group decreased pSmad3 protein expression levels and increased
Smad7 protein expression levels compared with TGF-β1-treated cells.
The expression levels of pSmad3 and Smad7 were reversed by
treatment with the B2R antagonist HOE-140 in BK+TGF-β1+HOE-140
group compared with BK+TGF-β1 group. These changes in pSmad3 and
Smad7 mRNA expression were verified by q-PCR (Fig. 3B). Immunofluorescence revealed that
BK+TGF-β1 group markedly downregulated the expression of pSmad3 and
upregulated the expression of Smad7 compared with TGF-β1-treated
ARPE-19 cells, molecular events that were antagonized by HOE140
(Fig. 3C). These results
demonstrated that BK had effects on TGF-β1-induced EMT by
upregulating the expression of Smad7 and downregulating the
expression of pSmad3 in the TGF-β/Smad signaling pathway.
Discussion
PVR is generally considered to be an afibrotic
process driven by EMT in RPE cells, causing traction of the retina
and leading to surgical failure (19). EMT widely exists in renal
interstitial fibrosis (20) and
liver fibrosis (21). EMT has a
vital role in the recurrence of liver cancer (22), in addition to the occurrence and
metastasis of lung cancer (23).
TGF may be divided into three subtypes in mammals: TGF-β1, TGF-β2
and TGF-β3. TGF-β1 is a multi-effective growth factor, and it is
the primary cytokine that mediates EMT in tumor cells (24). TGF-β1-induced EMT of RPE cells has
been acknowledged as a classic model to study the underlying
mechanisms of EMT (13,14). However, the optimal concentration
and time of treatment with TGF-β1 may differ (25–27).
In the present study, different concentrations of
TGF were used to treat ARPE-19 cells for 24 and 48 h, and it was
observed that 10 ng/ml TGF-β1 resulted in EMT of ARPE-19 cells. The
alteration in cell morphology was more marked when the TGF-β
treatment time was 48 h, compared with 24 h. This result was
consistent with the results of Yang et al (27). The integrity of cell-cell contacts
is an important regulator of TGF-β1-induced
epithelial-to-myofibroblast transition (28). E-cadherin is a principal component
of adhesive connections between cells. This suggests that
E-cadherin may be involved in TGF-β1-induced EMT. E-cadherin is
highly expressed in normal ARPE-19 cells (29). When E-cadherin transcription is
suppressed, it may affect adhesion between epithelial cells and
eventually result in EMT. Therefore, decreased E-cadherin is an
indicator of EMT in epithelial cells (30). In the present study, western blot
analysis was used to detect the expression levels of E-cadherin in
cells. The results revealed that the E-cadherin expression levels
were increased in normal ARPE-19 cells. With increased TGF-β1
concentration, the expression level of E-cadherin was gradually
reduced. This demonstrated that the TGF-β1-induced EMT model was
successfully established in vitro.
At present, there is no safe and effective clinical
drug used as a conventional treatment for PVR (31). Vitreous cavity injection of
polylactic acid, daunorubicin or 5-fluorouracil in rabbit eyes
following vitrectomy may effectively prevent the formation of PVR
membranes (32). In addition,
anti-inflammatory drugs, including triamcinolone acetonide, and
intravitreal injections as an adjunct to vitrectomy and silicone
oil tamponade, appear to be effective and safe in treating PVR
(grade C or D) (33).
Anti-inflammatory therapy in PVR may be a potential research
direction.
The complement and blood coagulation cascades are
the most important KEGG pathways in the PVR pathological process,
and the KKS is the intermediate process connecting the complement
pathways and the blood coagulation pathway (16). KKS serves an important role in
inflammation (34). In the KKS
system, activated kallikrein acts on kininogen. Plasma kininases
convert high molecular weight kininogen to BK and kallidin, whereas
tissue kininases convert low molecular weight kininogen to
kallidin, which may be converted into BK under the action of
enzymes (35). The BK amino acid
sequence is Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg, and it has a
series of complex physiological effects with kinin receptors in
tissues and organs (36). The BK
receptor is divided into B1R and B2R. B2R serves a primary role in
biological reactions. It is expressed in a variety of cell types,
including myocardial cells, pain sensitive neurons, endothelial
cells and smooth muscle cells (37). BK protects renal function via B2R
receptors in renal fibrosis, and this effect may be reversed by a
B2R receptor antagonist (38). To
examine whether BK has a similar effect in PVR disease, the present
study used BK to stimulate PVR in an in vitro cell model to
observe the influence of BK on PVR models. In the present study, it
was demonstrated that BK has effects on the protein expression
levels in TGF-β1-induced EMT and reverses EMT. It increased
epithelial cell marker protein levels and decreased interstitial
cell marker protein levels. Vitreous aspirates from patients with
PVR stimulate retinal pigment epithelial cell migration (39). Research has recently focused on
investigating RPE cell migration (40,41).
To further determine whether BK influences TGF-β1-induced EMT
migration, a wound healing assay and the Transwell migration method
was used. It was revealed that BK inhibited ARPE-19 cell migration.
With HOE-140, the cell migration ability was enhanced.
The TGF-β/Smad pathway is a major component of the
process of the phenotype alteration from epithelial cells to
mesenchymal cells (42,43). TGF-β combines with the TGF-β type
II receptor and forms a closely linked complex with TGF-β type I
receptor, which leads to Smad2 and Smad3 activation. The activated
Smad2 and Smad3 bind with Smad4 for target gene recognition and
transcriptional regulation (44–46).
Although the function of pSmad2 and pSmad3 is certain in this
process, a previous study demonstrated that Smad3 is the essential
mediator of TGF-β signaling and directly activates genes encoding
regulators of transcription and signal transducers (47). Smad7 negatively regulates Smad2/3,
and its overexpression inhibits EMT in RPE cells (48). Smad3 is required for the
de-differentiation of the retinal pigment epithelium following
retinal detachment. Blocking the Smad3 pathway may inhibit the
occurrence of EMT (14).
Upregulation of the BK-B2R pathway modulates the TGF-β/Smad
signaling cascade to reduce renal fibrosis (38). However, the function of BK in RPE
cells is unknown. In the present study, it was determined that BK
reduced pSmad3 expression levels and increased Smad7 expression
levels. The protective alterations were reversed by HOE-140.
However, the present results were only verified in vitro;
whether the same conclusion is observed in vivo requires
further investigation.
In conclusion, the results of the present study
suggested that BK contributes to the progression of TGF-β1-mediated
EMT in ARPE-19 cells via the TGF/Smad signaling pathway. These
findings may promote a future clinical therapeutic strategy for the
prevention or treatment of PVR.
Acknowledgements
The present study was financially supported by the
National Natural Science Foundation of China (grant no. 81470648).
The authors would like to thank the Central Laboratory of Shanghai
Tenth People's Hospital Affiliated with Tongji University for the
experimental facilities and equipment.
References
|
1
|
Pennock S, Haddock LJ, Eliott D, Mukai S
and Kazlauskas A: Is neutralizing vitreal growth factors aviable
strategy to prevent proliferative vitreoretinopathy? Prog Retin Eye
Res. 40:16–34. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Umazume K, Tsukahara R, Liu L, Fernandez
de Castro JP, McDonald K, Kaplan HJ and Tamiya S: Role of retinal
pigment epithelial cell β-catenin signaling in experimental
proliferative vitreoretinopathy. Am J Pathol. 184:1419–1428. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tosi GM, Marigliani D, Romeo N and Toti P:
Disease pathways in proliferative vitreoretinopathy: An ongoing
challenge. J Cell Physiol. 229:1577–1583. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li H, Wang H, Wang F, Gu Q and Xu X: Snail
involves in the transforming growth factor β1-mediated
epithelial-mesenchymal transition of retinal pigment epithelial
cells. PLoS One. 6:e233222011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang S, Li H, Li M and Wang F: Mechanisms
of epithelial-mesenchymal transition in proliferative
vitreoretinopathy. Discov Med. 20:207–217. 2015.PubMed/NCBI
|
|
7
|
Zhang J, Tian XJ, Zhang H, Teng Y, Li R,
Bai F, Elankumaran S and Xing J: TGF-β-induced
epithelial-to-mesenchymal transition proceeds through stepwise
activation of multiple feedback loops. Sci Signal. 7:ra912014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bochaton-Piallat ML, Kapetanios AD, Donati
G, Redard M, Gabbiani G and Pournaras CJ: TGF-beta1, TGF-beta
receptor II and ED-A fibronectin expression in myofibroblast of
vitreoretinopathy. Invest Ophthalmol Vis Sci. 41:2336–2342.
2000.PubMed/NCBI
|
|
9
|
Asaria RH, Kon CH, Bunce C, Sethi CS, Limb
GA, Khaw PT, Aylward GW and Charteris DG: Silicone oil concentrates
fibrogenic growth factors in the retro-oil fluid. Br J Ophthalmol.
88:1439–1442. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hoerster R, Muether PS, Vierkotten S,
Hermann MM, Kirchhof B and Fauser S: Upregulation of TGF-ß1 in
experimental proliferative vitreoretinopathy is accompanied by
epithelial to mesenchymal transition. Graefes Arch Clin Exp
Ophthalmol. 252:11–16. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liang CM, Tai MC, Chang YH, Chen YH, Chen
CL, Lu DW and Chen JT: Glucosamine inhibits
epithelial-to-mesenchymal transition and migration of retinal
pigment epithelium cells in culture and morphologic changes in a
mouse model of proliferative vitreoretinopathy. Acta Ophthalmol.
89:e505–e514. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nassar K and Grisanti S, Tura A, Lüke J,
Lüke M, Soliman M and Grisanti S: A TGF-β receptor 1 inhibitor for
prevention of proliferative vitreoretinopathy. Exp Eye Res.
123:72–86. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dvashi Z, Goldberg M, Adir O, Shapira M
and Pollack A: TGF-β1 induced transdifferentiation of RPE cells is
mediated by TAK1. PLoS One. 10:e01222292015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Saika S, Kono-Saika S, Tanaka T, Yamanaka
O, Ohnishi Y, Sato M, Muragaki Y, Ooshima A, Yoo J, Flanders KC and
Roberts AB: Smad3 is required for dedifferentiation of retinal
pigment epithelium following retinal detachment in mice. Lab
Invest. 84:1245–1258. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Garweg JG, Tappeiner C and Halberstadt M:
Pathophysiology of proliferative vitreoretinopathy in retinal
detachment. Surv Ophthalmol. 58:321–329. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu J, Peng R, Chen H, Cui C and Ba J:
Elucidation of the pathogenic mechanism of rhegmatogenous retinal
detachment with proliferative vitreoretinopathy by proteomic
analysis. Invest Ophthalmol Vis Sci. 53:8146–8153. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao HM, Yu J, Sheng MJ and Wang KS:
Experimental study of kallikrein-kinin system participating in
proliferative vitreoretinopathy procedure. Chin J Exp Ophthalmol.
29:591–595. 2011.(In Chinese).
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta DeltaC(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Saika S, Yamanaka O, Okada Y, Tanaka S,
Miyamoto T, Sumioka T, Kitano A, Shirai K and Ikeda K: TGF-beta in
fibroproliferative diseases in the eye. Front Biosci (Schol Ed).
1:376–390. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bai Y, Lu H, Lin C, Xu Y, Hu D, Liang Y,
Hong W and Chen B: Sonic hedgehog-mediated epithelial-mesenchymal
transition in renal tubulointerstitial fibrosis. Int J Mol Med.
37:1317–1327. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao YL, Zhu RT and Sun YL:
Epithelial-mesenchymal transition in liver fibrosis. Biomed Rep.
4:269–274. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu TJ, Chang SS, Li CW, Hsu YH, Chen TC,
Lee WC, Yeh CT and Hung MC: Severe hepatitis promotes
hepatocellular carcinoma recurrence via NF-κB pathway-mediated
epithelial-mesenchymal transition after resection. Clin Cancer Res.
22:1800–1812. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hong SK, Park JR, Kwon OS, Kim KT, Bae GY
and Cha HJ: Induction of integrin β3 by sustained ERK activity
promotes the invasiveness of TGFβ-induced mesenchymal tumor cells.
Cancer Lett. 376:339–346. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Massagué J: TGF-beta in cancer. Cell.
134:215–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Han L, Zhang HW, Zhou WP, Chen GM and Guo
KJ: The effects of genistein on transforming growth
factor-β1-induced invasion and metastasis in human pancreatic
cancer cell line Panc-1 in vitro. Chin Med J (Engl). 125:2032–2040.
2012.PubMed/NCBI
|
|
26
|
Koeck S, Amann A, Huber JM, Gamerith G,
Hilbe W and Zwierzina H: The impact of metformin and salinomycin on
transforming growth factor β-induced epithelial-to-mesenchymal
transition in non-small cell lung cancer cell lines. Oncol Lett.
11:2946–2952. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang S, Yao H, Li M, Li H and Wang F: Long
non-coding RNA MALAT1 mediates transforming growth factor
beta1-induced epithelial-mesenchymal transition of retinal pigment
epithelial cells. PLoS One. 11:e01526872016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Masszi A, Fan L, Rosivall L, McCulloch CA,
Rotstein OD, Mucsi I and Kapus A: Integrity of cell-cell contacts
is a critical regulator of TGF-beta 1-induced
epithelial-to-myofibroblast transition: Role for beta-catenin. Am J
Pathol. 165:1955–1967. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Palma-Nicolás JP and López-Colomé AM:
Thrombin induces slug-mediated E-cadherin transcriptional
repression and the parallel up-regulation of N-cadherin by a
transcription-independent mechanism in RPE cells. J Cell Physiol.
228:581–589. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cano A, Pérez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Priglinger CS and Priglinger S:
Pharmacological approach to treatment of proliferative
vitreoretinopathy. Ophthalmology. 110:948–959. 2013.(In German).
View Article : Google Scholar
|
|
32
|
Mandava N, Blackburn P, Paul DB, Wilson
MW, Read SB, Alspaugh E, Tritz R, Barber JR, Robbins JM and Kruse
CA: Ribozyme to proliferating cell nuclear antigen to treat
proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci.
43:3338–3348. 2002.PubMed/NCBI
|
|
33
|
Chen W, Chen H, Hou P, Fok A, Hu Y and Lam
DS: Midterm results of low-dose intravitreal triamcinolone as
adjunctive treatment for proliferative vitreoretinopathy. Retina.
31:1137–1142. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yarovaya GA and Neshkova EA:
Kallikrein-Kinin system. Long history and present. (To 90th
Anniversary of Discovery of the System). Bioorg Khim. 41:275–291.
2015.(In Russian).
|
|
35
|
Wang T, Tang Y and Lou JS: Advances in
research on kallikrein-kinin system in cardiovascular system. Chin
J Pharmacol Toxicol. 17:466–470. 2003.
|
|
36
|
Yu J, Peng R, Chen H, Cui C, Ba J and Wang
F: Kininogen 1 and insulin-like growth factor binding protein 6:
Candidate serum biomarkers of proliferative vitreoretinopathy. Clin
Exp Optom. 97:72–79. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Leeb-Lundberg LM, Marceau F, Müller-Esterl
W, Pettibone DJ and Zuraw BL: International union of pharmacology.
XLV. Classification of the kinin receptor family: From molecular
mechanisms to pathophysiological consequences. Pharmacol Rev.
57:27–77. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cárdenas A, Campos J, Ehrenfeld P, Mezzano
S, Ruiz-Ortega M, Figueroa CD and Ardiles L: Up-regulation of the
kinin B2 receptor pathway modulates the TGF-β/Smad signaling
cascade to reduce renal fibrosis induced by albumin. Peptides.
73:7–19. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Campochiaro PA, Jerdan JA, Glaser BM,
Cardin A and Michels RG: Vitreous aspirates from patients with
proliferative vitreoretinopathy stimulate retinal pigment
epithelial cell migratio. Arch Ophthalmol. 103:1403–1405. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
He S, Kuma SR, Zhou P, Krasnoperov V, Ryan
SJ, Gill PS and Hinton DR: Soluble EphB4 inhibition of PDGF-induced
RPE migration in vitro. Invest Ophthalmol Vis Sci. 51:543–552.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chan CM, Huang JH, Chiang HS, Wu WB, Lin
HH, Hong JY and Hung CF: Effects of (−)-epigallocatechin gallate on
RPE cell migration and adhesion. Mol Vis. 16:586–595.
2010.PubMed/NCBI
|
|
42
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Saitoh M and Miyazawa K: Transcriptional
and post-transcriptional regulation in TGF-β-mediated
epithelial-mesenchymal transition. J Biochem. 151:563–571. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Choi K, Lee K, Ryu SW, Im M, Kook KH and
Choi C: Pirfenidone inhibits transforming growth factor-β1-induced
fibrogenesis by blocking nuclear translocation of Smads in human
retinal pigmentepithelial cell line ARPE-19. Mol Vis. 18:1010–1020.
2012.PubMed/NCBI
|
|
45
|
Feng XH and Derynck R: Specificity and
versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev
Biol. 21:659–693. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Massagué J: TGF-β signalling in context.
Nat Rev Mol Cell Biol. 13:616–630. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yang YC, Piek E, Zavadil J, Liang D, Xie
D, Heyer J, Pavlidis P, Kucherlapati R, Roberts AB and Böttinger
EP: Hierarchical model of gene regulation by transforming growth
factor beta. Proc Natl Acad Sci USA. 100:pp. 10269–10274. 2003;
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Saika S, Yamanaka O, Nishikawa-Ishida I,
Kitano A, Flanders KC, Okada Y, Ohnishi Y, Nakajima Y and Ikeda K:
Effect of Smad7 gene overexpression on transforming growth factor
beta-induced retinal pigment fibrosis in a proliferative
vitreoretinopathy mouse model. Arch Ophthalmol. 125:647–654. 2007.
View Article : Google Scholar : PubMed/NCBI
|