Introduction
Hearing loss (HL), which is a common sensory
disorder, may affect humans of all ages, from newborns to the
elderly. According to the World Health Organization, >5% of the
world's population, 360 million people, have disabling HL (328
million adults and 32 million children) (1). In China, the office of the Second
Sampling Survey on Disabled People reported that there were
27,800,000 people with this disorder in 2006; this disorder is a
birth defect that affects ~1 in 1,000 children in China (2).
HL may be caused by genetic or environmental
factors, or a combination of the two, and >50% of cases are
caused by genetic factors (3).
With respect to the mode of inheritance, this disorder may be
classified as autosomal recessive (77%), autosomal dominant (22%),
or X- or Y-linked and mitochondrial inheritance (1%) (4). There are two clinical types:
Syndromic (S)HL and nonsyndromic (NS)HL. SHL is usually
distinguished via systemic medical examination (5); however, NSHL is frequently classified
as SHL by doctors, as NSHL does not display other associated
clinical features (6). NSHL
accounts for ~70% of hereditary HL (6). At present, >80 genes and >100
genetic loci have been implicated in NSHL (7). In China, the most frequent causative
genes, in order of frequency, are gap junction protein β (GJB)2,
GJB3, solute carrier family 26 member 4 (SLC26A4) and mitochondrial
(mt)DNA for HL (3), and these four
genes are screened by doctors in persons with HL. However, certain
patients with NSHL do not have mutations in these four genes;
certain patients, therefore, remain genetically unexplained.
Traditionally, studies into a family pedigree were performed by
linkage analysis and candidate gene Sanger sequencing. When the
pedigree is very large or very small, it is difficult to identify
causative mutations via regular Sanger sequencing for numerous
cases of NSHL (8). Whole exome
sequencing (WES), a technique for sequencing all of the expressed
genes in a genome, has become an effective alternative strategy
(9).
In the present study, to investigate the genetic
causes of deafness in children in the Yunnan province, which has
been inhabited by 26 nationalities throughout history, mutations in
two deaf families were investigated. A novel mutation in the
melanogenesis associated transcription factor (MITF) gene was
identified in one family from Yunnan-Guizhou Plateau with two
siblings affected by congenital sensorineural HL and heterozygous
GJB2 c.235delC del/del variant was detected in another family from
Yunnan-Guizhou Plateau with two siblings affected by congenital
sensorineural HL.
Case report
Subjects
A total of two consanguineous Chinese Han families
from Yunnan-Guizhou Plateau were recruited by the Children's
Hospital, Kunming Medical University (Kunming, China), for genetic
diagnosis. Within these two families, two siblings suffered from
bilateral prelingual deafness, although the hearing of the parents
was normal. The present study was approved by the ethics committee
of the Children's Hospital, Kunming Medical University; written
informed consent was obtained from participants or their
guardians.
Clinical evaluations
All clinical and audiometric assessments were
performed in the Children's Hospital and First Affiliated Hospital
of Kunming Medical University. Pure tone audiometry, auditory
brainstem response and distortion product otoacoustic emission were
performed. To examine the inner malformation, computed tomography
or magnetic resonance imaging (MRI) were employed in the present
study. The 250, 500, 1,000, 2,000, 4,000 and 8,000 Hz settings of
pure tone audiometry were used to assess the hearing level as
follows: Normal [<20 decibels hearing level (dBHL)], mild (20–40
dBHL), moderate (41–70 dBHL), severe (71–95 dBHL), and profound
(>95 dBHL) deafness (10).
WES and variant analysis
Peripheral blood samples (2 ml) in tubes containing
0.2 M EDTA were collected from the siblings and their parents. The
peripheral blood samples for each subject were extracted using a
blood DNA extraction kit, QIAamp DNA (Qiagen GmbH, Hilden,
Germany). Following extraction, DNA was randomly fragmented with
DNA enzymes (Tagment DNA Enzyme 1, TruSight One Sequencing Panel
Series, Illumina Inc., San Diego, CA, USA) and purified with
magnetic beads as described (11).
DNA fragments (80.4 ng/µl) were amplified by ligation-mediated
polymerase chain reaction (PCR) as described (11). Sequences of ‘TAAGGCGA’ and
‘CTCTCTAT’ were added using a 10-cycle PCR program. The
thermocycling conditions were: An initial denaturation of 98°C for
30 sec, 10 cycles of denaturation at 98°C for 10 sec, annealing at
60°C for 30 sec, extension at 72°C for 30 sec and a final extension
of 72°C for 5 min (11). The DNA
fragments were captured and purified twice using a TruSight One
Sequencing panel according to the manufacturer's protocols
(Illumina China, Shanghai, China).
Size-selected DNA fragments were amplified by PCR as
described (11). A total of 5 µl
PCR Primer Cocktail was added to each well, and 20 µl Enrichment
Amp Mix was added. The total volume per well was 50 µl.
Subsequently, the mixture was centrifuged at 1,200 × g for 1 min at
4°C and then at 280 × g for 1 min at 4°C. The thermocycling
conditions were described as aforementioned. Samples were purified
to obtain a qualified captured library.
Each resulting qualified captured library was loaded
on MiSeq Next 500 sequencing platforms (Illumina Inc.), and
high-throughput sequencing for each captured library was performed
to ensure that each sample met the desired average sequencing
coverage. As an autosomal recessive pattern of inheritance was
suspected, the candidates were two affected siblings and their
parents who were expected to be heterozygous for the variant.
Mutation analysis
The debased reads or clean reads without adapters
were mapped to the human reference genome (UCSC hg19; genome.ucsc.edu) by Burrows-Wheeler Alignment. All
variants were screened with the single nucleotide polymorphism
database version 142 (www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi?build_id=142),
the Human Gene Mutation Database (www.hgmd.cf.ac.uk/ac/index.php), ClinVar (www.ncbi.nlm.nih.gov/clinvar), the 1,000 Genomes
Project (www.internationalgenome.org), UniProt (www.uniprot.org/uniprot) and the National Heart,
Lung and Blood Institute Exome Sequencing Project 6500 (https://esp.gs.washington.edu/drupal/)
Functional prediction was performed via Sorting Intolerant from
Tolerant (http://sift.jcvi.org/), a likelihood
ratio test (12), Mutation Taster
(www.mutationtaster.org), Mutation
Assessor (mutationassessor.org/r3), Functional Analysis through
Hidden Markov Models (fathmm.biocompute.org.uk), Genomic Evolutionary Rate
Profiling (mendel.stanford.edu/SidowLab/downloads/gerp),
PhyloP-2 (http://genetics.bwh.harvard.edu/pph2/), SiPhy
(portals.broadinstitute.org/genome_bio/siphy/index.html)
and Polymorphism Phenotyping-2 (genetics.bwh.harvard.edu/pph2). Candidate variants
were annotated using Annotate Variation software (http://annovar.openbioinformatics.org/en/latest/)
(13).
Mutation validation
Sanger sequencing was performed with an ABI3500
sequencer (Applied Biosystems, Thermo Fisher Scientific Inc.,
Waltham, MA, USA) and PCR; thermocycling conditions: An initial
denaturation of 94°C for 4 min, 32 cycles of denaturation at 94°C
for 30 sec, annealing at 60°C for 30 sec, extension at 72°C for 30
sec and a final extension of 72°C for 7 min (14). The sequences of forward and reverse
primers are presented in Table I
and were used to confirm potential causative variants in this
family.
 | Table I.PCR primers for amplification. |
Table I.
PCR primers for amplification.
| Gene | Exon | Forward primers | Reverse primers |
|---|
| MITF | 8 |
TACACGGCTTGGGTGGTG |
GCACATGTCCAAGAATGACTG |
| CHD7 | 29 |
AATCTTAATGAGTCATCCTGTTTG |
ACTTGGGGAGAATTCAAGGG |
Clinical findings
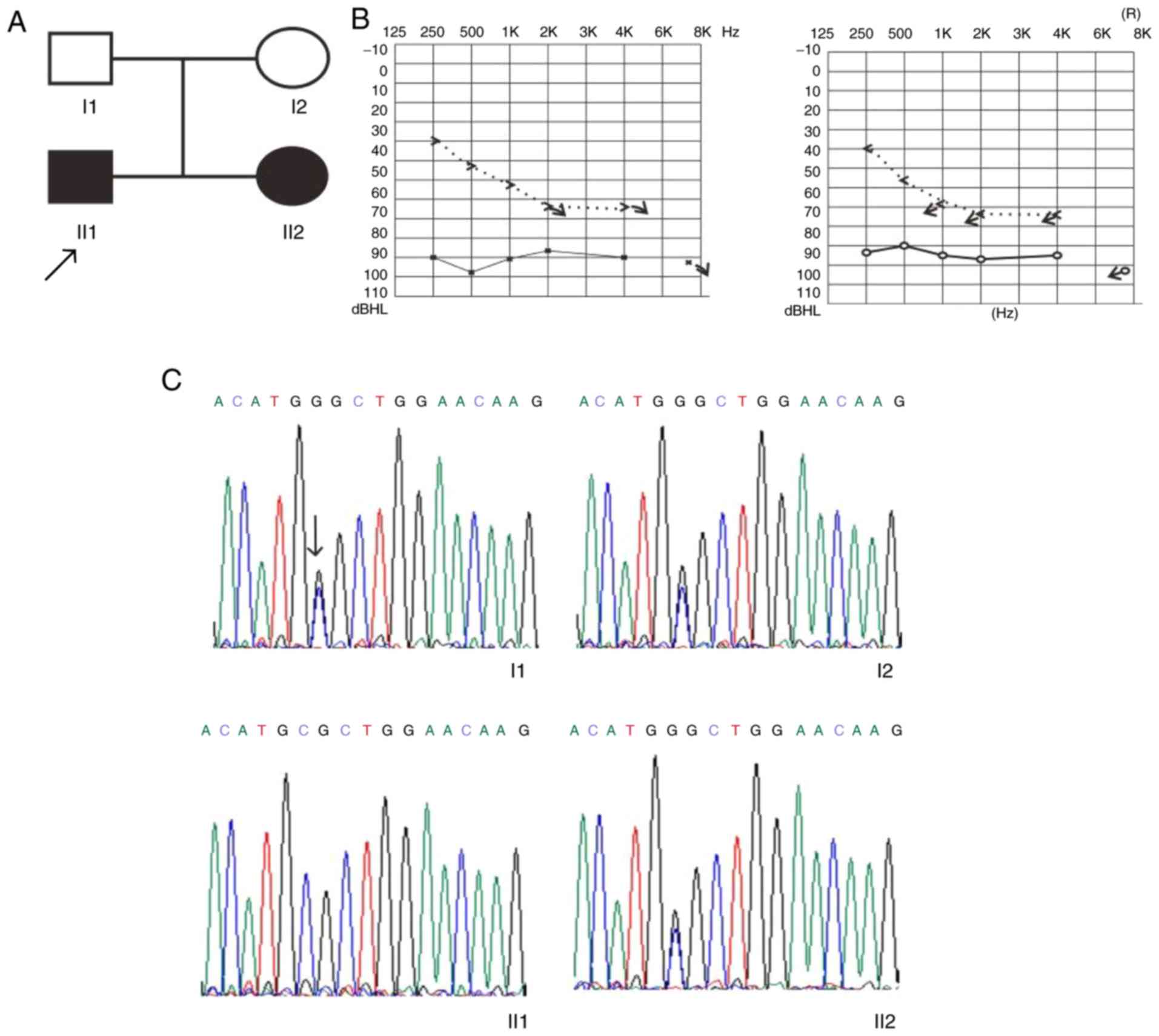
Clinical presentation of family 1
The family included two affected siblings and two
unaffected parents. Audiogram analysis of this family revealed that
the siblings had bilateral HL. The proband was a 12-year-old boy
(II1; Fig. 1A) with deafness,
although MRI analysis demonstrated that the inner ear was normal.
When the proband was 6 years old, pure tone audiometry revealed
profound bilateral sensorineural HL. The results of the acoustic
immittance measurement demonstrated a type A tympanometric curve.
The auditory brainstem response at 95 dB and distortion product
otoacoustic emission were absent in both ears of the proband. The
younger affected sibling, a 2-year-old girl (II2; Fig. 1A) with severe sensorineural HL, had
a normal inner ear and a type A tympanometric curve. The younger
sibling had bilaterally absent distortion product otoacoustic
emission and a 90-dB auditory brainstem response. The father and
mother (45 and 35 years old, respectively) had no HL (I1 and 2;
Fig. 1A). The clinical information
for this family is summarized in Table II.
| Table II.Phenotypes and genotypes of family
1. |
Table II.
Phenotypes and genotypes of family
1.
| Subject | Age, years | Hearing loss | ABR | DPOAE | CT | MRI | Genotype |
|---|
| Father | 45 | N/A | Present | Present | N/A | Normal | Wild-type |
| Mother | 35 | N/A | Present | Present | N/A | Normal | Mutation |
| Boy | 12 | L (profound), R
(severe) | Absent | Absent | Normal | Normal | Mutation |
| Girl | 2 | Bil (severe) | Absent | Absent | Normal | Normal | Mutation |
The genes GJB2, GJB3, SLC26A4 and mtDNA were
sequenced in this family to exclude mutations in the four genes
known to be associated with hereditary HL; however, no mutations
were detected in these four genes (Table III).
| Table III.Gene mutations of family 1 and 2. |
Table III.
Gene mutations of family 1 and 2.
|
| Family 1 | Family 2 |
|---|
|
|
|
|
|---|
| Gene or
mitochondria | Proband | Younger sister | Father | Mother | Proband | Younger sister | Father | Mother |
|---|
| GJB2 | Normal | Normal | Normal | Normal | c.235delC
(homozygous mutation) | c.235delC
(homozygous mutation) | c.235delC
(heterozygous mutation) | c.235delC
(heterozygous mutation) |
| GJB3 | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal |
| SLC26A4 | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal |
| mtDNA | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal |
Clinical presentation of family 2
This family included two affected siblings (a
3-month-old and a 3-year-old, both girls) and two unaffected
parents. Sanger sequencing demonstrated a homozygous and
heterozygous GJB2 c.235delC del/del variant in the two siblings and
unaffected parents, respectively (Table III).
WES
WES was used to sequence the proband of family 1,
and heterozygous MITF c.718C>G and chromodomain DNA helicase
binding protein 7 (CHD7) c.5770T>C variants were detected in
this proband.
Identification of pathogenic
mutation
In family 1, the heterozygous MITF c.718C>G and
CHD7 c.5770T>C variants were confirmed by Sanger sequencing. The
heterozygous MITF c.718C>G variant was identified in the
affected sibling and unaffected mother (Fig. 1C).
Discussion
In the present study, GJB2, GJB3, SLC26A4 and mtDNA
were screened in two families with NSHL, and a novel c.718C>G
variation in the MITF gene in family 1 was detected by WES. The
MITF gene was named after mice with a mutation in this particular
gene (15), and Hemesath et
al (16) demonstrated that
MITF functions in melanocyte survival; pigmentation is mediated by
MiT family interactions and transcriptional activities. In 1994, a
mutation in the MITF gene was detected in humans with HL (17). According to the study of Yang et
al (14) proposed that a
MITF mutation (Mitf) database should be created for those
with Waardenburg syndrome in the Chinese population. The locus of
human and mouse MITF covers 229 kb and starts at ~214 kb from the
beginning of exon 1A to the end of exon 9 (including the extended
3′ untranslated region), respectively. The MITF/Mitf locus is over
200 kb inlength, with strong but imperfect exon conservation
between human and mouse (18). A
total of ≥9 isoforms of MITF/Mitf have been identified, and the
expression profile of each isoform varies (19,20).
MITF/Mitf functions are associated with numerous inherited
disorders of humans and mice (21). Elimination of the MITF-M isoform
alone is sufficient to cause deafness and depigmentation (22).
According to genome databases (grenada.lumc.nl/LOVD2/ for humans, and www.informatics.jax.org for mice), ~89 MITF
mutations in human and mouse alleles have been reported. A total of
>40 MITF mutations have been identified in a number of people
with Waardenburg's syndrome type 2 and in Tietz syndrome families;
9 mutations of the MITF gene were identified in Chinese patients
with Waardenburg's syndrome type 2 (14).
At present, severe or profound HL is frequently
treated with a cochlear implant (23). In particular, when those with HL
secondary to GJB2 (or Cx26) (OMIM: 121011) mutations were fitted
with cochlear implants, excellent speech and language performance
was observed (24). However, the
cochlear implant was only used for analysis study in a large mammal
Rongchang pigs with mutations in the MITF gene (25).
In the present study, the heterozygous c.718C>G
variant in the MITF gene was identified in two affected siblings,
although it was absent in one unaffected parent. This variant of
MITF may be the deafness disease-causing mutation in this
particular family.
Congenital causes led to HL in the second child of
the family again, although the affected parents were told that the
first child has hearing loss. These might be that the parents did
not initially receive a genetic diagnosis for the first child, and
were thus unaware of the possible inherited nature of this HL.
Therefore, the present study aimed to characterize the severity of
HL and advise on the use of hearing aids, early identification and
prenatal genetic diagnosis to parents in Yunnan.
Acknowledgements
The authors would like to thank Mrs. Yu Wang
(Beijing Myogenostics Co., Ltd., Beijing, China) for advice
regarding data analysis. We would also like to thank the Miss Yali
Guo of the Department of Otolaryngology-Head Neck Surgery, Kunming
Children's Hospital, Kunming Medical University (Kunming, China)
for the clinical data collection.
Funding
The present study was supported by funds from the
Health and Family Planning Commission of Kunming and Yunnan,
respectively [grants nos. 2015-1-S-00967, 2016-SW (Sheng)-30,
2017-SW(Yang)-02, 2016NS125 and D-201254].
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding authors upon reasonable
request.
Authors' contributions
Conceptualization of the present study was performed
by ZZ, HG, WJH and QC, ZZ, JM, YFL and MFW conducted data curation.
Acquisition of funding was carried out by ZZ and QDC. ZZ, TSZ, JXP,
YFL, MFW and LPZ conducted the investigations. Methodologies were
suggested by ZZ, QDC, LPZ, APW, LT and LJL. Project administration
was performed by HG and WJH. Software were suggested by APW, LT and
ZZ. Supervision was performed by WJH and HG. ZZ prepared the
manuscript, ZZ and HG reviewed and edited the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the ethics
committee of the Children's Hospital, Kunming Medical University;
written informed consent was obtained from participants or their
guardians.
Consent for publication
Written informed consent for publication of their
clinical details and data was obtained from participants or their
guardians.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Deafness and hearing loss fact sheet No.
300. WHO. http://www.who.int/mediacentre/factsheets/fs300/en/2015.
|
|
2
|
Li L, Lu JGM, Liu Y and Xiao YL: Common
deafness genes detection and hot mutation spots analysis in
patients with congenital non-syndromic hearing loss. Chinese
Journal of otology. 10:246–251. 2012.
|
|
3
|
Jiang L, Ling Y, Cai G and Wang R:
Research on the clinical application of DNA microarray on deafness
gene mutations. J Mol Diag Ther. 3:170–172. 2011.
|
|
4
|
Smith RJ: Clinical application of genetic
testing for deafness. Am J Med Genet A. 130A:1–12. 2004. View Article : Google Scholar
|
|
5
|
Petersen MB and Willems PJ: Non-syndromic,
autosomal-recessive deafness. Clin Genet. 69:371–392. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen PH and Yang T: Whole exome sequencing
helps diagnose Muckle-Wells syndrome in a Chinese family with
autosomal dominant hearing loss. J Shanghai Jiao Tong Univ (Med
Sci). 36:1135–1139. 2016.
|
|
7
|
Li S, Peng Q, Liao S, Li W, Ma Q and Lu X:
A reverse dot blot assay for the screening of twenty mutations in
four genes associated with NSHL in a Chinese population. PLoS One.
12:e01771962017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shearer AE, DeLuca AP, Hildebrand MS,
Taylor KR, Gurrola J II, Scherer S, Scheetz TE and Smith RJ:
Comprehensive genetic testing for hereditary hearing loss using
massively parallel sequencing. Proc Natl Acad Sci USA. 107:pp.
21104–21109. 2010; View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Choi M, Scholl UI, Ji W, Liu T, Tikhonova
IR, Zumbo P, Nayir A, Bakkaloğlu A, Ozen S, Sanjad S, et al:
Genetic diagnosis by whole exome capture and massively parallel DNA
sequencing. Proc Natl Acad Sci USA. 106:pp. 19096–19101. 2009;
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bae SH, Baek JI, Lee JD, Song MH, Kwon TJ,
Oh SK, Jeong JY, Choi JY, Lee KY and Kim UK: Genetic analysis of
auditory neuropathy spectrum disorder in the Korean population.
Gene. 522:65–69. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
TruSight One Sequencing Panel Series
Reference Guide. https://support.microsoft.com/en-us/help/17621/internet-explorer-downloadsAugust.
2017
|
|
12
|
Chun S and Fay JC: Identification of
deleterious mutations within three human genomes. Genome Res.
19:1553–1561. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang SZ, Dai P, Liu X, Kang D, Zhang X,
Yang W, Zhou C, Yang S and Yuan H: Genetic and phenotypic
heterogeneity in Chinese patients with Waardenburg syndrome type
II. PLoS One. 8:e771492013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hertwig P: Neue mutationen und
Kopplungsgruppen bei der Hausmaus. Z Induct
Abstammungs-Vererbungsl. 80:220–246. 1942.
|
|
16
|
Hemesath TJ, Steingrímsson E, McGill G,
Hansen MJ, Vaught J, Hodgkinson CA, Amheiter H, Copeland NG,
Jenkins NA and Fishe DE: Microphthalmia, a critical factor in
melanocyte development, defines a discrete transcription factor
family. Genes Dev. 8:2770–2780. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tassabehji M, Newton VE and Read AP:
Waardenburg syndrome type 2 caused by mutations in the human
microphthalmia (MITF) gene. Nat Genet. 8:251–255. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hershey CL and Fisher DE: Genomic analysis
of the Microphthalmia locus and identification of the MITF-J/Mitf-J
isoform. Gene. 347:73–82. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Steingrímsson E, Moore KJ, Lamoreux ML,
Ferré-D'Amaré AR, Burley SK, Zimring DC, Skow LC, Hodgkinson CA,
Arnheiter H, Copeland NG, et al: Molecular basis of mouse
microphthalmia (mi) mutations helps explain their developmental and
phenotypic consequences. Nat Genet. 8:256–263. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Steingrímsson E, Copeland NG and Jenkins
NA: Melanocytes and the microphthalmia transcription factor
network. Annu Rev Genet. 38:365–411. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kawakami A and Fisher DE: The master role
of microphthalmia-associated transcription factor in melanocyte and
melanoma biology. Lab Invest. 97:649–656. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen L, Guo W, Ren L, Yang M, Zhao Y, Guo
Z, Yi H, Li M, Hu Y, Long X, et al: A de novo silencer causes
elimination of MITF-M expression and profound hearing loss in pigs.
BMC Biol. 14:522016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu CC, Lee YC, Chen PJ and Hsu CJ:
Predominance of genetic diagnosis and imaging results as predictors
in determining the speech perception performance outcome after
cochlear implantation in children. Arch Pediatr Adolesc Med.
162:269–276. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sinnathuray AR, Toner JG, Clarke-Lyttle J,
Geddis A, Patterson CC and Hughes AE: Connexin 26 (GJB2)
gene-related deafness and speech intelligibility after cochlear
implantation. Otol Neurotol. 25:935–942. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen W, Yi H, Zhang L, Ji F, Yuan S, Zhang
Y, Ren L, Li J, Chen L, Guo W and Yang S: Establishing the standard
method of cochlear implant in Rongchang pig. Acta Otolaryngol.
137:503–510. 2017. View Article : Google Scholar : PubMed/NCBI
|