Introduction
Ataxia-telangiectasia (A-T; Online Mendelian
Inheritance in Man entry no. 208900) is a rare progressive
neurological disease mainly caused by mutations in the A-T mutated
(ATM) gene. A-T patients initially present with the typical
symptoms of cerebellar degeneration, telangiectasia, sensitivity to
ionizing radiation and immune defects. With time, the patients
display premature aging and stunting, while certain neurological
symptoms appear, including oculomotor apraxia, neuropathy and
nystagmus (1–4). Certain patients also exhibit gonad
retardation and growth factor deficiency (5,6). An
increased risk of malignancy and recurrent sinopulmonary infections
are two major causes of mortality (7). Of note, certain patients may exhibit
atypical symptoms, including generalized dystonia and other
unassociated cerebellar disorders (8,9). To
date, no effective treatments exist to delay or stop
neurodegeneration. However, other manifestations of A-T, such as
immunodeficiency, lung disease, hypoplasia and diabetes, can be
effectively treated (10).
ATM, as a causative gene for the A-T disorder, is
located in the autosomal region 11q22.3 containing 66 exons
(11) and encodes the ATM protein,
which has a critical role in controlling cell cycle checkpoints and
responses to DNA damage after DNA double-strand breaks caused by
oxidative damage, endogenous sources or ionizing radiation
(12). In addition, activated ATM
regulates numerous downstream proteins, including the tumor
suppressor proteins p53 and BRCA1, checkpoint kinase 2, the
checkpoint protein RAD17 and the DNA repair protein NBS1 (13). Therefore, the ATM protein is
necessary for cells to maintain genomic stability (10). To date, >600 pathogenic variants
have been reported for the ATM gene (14).
However, few studies have assessed ATM mutations in
Chinese populations, and even fewer have examined their association
with a family history of A-T (15,16).
In the present study, a Chinese pedigree affected by A-T was
subjected to genetic testing by whole-exome sequencing (WES), and a
novel ATM mutation was identified, which is likely to be associated
with the A-T syndrome.
Patients and methods
Subjects and clinical evaluation
The proband was an 11-year-old Chinese male, who was
hospitalized at the Department of Cardiothoracic Surgery of the
Children's Hospital of Fudan University (Shanghai, China) due to
pyothorax along with typical symptoms of limb and truncal ataxia in
October 2016. The proband's 10-year-old brother and his 9-year-old
sister exhibited the same symptoms of limb and truncal ataxia.
Their parents were healthy and non-consanguineous. Analysis of the
medical family history indicated that no other members of the
pedigree had presented with any A-T-like symptoms. A total of 3
healthy volunteers, including 2 25-year-old women and a 26-year-old
man, were recruited as control. Blood samples of the proband, his
two siblings and parents and controls were collected for further
analysis.
The proband and his two siblings underwent a series
of clinical examinations to evaluate the presentation of A-T,
including neurological examination, eye checks, biochemical blood
analysis [including detection of immunoglobulin, α-fetoprotein
(AFP) and carcinoembryonic antigen (CEA) levels] and brain magnetic
resonance imaging (MRI) examination.
The present study was approved by the local Ethics
Committee of Fudan University (Shanghai, China). Written informed
consent was obtained from the parents of proband and the controls
of the study subjects.
Genetic testing
Genomic DNA was extracted from the blood samples
using the TIANamp Genomic DNA Kit (DP304-03; Tiangen Biotech Co.,
Ltd., Beijing, China) according to the product protocol. Initially,
gene chip analysis of the DNA from the proband was performed by
Gemple Biotech Co., Ltd. (Shanghai, China), and subsequently, WES
was performed for the two siblings and parents of the proband, also
conducted by Gemple Biotech Co., Ltd. A whole-exome library was
constructed on the KAPA platform using the KAPA Hyper Prep kit
(KK8514 and KK8515; Roche, Basel, Switzerland). All of the exomes
were captured by a custom-designed SeqCap EZ MedExome kit
(7681330001; NimbleGen; Roche, Madison, WI, USA) and sequenced with
a HiSeq X Ten instrument (Illumina, Inc., San Diego, CA, USA). The
quality of the original data was evaluated using FASTQC (version
0.11.5; Illumina, Inc.), and the linker sequences were removed by
Cutadapt (version 1.10; http://github.com/marcelm/cutadapt/). The reads were
then aligned to the genome using the software BWA (version 0.7.15)
(17). SAMtools (version 1.3.1;
http://samtools.sourceforge.net/) was
used for format conversion of the comparison results, and the
Picard software package (http://broadinstitute.github.io/picard/) was used for
sorting and deduplication. Finally, mutation detection was
performed with GATK Haplotype Caller (version 3.6) (18). Mutation data were annotated and
categorized according to the American College of Medical Genetics
and Genomics (ACMG) guidelines for analysis of phenotype-associated
mutation sites (19).
Sanger sequencing
Sanger sequencing was performed for all 5 subjects
with an ABI 3730 XL automated sequencer (Applied Biosystems; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) to validate the
candidate variants reported by WES. Primer pairs were designed with
the online software Primer3 (http://frodo.wi.mit.edu/primer3) (forward,
5′-ACAGTGATGTGTGTTCTGAAATTGT-3′, and reverse,
5′-TCTCGAATCAGGCGCTTAAA-3′) to amplify fragments including the
variants. The ATM gene reference sequence was obtained from the
National Institutes for Biotechnology Information GenBank. The
pathogenicity of candidate variants was evaluated based on the ACMG
guidelines for analysis of phenotype-associated mutation sites
(19).
Reverse transcription-polymerase chain
reaction (RT-PCR) and complementary DNA (cDNA) sequencing
Blood was collected from all family members for mRNA
extraction. Total RNA from peripheral blood mononuclear cells of
the fresh blood was extracted with TRIzol reagent (Thermo Fisher
Scientific, Inc.). The RNA concentration/quality was determined
using a NanoDrop spectrophotometer (ND-1,000, Thermo Fisher
Scientific, Inc.). cDNA synthesis was performed with PrimeScript™
RT Master Mix (RR036A-1; Takara Biotechnology Co., Ltd., Dalian,
China) using 500 ng total RNA according to the manufacturer's
protocol. To determine the mRNA sequence of ATM, the target
fragments of the cDNA were amplified by PrimeSTAR Max DNA
Polymerase (R045; Takara Biotechnology Co., Ltd.) according to the
product protocol. The PCR primer pair used, which was designed with
the online software Primer3, was as follows: Forward,
5′-CAGTGATGTGTGTTCTGAAA-3′, and reverse,
5′-TTGTGTTGAGGCTGATACAT-3′.
The PCR thermal cycling conditions is as follows:
Activation of polymerase, 98°C, 5 min; thermal cycling, 30 cycles;
denaturation, 98°C, 10 sec; Annealing: 55°C, 15 sec; elongation,
72°C, 30 sec; extension, 72°C, 5 min; and storage, 4°C.
The Axygen® AxyPrep™ DNA Gel Extraction
kit (AP-GX-250; Axygen; Corning Inc., Corning, NY, USA) was used to
extract the target RT-PCR product separated by agarose gel
electrophoresis with a gel percentage of 4%. PCR was then used to
amplify the target fragments using the same primer pair as that
stated earlier. Subsequently, the PCR products were subjected to
direct Sanger sequencing. The cDNA nucleotide sequence analysis was
based on the GenBank sequence for wild-type ATM (NM_000051.3). The
novel variant was named following the recommendations of the Human
Genome Variation Society (http://varnomen.hgvs.org/).
Three-dimensional (3D) structure of
the ATM protein
The 3D structure of the protein was predicted by
Swiss Model (https://www.swissmodel.expasy.org/), and that of its
mutated form was predicted based on the cDNA nucleotide sequences
of the patients. The wild-type and mutated form were compared to
determine the effect of the mutation on its structure.
Results
Clinical features
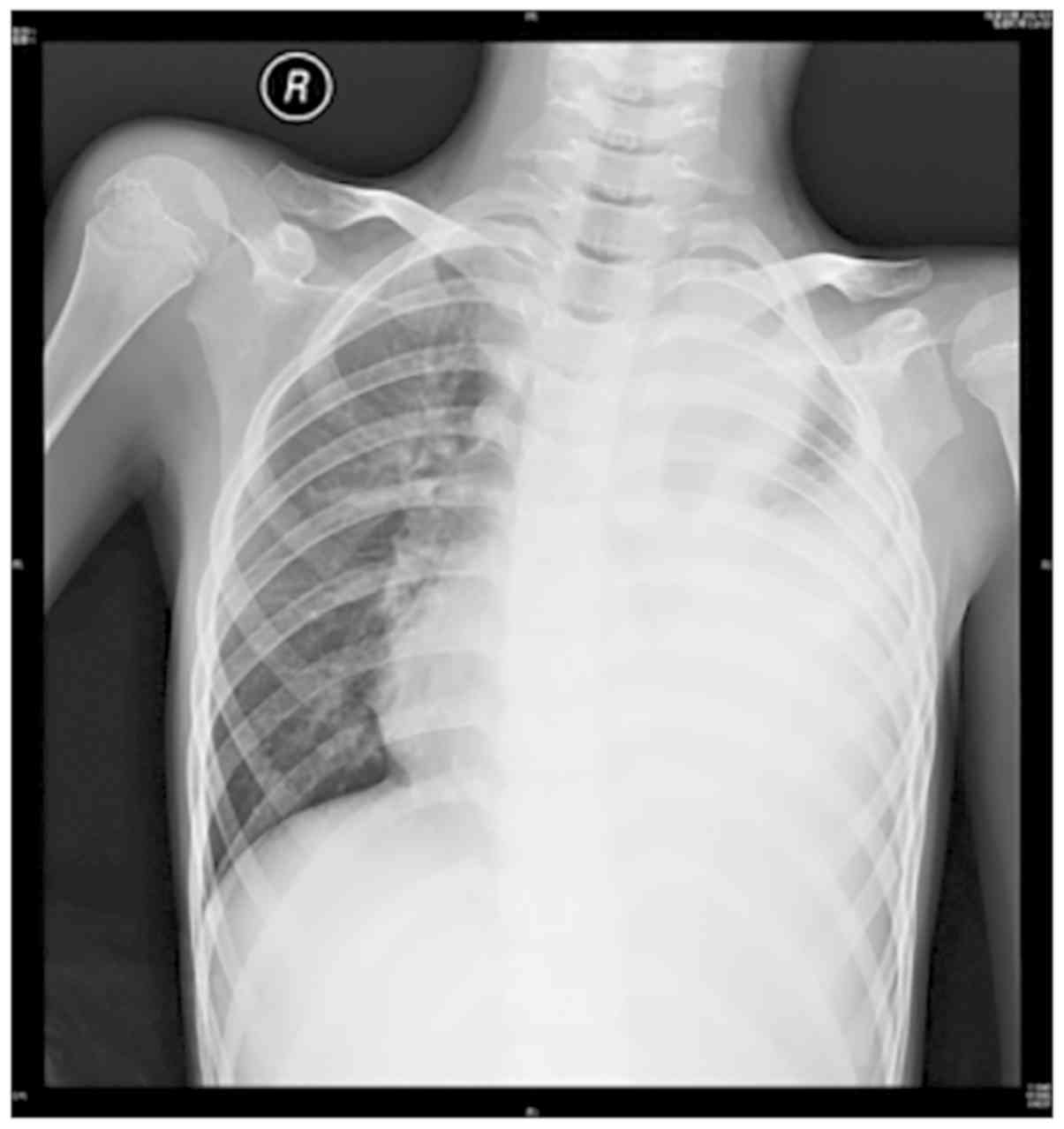
The proband (patient II:1) was diagnosed with
pneumonia and pyothorax diagnosed by chest X-ray (Fig. 1). Neurological examination revealed
that the proband exhibited progressive limb and truncal ataxia,
diagnosed based on the symptoms of difficulties in walking and
gasping, choking frequently while swallowing, delay in language
development, diminished limb strength and decrease of deep tendon
reflexes. The proband's brother (patient II:2) and sister (patient
II:3) exhibited the same symptoms of limb and truncal ataxia, but
to a less severe extent compared with the proband. The proband
initially developed symptoms of ataxia at the age of 2 years,
manifesting as gait instability and frequent falls. His two
siblings both had developed similar symptoms when they were 6.5
years old. Their parents were healthy and exhibited a normal
phenotype.
Physical examination, laboratory tests and imaging
tests were performed on the proband and his siblings, and all three
patients were found to exhibit the typical symptoms of A-T
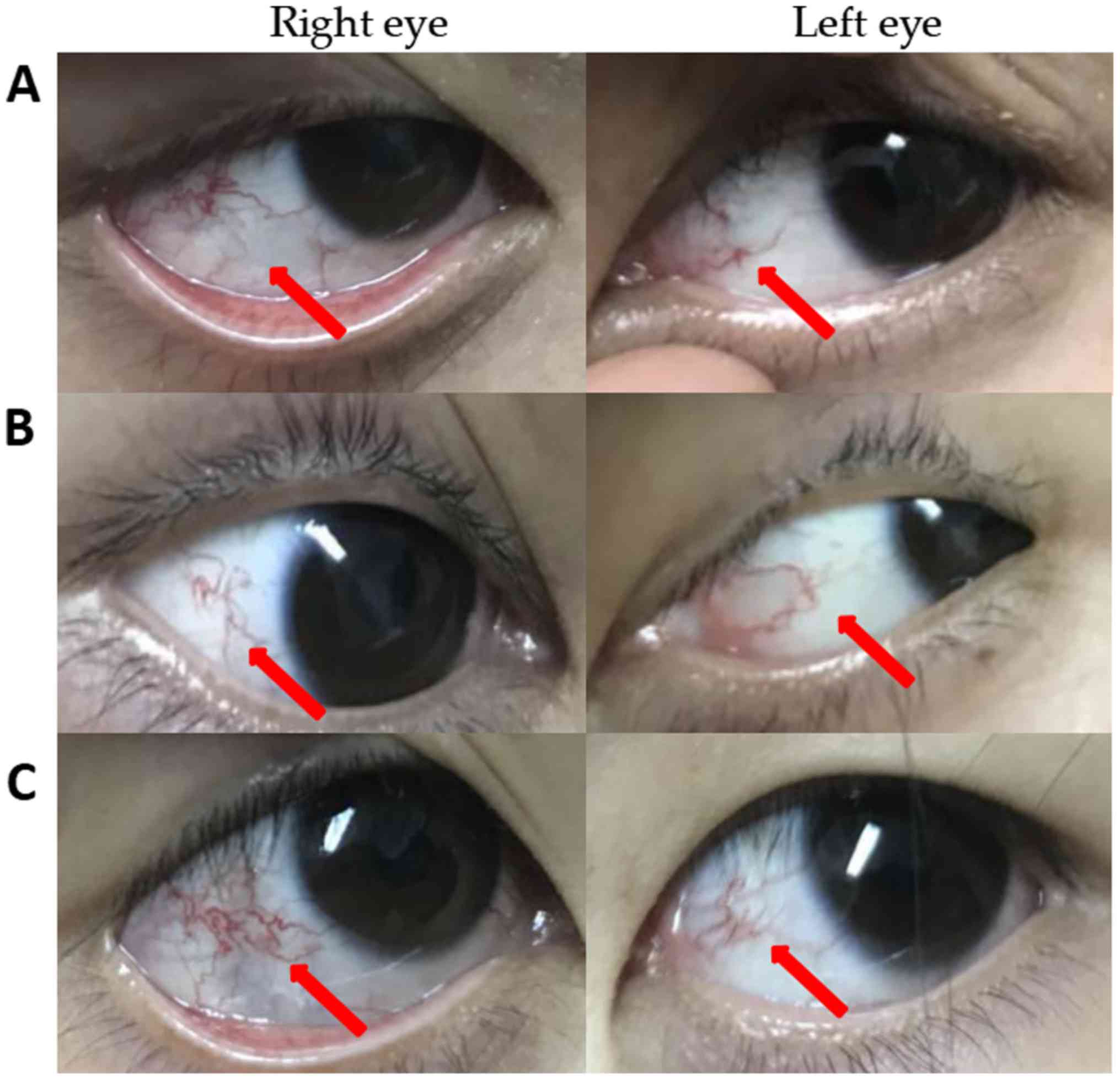
(20). Ocular telangiectasia was
observed in all three patients (Fig.
2). The laboratory test results are summarized in Table I. The serum AFP levels were
significantly increased in all three patients, whereas the serum
CEA levels were within the normal levels. All three patients
exhibited normal or slightly increased serum levels of
immunoglobulin (Ig)A, IgE and IgM. Biochemical tests indicated no
evident abnormalities, including the triglyceride, creatinine,
alkaline phosphatase and lactate dehydrogenase levels. Furthermore,
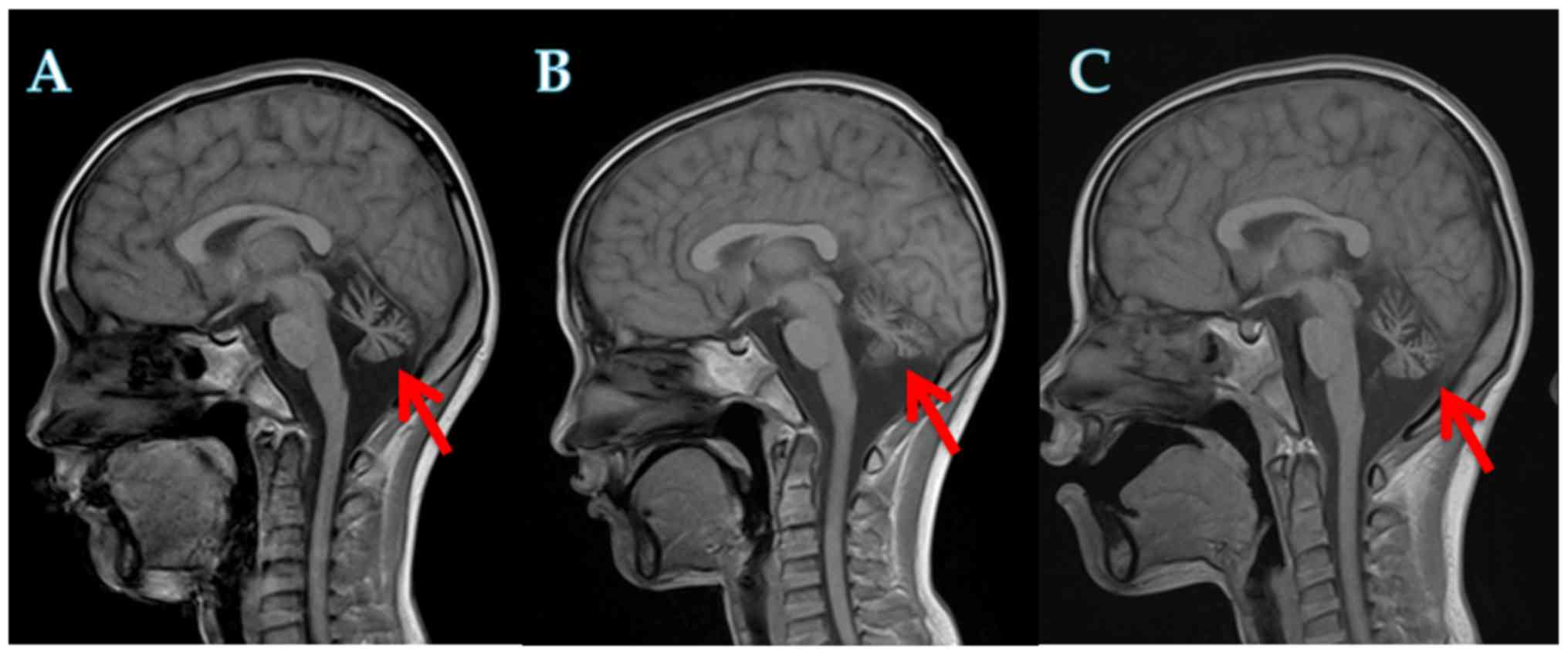
cerebellar atrophy was detected in all three patients by cranial
MRI (Fig. 3).
 | Table I.Clinical and laboratory features of
three ataxia-telangiectasia patients. |
Table I.
Clinical and laboratory features of
three ataxia-telangiectasia patients.
|
|
|
|
|
|
|
| Immunoglobulins | Biochemical
examination |
|---|
|
|
|
|
|
|
|
|
|
|---|
| Patient ID | Sex | Age (years) | Age at onset
(years) | Cerebellar
atrophy | α-fetoprotein
(ng/ml) | Carcinoembryonic
antigen (ng/ml) | IgA (g/l) | IgG (g/l) | IgM (g/l) | IgE (kU/l) | TG (mmol/l) | Cr (µmol/l) | ALP (U/l) | LDH (U/l) |
|---|
| Patient (II:1) | M | 13 | 2.5 | Atrophied | ↑ 1,836.00 | 2.8 | 1.01 | 8.4 | ↑2.69 | <2.0 | 0.92 | 34 | 286 | 229 |
| Patient (II:2) | M | 12 | 6.5 | Atrophied | ↑ 1,034.00 | 1.87 | ↓0.17 | ↑ 14.90 | ↑ 14.90 | 6.62 | 1.14 | 34 | 260 | 256 |
| Patient (II:3) | F | 11 | 6.5 | Atrophied | ↑ 629.90 | 1.86 | ↓0.08 | ↑ 14.10 | ↑ 3.57 | <2.0 | 0.82 | 34 | 242 | 255 |
| Normal value |
|
|
|
| <3.7 | <5.0 | 0.52–2.16 | 6.09–12.85 | 0.67–2.01 | <100 | 0.56–1.7 | 21–65 | 42–383 | 110–290 |
Genetic analysis
WES was performed for the proband, his two siblings
and parents against the exons and exon-intron boundaries of the ATM
gene. In total, >95% of the ATM gene was examined on the test
platform with a sensitivity of >99%. Point mutations and
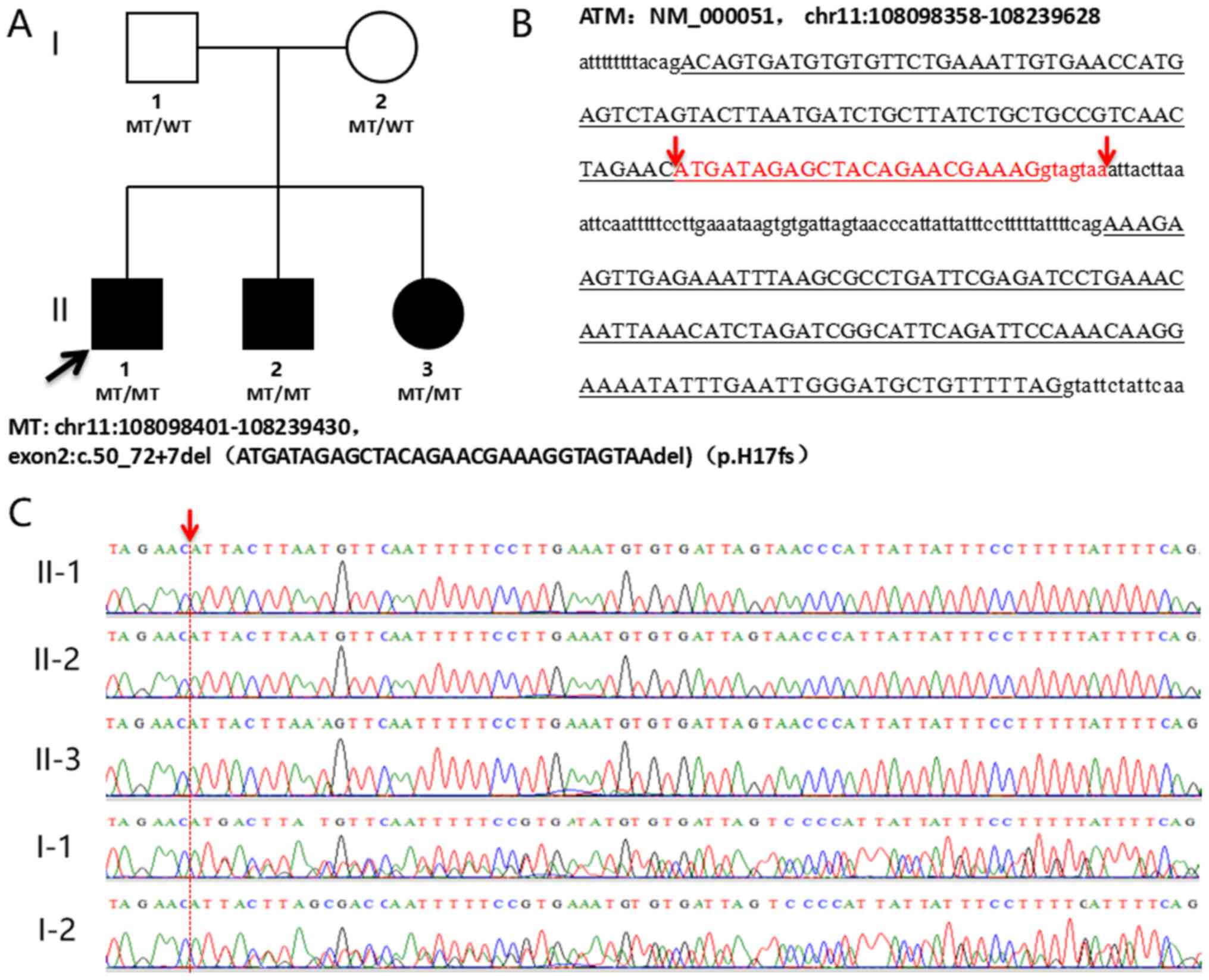
deletions were detected simultaneously. A 30-bp homozygous deletion
mutation, namely NM_000051.3:c.50_72+7del,p.Asp18_Lys24delins(23),
was detected in the three affected siblings, which was inherited
from their carrier parents, who both had a normal phenotype. This
mutation spans the exon 2 and intron 2 regions of the ATM gene,
which is predicted to cause a splicing aberration, resulting in a
30-bp deletion of exon 2 and intron 2, as well as a 71-bp insertion
of intron 2 in the splicing (Fig.
4). According to the ACMG guidelines, the detected mutation
[NM_000051.3:c.50_72+7del,p.Asp18_Lys24delins(23)] is a suspected
pathogenic mutation. However, the mutation was not included in the
1000 Genomes Project (http://www.internationalgenome.org/), the Exome
Aggregation Consortium (http://exac.broadinstitute.org/), the Human Gene
Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) or the ClinVar
database (https://www.clinicalgenome.org/data-sharing/clinvar/),
thereby confirming its novelty.
Mutation leading to splicing
abnormality
To verify the influence of the deletion variant in
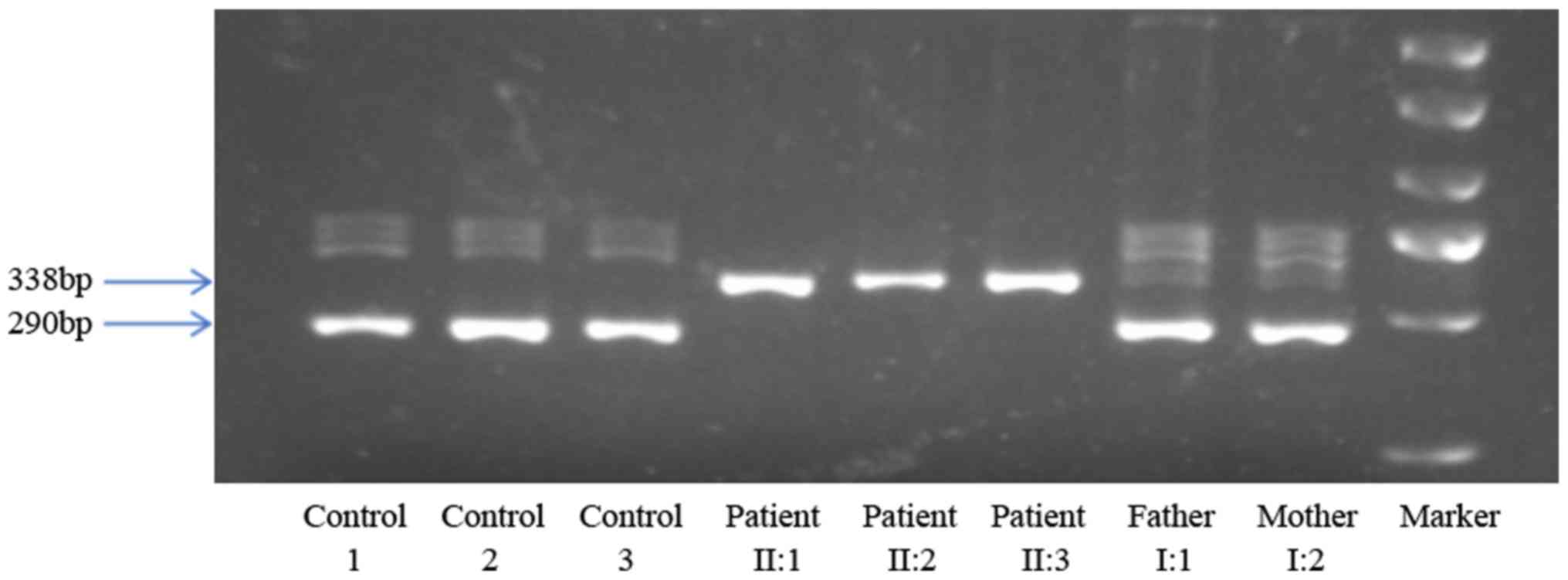
the ATM gene on the splicing, cDNA was synthesized by RT-PCR using
total RNA as the template, followed by amplification of the 290-bp
region spanning exons 2 and 3 using the cDNA as template. As
expected, the controls exhibited a 290-bp band in the gel, while
the three affected siblings had a 338-bp band, and the parents had
two bands in the gel, including a prominent one at 290 bp and a
faint one at 338 bp (Fig. 5). The
DNA of the 290- and 338-bp bands in the gel was extracted and
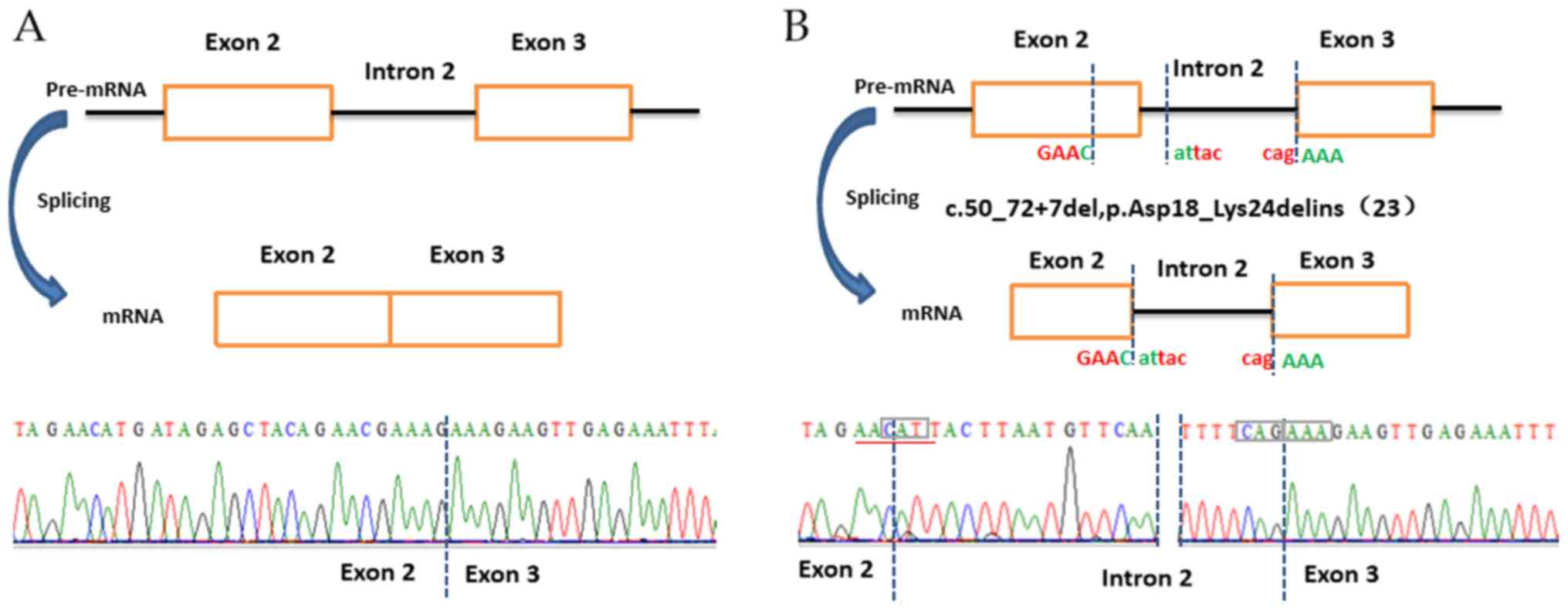
subjected to direct Sanger sequencing. As indicated in Fig. 6A, the normal controls exhibited
normal precursor mRNA (pre-mRNA) splicing; however, all the
patients exhibited splicing abnormality. Between exons 2 and 3, a
23-bp fragment of exon 2 and 7-bp fragment of intron 2 were
deleted, while a 71-bp fragment of intron 2 was inserted in the
cDNA sequence (Fig. 6B). These
results indicated that the deletion mutation in the ATM gene
influenced the pre-mRNA splicing.
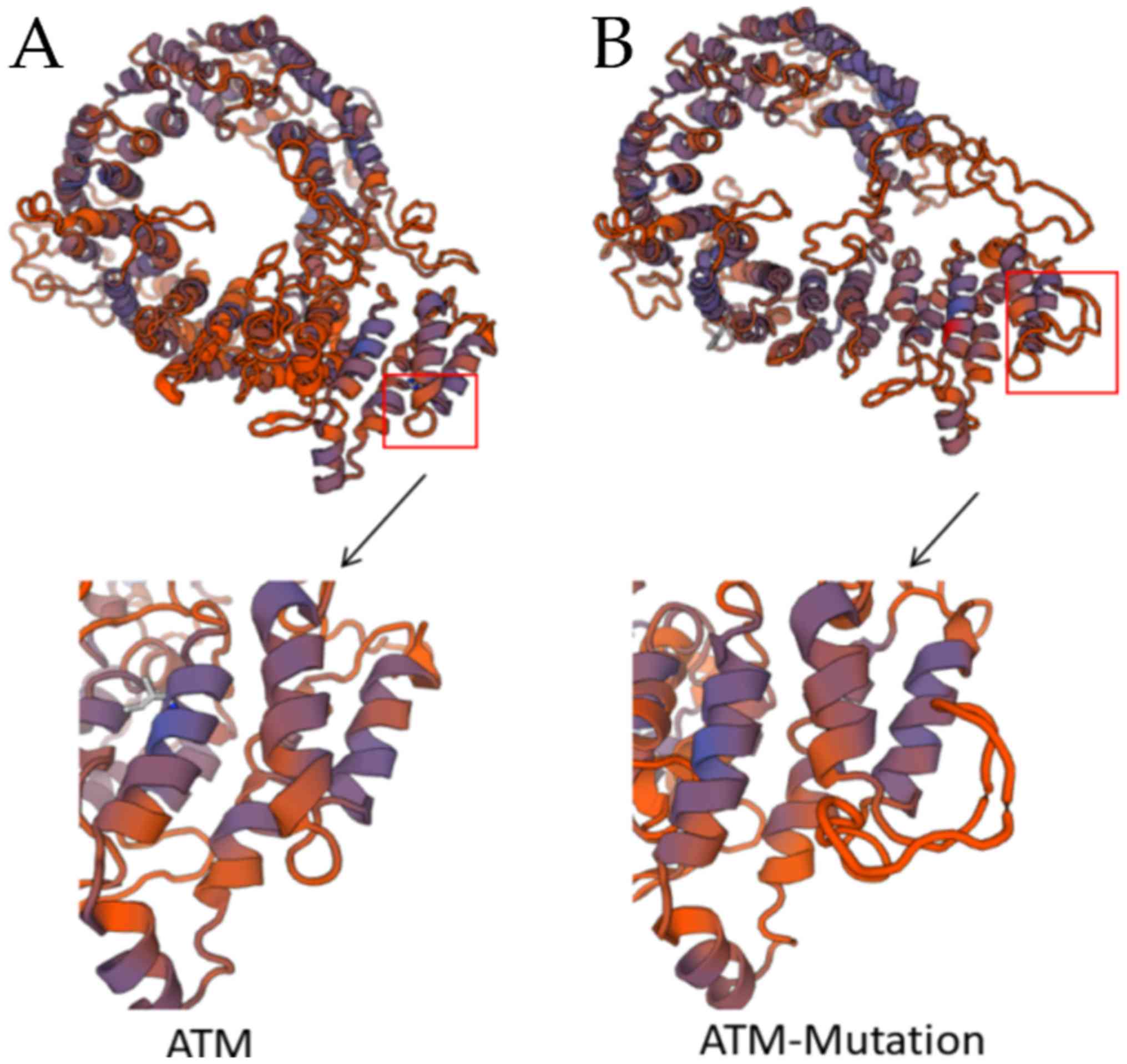
3D structure of ATM and its variant
form
Based on the cDNA sequence of patient, in which the
first two bases are identical to the normal sequence, it was found
that the deletion variant eventually leads to the insertion of 69
bases, replacing the 21 bp of the normal sequence. However, no new
termination codon was formed, which resulted in a normal sequence
of the first 17 amino acids, and an insertion of a new 23 amino
acid sequence (YLMFNFSLKCVISNPLLFPFYFQ), replacing the 7 normal
amino acids. The sequence returned to normal from the 34th amino
acid and remained normal from exon 3 onwards. The structure of the
protein predicted by Swiss Model indicated that the mutation
resulted in extra secondary structures and a negative effect on the
normal formation of tertiary structures (Fig. 7). According to the Smart
(smart.embl-heidelberg.de/) and Pfam
(http://pfam.xfam.org/) databases, the function of
this area is associated with telomere length maintenance and DNA
damage repair, which may be impaired in the deletion variant.
Discussion
The estimated incidence of A-T is between 1 in
40,000 and 1 in 100,000 live births (21). At birth, affected individuals have
no evident symptoms; however, serious symptoms usually arise within
the next years and deteriorate rapidly. The majority of A-T
patients die of malignancy or recurrent sinopulmonary infections.
Prior to the appearance of typical symptoms, the patients appear as
healthy, which often results in missed diagnosis and treatment
delay. Furthermore, when symptoms appear, it is difficult to
diagnose A-T syndrome due to insufficient knowledge and awareness
of clinicians regarding this condition, and variable clinical
manifestations. In this light, genetic testing may be a valuable
tool to improve the diagnostic efficacy for A-T.
As reported previously, A-T is caused by loss of
function of the ATM gene in nearly 75% of cases (22,23).
In the present study, a novel deletion variant was identified by
WES in a family with A-T syndrome. All three patients were
homozygous for the deletion mutation, while the two parents were
heterozygous. cDNA sequence analysis by RT-PCR and Sanger
sequencing indicated that the deletion mutation in ATM gene causes
the production of pre-mRNA, which contains parts of exon 2 and
intron 2 in between the exon 2 and 3 regions. of The parents, as
heterozygous carriers of the mutations, exhibited two bands with
uniformity in brightness in the gel of RT-PCR products, including a
normal band (290 bp) and a mutant band (338 bp); however, the
actual mutant band was faint. As intron retention in mRNAs may be
regarded as a consequence of mis-splicing (24,25),
transcripts retaining introns are often removed by
nonsense-mediated decay (NMD), or removed by nuclear retention and
exosome degradation, which may prevent the translation of
intron-retaining transcripts into potentially harmful proteins
(25,26). In the present study, the structure
of the mutant mRNA may have been unstable due to intron retention,
which may have induced an NMD-like mRNA regulation mechanism,
leading to the degradation of mutant mRNA and resulting in the
338-bp fragment being faint. Furthermore, the 3D structure of the
protein predicted by Swiss Model indicated that the likely cause of
the disease may be that the mutation affected the protein
structure, impairing its function.
In the present study, a systematic analysis of a
Chinese family including three A-T patients was performed, and a
novel pathogenic deletion variant in the ATM gene was identified
that contributes to the spectrum of known causative mutations and
phenotypes of A-T. Furthermore, the three pediatric patients
exhibited almost the same symptoms, which points to a familial
inherited disease. The parents of the patients were from two
adjacent counties in Chongqing, China, and were not consanguineous
according to their narrative. Although the parents had taken their
children to several hospitals to obtain a definite diagnosis, A-T
was not confirmed until the proband was 13 years old. A-T is so
rare that its current understanding is insufficient, while ocular
telangiectasia and cerebellar ataxia, which are the most common
clinical manifestations of this disease (27), are not exclusive clinical features
based on which A-T may be suspected. Furthermore, the variable
clinical manifestation of the disease increases the difficulty of
recognition, which emphasizes the importance of genetic testing in
the diagnosis of A-T, particularly in those cases where the
clinical symptoms are not typical. Although A-T cannot be currently
cured with any of the available treatment methods, the rapid
development of mutation-targeted therapeutic approaches may bring
hope for potential treatments for A-T patients (16). In addition, parents with a family
history of A-T wishing to conceive may be subjected to genetic
testing and prenatal genetic counseling to contribute to
eugenics.
In conclusion, a Chinese pedigree affected by A-T
was subjected to WES in the present study, revealing a novel
homozygous deletion mutation in ATM [namely,
NM_000051.3:c.50_72+7del, p.Asp18_Lys24delins (23)] in three affected siblings, which
was inherited from their carrier parents who exhibited a normal
phenotype. The deletion mutation in the ATM gene affected pre-mRNA
splicing and resulted in a change of the 3D structure of the
protein, which may have impaired the functions associated with
telomere-length maintenance and DNA damage repair. In the Chinese
non-consanguineous family, the three affected children all carried
the same homozygous mutation and the parents were heterozygous,
which is rare and has never been reported in China previously.
Furthermore, the present study contributed to the spectrum of known
pathogenic variants of ATM, expanded the current understanding on
the A-T syndrome and highlighted the importance of genetic testing
in the diagnosis of this disease.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Key
Research and Development Program of China (grant no.
2016YFC1000500), the National Natural Science Foundation of China
(grant nos. 81570282 and 81570283), and the Science and Research
Foundation of Shanghai Municipal Commission of Health and Family
Planning for Young Scientists (grant no. 20144Y0057).
Availability of data and materials
All data generated and analyzed during the present
study are included in this published article.
Authors' contributions
GH and WS conceived and designed the study. WC, GC,
SZ and BJ were responsible for the acquisition of patient
information and communication with the patients' families. SL
collected clinical data from the A-T family. HH performed molecular
genetics experiments on the family and the controls. HH drafted the
manuscript, which was edited and revised by WC and WS.
Ethics approval and consent to
participate
Informed consent for this investigation was obtained
from all participating A-T patients and parents, and the principles
outlined in the Declaration of Helsinki were followed. The study
was conducted in agreement with the Ethical Committee of the Center
for Medical Genetics, Children's Hospital of Fudan University
(Shanghai, China).
Patient consent for publication
The publication of information from these three A-T
patients have their support and informed consent.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Levy A and Lang AE: Ataxia-telangiectasia:
A review of movement disorders, clinical features, and genotype
correlations-Addendum. Mov Disord. 33:13722018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gatti RA, Shaked R, Wei S, Koyama M,
Salser W and Silver J: DNA polymorphism in the human Thy-1 gene.
Hum Immunol. 22:145–150. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Savitsky K, Bar-Shira A, Gilad S, Rotman
G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, et al: A
single ataxia telangiectasia gene with a product similar to PI-3
kinase. Science. 268:1749–1753. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lavin MF and Shiloh Y: The genetic defect
in ataxia-telangiectasia. Annu Rev Immunol. 15:177–202. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schubert R, Reichenbach J and Zielen S:
Growth factor deficiency in patients with ataxia telangiectasia.
Clin Exp Immunol. 140:517–519. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kieslich M, Hoche F, Reichenbach J,
Weidauer S, Porto L, Vlaho S, Schubert R and Zielen S:
Extracerebellar MRI-lesions in ataxia telangiectasia go along with
deficiency of the GH/IGF-1 axis, markedly reduced body weight, high
ataxia scores and advanced age. Cerebellum. 9:190–197. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Micol R, Ben Slama L, Suarez F, Le Mignot
L, Beauté J, Mahlaoui N, Dubois d'Enghien C, Laugé A, Hall J,
Couturier J, et al: Morbidity and mortality from
ataxia-telangiectasia are associated with ATM genotype. J Allergy
Clin Immunol. 128:382–389.e1. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kuhm C, Gallenmüller C, Dörk T, Menzel M,
Biskup S and Klopstock T: Novel ATM mutation in a German patient
presenting as generalized dystonia without classical signs of
ataxia-telangiectasia. J Neurol. 262:768–770. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Piane M, Molinaro A, Soresina A, Costa S,
Maffeis M, Germani A, Pinelli L, Meschini R, Plebani A, Chessa L
and Micheli R: Novel compound heterozygous mutations in a child
with Ataxia-Telangiectasia showing unrelated cerebellar disorders.
J Neurol Sci. 371:48–53. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rothblum-Oviatt C, Wright J, Lefton-Greif
MA, McGrath-Morrow SA, Crawford TO and Lederman HM: Ataxia
telangiectasia: A review. Orphanet J Rare Dis. 11:1592016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Platzer M, Rotman G, Bauer D, Uziel T,
Savitsky K, Bar-Shira A, Gilad S, Shiloh Y and Rosenthal A:
Ataxia-telangiectasia locus: Sequence analysis of 184 kb of human
genomic DNA containing the entire ATM gene. Genome Res. 7:592–605.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ditch S and Paull TT: The ATM protein
kinase and cellular redox signaling: Beyond the DNA damage
response. Trends Biochem Sci. 37:15–22. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Foray N, Marot D, Gabriel A,
Randrianarison V, Carr AM, Perricaudet M, Ashworth A and Jeggo P: A
subset of ATM- and ATR-dependent phosphorylation events requires
the BRCA1 protein. Embo J. 22:2860–2871. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Telatar M, Teraoka S, Wang Z, Chun HH,
Liang T, Castellvi-Bel S, Udar N, Borresen-Dale AL, Chessa L,
Bernatowska-Matuszkiewicz E, et al: Ataxia-telangiectasia:
Identification and detection of founder-effect mutations in the ATM
gene in ethnic populations. Am J Hum Genet. 62:86–97. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang H, Tang B, Xia K, Hu Z, Shen L, Tang
J, Zhao G, Zhang Y, Cai F, Pan Q, et al: Mutation analysis of the
ATM gene in two Chinese patients with ataxia telangiectasia. J
Neurol Sci. 241:1–6. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang Y, Yang L, Wang J, Yang F, Xiao Y,
Xia R, Yuan X and Yan M: Twelve novel Atm mutations identified in
Chinese ataxia telangiectasia patients. Neuromolecular Med.
15:536–540. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The Genome Analysis Toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Teive HA, Moro A, Moscovich M, Arruda WO,
Munhoz RP, Raskin S and Ashizawa T: Ataxia-telangiectasia-A
historical review and a proposal for a new designation: ATM
syndrome. J Neurol Sci. 355:3–6. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tabatabaiefar MA, Alipour P, Pourahmadiyan
A, Fattahi N, Shariati L, Golchin N and Mohammadi-Asl J: A novel
pathogenic variant in an Iranian Ataxia telangiectasia family
revealed by next-generation sequencing followed by in silico
analysis. J Neurol Sci. 379:212–216. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Concannon P and Gatti RA: Diversity of ATM
gene mutations detected in patients with ataxia-telangiectasia. Hum
Mutat. 10:100–107. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jacquemin V, Rieunier G, Jacob S,
Bellanger D, D'Enghien CD, Laugé A, Stoppa-Lyonnet D and Stern MH:
Underexpression and abnormal localization of ATM products in ataxia
telangiectasia patients bearing ATM missense mutations. Eur J Hum
Genet. 20:305–312. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Roy SW and Irimia M: Intron mis-splicing:
No alternative? Genome Biol. 9:2082008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jaillon O, Bouhouche K, Gout JF, Aury JM,
Noel B, Saudemont B, Nowacki M, Serrano V, Porcel BM, Ségurens B,
et al: Translational control of intron splicing in eukaryotes.
Nature. 451:359–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gudipati RK, Xu Z, Lebreton A, Séraphin B,
Steinmetz LM, Jacquier A and Libri D: Extensive degradation of RNA
precursors by the exosome in wild-type cells. MolCell. 48:409–421.
2012.
|
|
27
|
Greenberger S, Berkun Y, Ben-Zeev B, Levi
YB, Barziliai A and Nissenkorn A: Dermatologic manifestations of
ataxia-telangiectasia syndrome. J Am Acad Dermatol. 68:932–936.
2013. View Article : Google Scholar : PubMed/NCBI
|