Introduction
Acute lung injury (ALI) or its severe form, acute
respiratory distress syndrome (ARDS), is characterized by an
excessive and uncontrolled inflammatory response, which results in
increased permeability of the alveolar-capillary barrier, alveolar
flooding and acute respiratory failure (1,2).
Type II alveolar epithelial cells (AEC II), the progenitor cells in
the corners of alveoli, are thought to play a central role in the
pathogenesis of ALI by synthesizing, secreting and reutilizing
surfactant (3). In addition, the
apoptosis of AEC II directly accelerates the progression of ALI,
which is the leading cause of mortality in patients with ARDS
(4–6).
The nephroblastoma overexpressed protein (NOV/CCN3),
is a cysteine-rich protein that belongs to the CCN (Cyr61, CTGF,
Nov) family of matricellular proteins with a variety of functions
(7,8). CCN3 has been previously shown to be
involved in regulating a variety of chronic inflammatory diseases,
such as atherosclerosis, rheumatoid arthritis and liver disease
(7,9,10).
In addition, studies have also shown that CCN proteins are key
signaling and regulatory molecules involved in the pathophysiology
of various lung diseases, including lung cancer, chronic
obstructive pulmonary disease and ventilator-induced lung injury
(11–15). By integrating the proteomic
profiles of inflammatory mediators with clinical informatics, our
previous study (16) indicated
that the plasma levels of CCN3 and other inflammatory mediators
were significantly increased in patients with severe
pneumonia-induced ARDS compared to the healthy controls. Thus, we
hypothesized that CCN3 could play a role in the pathophysiology of
ALI/ARDS.
Proteins involved in transcriptional regulation,
such as nuclear factor (NF)-κB and transforming growth factor
(TGF)-β, have been implicated in the development and progression of
ALI and ARDS (17–21). Human A549 alveolar epithelial cells
have been widely used as an epithelial cell injury model to examine
lipopolysaccharide (LPS)-induced acute lung inflammatory response
(22–28). The objective of this study was to
reveal the potential role and underlying mechanism of CCN3 in lung
dysfunction from the perspective of inflammation and apoptosis
using siRNA-mediated transfection approach.
Materials and methods
Reagents and chemicals
Human lung alveolar type II epithelial A549 cells
were obtained from the Cell Bank of the Chinese Academy of Science
(Shanghai, China). LPS (Sigma-Aldrich; Merck KGaA), anti-TGF-β1
antibody (cat. no. ab64715, Abcam), ALK5 inhibitor (TP0427736; cat.
no. S8700, Selleck Chemicals), pyrrolidine dithiocarbamate (PDTC;
cat. no. S363302, Selleck Chemicals) and immunohistochemistry
reagents and kits (Beyotime, China) were used in this study. Other
reagents and chemicals were obtained from Western Biotechnology
(China).
Lung alveolar epithelial cell
culture
Human A549 cells were cultured in F12K medium
(Gibco; Thermo Fisher Scientific, Inc.), containing 10% FBS, 100
U/ml penicillin, and 100 mg/ml streptomycin, in a 37°C/5%
CO2 atmosphere. The medium was routinely changed every 3
days to remove non-adherent cells.
siRNA transfection
Cells were transfected with 100 nM negative control
(NC)-siRNA or CCN3-siRNA (Invitrogen; Thermo Fisher Scientific,
Inc.) using Lipofectamine 2000 (cat. no. 11668019; Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions (29).
Enzyme-linked immunosorbent assay
(ELISA)
The levels of tumor necrosis factor (TNF)-α, IL-1β
and TGF-β1 in the supernatant of cultured cells were analyzed using
sandwich ELISA kits (cat. nos. F02810, F01220 and F02750; Bio-Tek
Inc., USA), respectively, according to the manufacturer's
instructions. The optical density of each well was assayed at 450
nm on a microplate reader.
Flow cytometric analysis
BD Pharmingen™ FITC Annexin V Apoptosis Detection
Kit I (cat. no. 556547; BD Biosciences) was used to quantify
apoptotic cells according to the manufacturer's instructions. After
the treatments, cells were harvested, trypsinized, and rinsed twice
with PBS. The suspended cells were incubated with 5 µl Annexin
V-FITC solution and 5 µl propidium iodide (PI) solution at 4°C for
15 min. The cells were then suspended in 400 µl binding buffer and
subjected to flow cytometry (BD Biosciences), and the rates of
apoptotic and necrotic cells were determined using CytExpert
2.3.0.84 (Beckman Coulter, Inc.).
Real-time quantitative PCR (qPCR)
Total RNA was extracted from A549 cells using Trizol
reagent (Beyotime, China), and then used for first-strand cDNA
synthesis. qPCR was performed using an Applied Biosystems™ SYBR
Green I Real-Time PCR Master Mix system (Funglyn Biotech, Canada).
The amplification program was set to 94°C for 4 min, 35 cycles of
94°C for 20 sec and 60°C for 30 sec, and 72°C for 30 sec as a final
elongation step. The gene expression level was normalized to GAPDH
as the internal gene using the 2−ΔΔCq method (30). The primer sequences used in this
study are shown in Table I.
 | Table I.Sequences of primers used for reverse
transcription-quantitative PCR. |
Table I.
Sequences of primers used for reverse
transcription-quantitative PCR.
| Genes | Primer sequences
(5′-3′) |
|---|
| CCN3 | Forward
GGAGGATTCACTGGGAGGC |
| (153 bp) | Reverse
ATTGACGGTTCCTATTGGTGAC |
| Bcl-2 | Forward
AGGGACGGGGTGAACTGG |
| (175 bp) | Reverse
CTACCCAGCCTCCGTTATCC |
| TGF-β1 | Forward
CGTGGAGGGGAAATTGAGG |
| (185 bp) | Reverse
GCCATGAGAAGCAGGAAAGG |
| GAPDH | Forward
ATCCCATCACCATCTTCCAGG |
| (146 bp) | Reverse
GATGACCCTTTTGGCTCCC |
Western blot analysis
A549 cells were seeded at a density of
1×106 cells with 0.1 ml RIPA buffer. After passaging and
centrifugation (12,000 × g for 15 min), the supernatants were
collected and protein concentrations were assessed using the
Bradford assay. An equal amount of protein (20 µg) was loaded onto
10% SDS-PAGE gels. Proteins were resolved through SDS-PAGE and
transferred to polyvinylidene difluoride (PVDF) membranes
(Bio-Rad). Nonspecific sites were blocked with 5% nonfat milk at
room temperature for 2 h. The blots were incubated with antibodies
against Bcl-2 (dilution 1:500, cat. no. ab182858; Abcam), caspase-3
(dilution 1:500, cat. no. ab184787), TGF-β receptor (R)II (dilution
1:500, cat. no. ab113670, Abcam), p-Smad2/3 [dilution 1:500, cat.
no. 8828S, Cell Signaling Technology, Inc. (CST)], CCN3 (dilution
1:500; cat. no. ab191425; Abcam) and β-actin (dilution 1:1,000;
cat. no. ab8226; Abcam) at room temperature for 1.5 h or 4°C
overnight. After washing with TBST three times, the blots were
incubated with the secondary goat anti-rat IgG antibody (dilution
1:1,000; cat. no. AP156P; Sigma-Aldrich; Merck KGaA) at room
temperature for 1.5 h, and visualized using an enhanced
chemiluminescence (ECL) kit (cat. no. 34580; Thermo Fisher
Scientific, Inc.) in the ImageQuant Tanon-4200 system (Tanon
Science and Technology Co., Ltd., Shanghai, China). The rat
anti-β-actin Ab was used as an internal control for western
blotting. The densities of the detected bands were quantified in
triplicate using Labworks™ Analysis Software (UVP, LLC,
USA).
Immunofluorescence staining
Cells were grown on 12-mm glass coverslips. After
transfection with CCN3-siRNA or NC-siRNA, the cells were fixed in
4% paraformaldehyde at room temperature for 15 min, permeabilized
in PBS with 0.5% Triton X-100 for 15 min, and blocked with 6% goat
serum for 30 min. Next, the cells were incubated with the primary
NF-κB p65 antibody (dilution 1:100, cat. no. ab32536, Abcam)
solution at 4°C overnight. After washing, the cells were incubated
with the secondary antibody, goat anti-rabbit IgG (Cy3; dilution
1:800, cat. no. ab6939, Abcam) at room temperature for 30 min, and
DAPI was added for nuclear staining for 5 min. Finally, the cells
were visualized using a confocal laser scanning microscope (Leica,
Germany). The data are presented as relative fluorescence
intensity.
Statistical analysis
Statistical analysis was performed with Statistical
Product and Service Solutions (SPSS) v24.0 (SPSS, Inc.) and Prism 6
(GraphPad Software). Data are presented as means ± SEM. Statistical
significance was assessed with Student's t-test or one-way ANOVA
with Bonferroni significant analysis. Results were considered
statistically significant when P<0.05.
Results
Effect of LPS on the expression level
of CCN3 in human lung alveolar epithelial cells
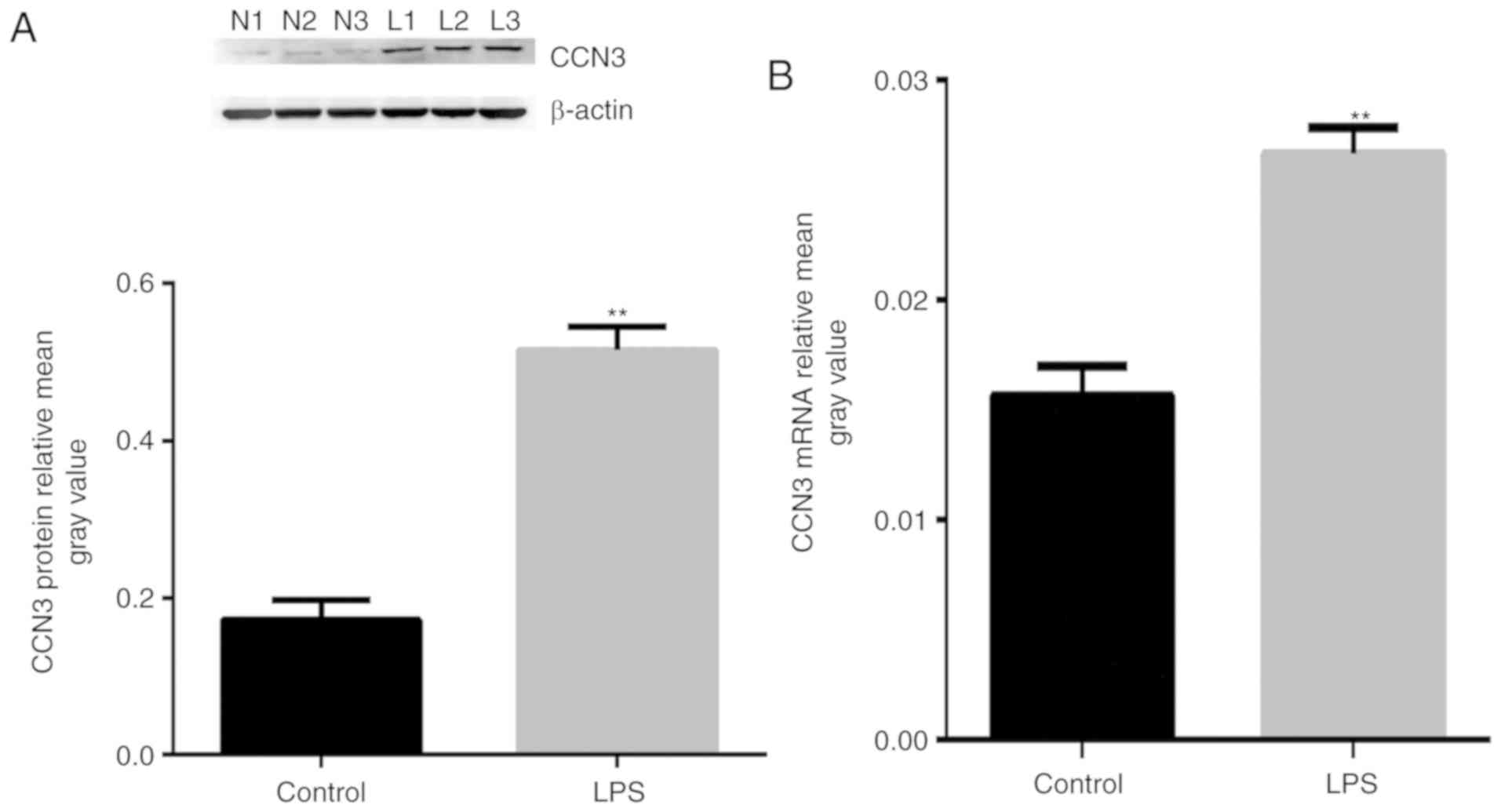
To assess the effects of LPS on CCN3 expression, we
analyzed mRNA expression by qPCR and protein expression by western
blot analysis. The results revealed that the levels of CCN3 mRNA
and CCN3 protein were significantly upregulated following treatment
with 0.1 µg/ml LPS for 12 h (Fig. 1A
and B) (P<0.01). Our findings suggest that CCN3 is
associated with LPS-induced lung injury.
Reduction of inflammatory cytokines by
CCN3-siRNA
To assess the function of CCN3 in the regulation of
inflammatory cytokines, we inhibited CCN3 expression by siRNA and
measured the levels of TNF-α, IL-1β and TGF-β1 by ELISA. As shown
in Fig. 2A and B, it was
demonstrated that inhibition of CCN3 by siRNA significantly
downregulated the levels of inflammatory cytokines, such as IL-1β
(P<0.01) and TGF-β1 (P<0.05), compared with the vehicle
control (NC). Similar effects were observed after LPS treatment.
Surprisingly, there were no significant changes in the levels of
TNF-α after CCN- siRNA treatment compared with both the NC group
and following LPS treatment (Fig.
2C).
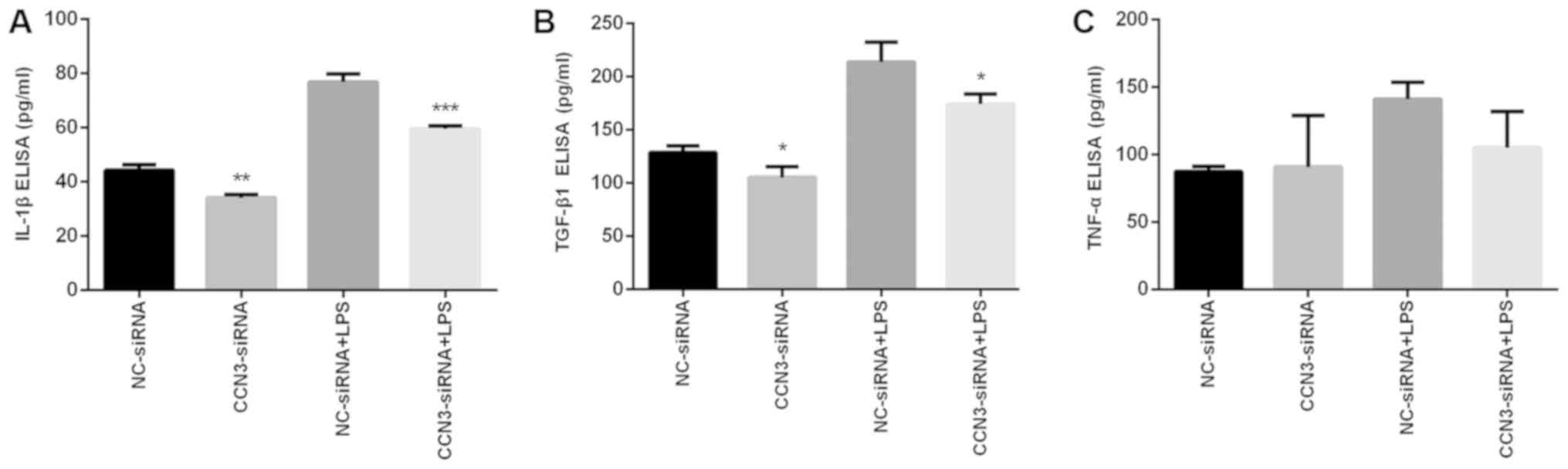 | Figure 2.Effects of CCN3 siRNA treatment on
the expression of inflammatory cytokines. A549 cells were treated
with NC-siRNA or CCN3-siRNA for 36 h, and then incubated with or
without 0.1 µg/ml LPS for an additional 12 h. The expression levels
of (A) IL-1β, (B) TGF-β1 and (C) TNF-α were assessed in
vitro with ELISA. Data are presented as means ± SEM.
*P<0.05, **P<0.01, ***P<0.001 vs. respective siRNA-NC.
NC-siRNA, siRNA-negative control group; CCN3-siRNA, CCN3
siRNA-transfected group; LPS, lipopolysaccharide; CCN3,
nephroblastoma overexpressed (also known as NOV); IL, interleukin;
TGF, transforming growth factor; TNF, tumor necrosis factor. |
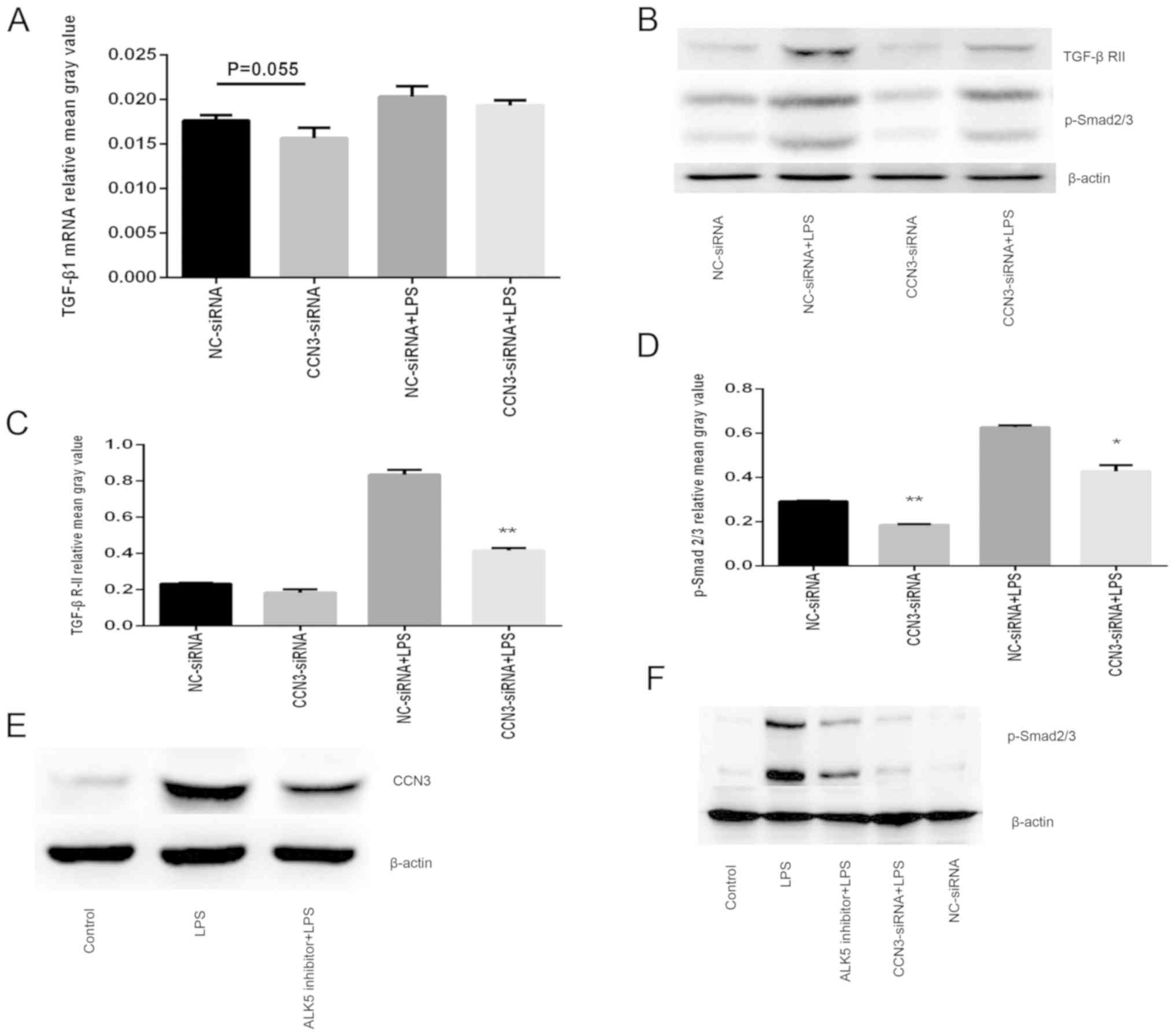
Anti-inflammatory activity of
CCN3-siRNA through modulation of TGF-β/p-Smad signaling
Activation of TGF-β though interaction with
receptor-regulated Smad (Smad2/3) signaling plays a
pro-inflammatory role in the resolution of lung alveolar epithelial
cell injury (18,31). We next sought to explore whether
the CCN3-induced pro-inflammatory effects are mediated through the
TGF-β/p-Smad signaling pathway. To investigate this, A549 cells
were transfected with CCN3-siRNA for 36 h and then stimulated with
or without LPS for 12 h. The mRNA level of TGF-β1, and the protein
levels of TGF-β RII and p-Smad2/3 were detected by qPCR and western
blot analysis, respectively. We found that knockdown of CCN3 by
siRNA largely downregulated the protein levels of TGF-β RII and
p-Smad2/3 (Fig. 3B-D). However,
the mRNA level of TGF-β1 was slightly decreased, without
statistical significance (P=0.055) (Fig. 3A). In addition, we confirmed that
pretreatment of the cells with TP0427736 (an ALK5 inhibitor), which
inhibits the TGF-β/p-Smad signaling pathway, greatly prevented the
overexpression of CCN3 induced by LPS treatment, while knockdown of
CCN3 by siRNA effectively attenuated the LPS-induced p-Smad2/3
expression (Fig. 3E and F). To
conclude, the anti-inflammatory activity of CCN3-siRNA in
LPS-induced lung injury is modulated by TGF-β/p-Smad signaling.
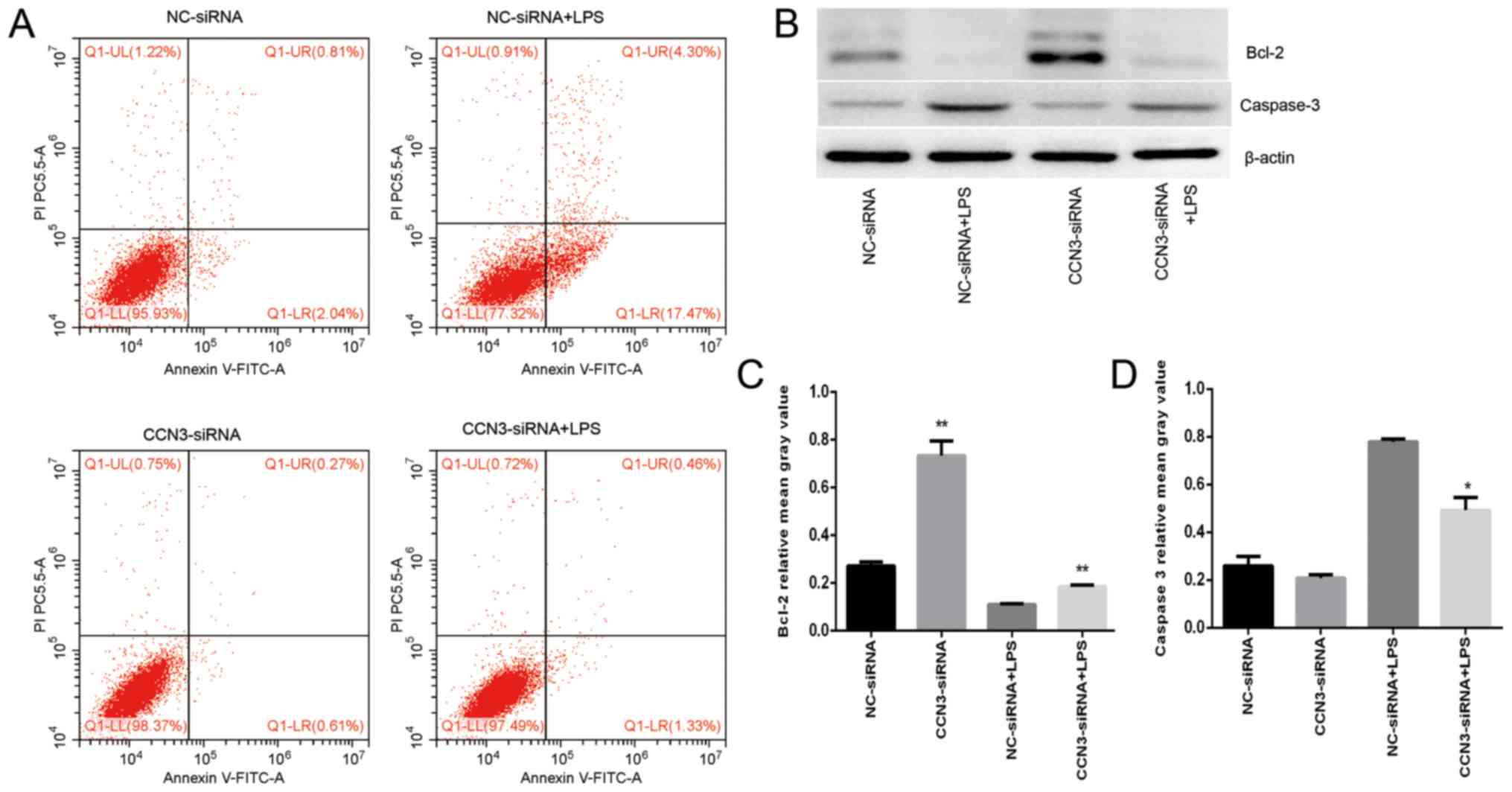
Suppression of CCN3 inhibits apoptosis
in A549 cells through the Bcl-2/caspase-3 pathway
Lung epithelial cell apoptosis is increased after
LPS treatment (24). Bcl-2 is an
important anti-apoptotic factor, and decreased expression of Bcl-2
can lead to the activation of caspase-3 and apoptosis (32). To assess the effect of CCN3 on
apoptosis, we performed flow cytometry using an apoptosis detection
kit. Our observations indicated that the sum of the proportions of
early apoptosis and late apoptosis was significantly decreased in
the CCN3 knockdown group with or without LPS treatment, but
increased after LPS treatment compared with the negative group
(P<0.001; Fig. 4A). In
addition, the western blot assays revealed that silencing of CCN3
expression upregulated Bcl-2 protein levels and downregulated the
expression of caspase-3 protein, also after LPS treatment (Fig. 4B-D). Our data suggest that CCN3 is
associated with the apoptosis of lung epithelial cells.
CCN3-siRNA knockdown reduces the
activation of the NF-κB signaling pathway
A previous study demonstrated that LPS could cause
the activation of NF-κB, which is involved in the process of
apoptosis in ALI/ARDS (33). In
order to establish whether the CCN3-induced pro-apoptotic effects
are mediated through the NF-κB signaling pathway, we used confocal
microscopy to analyze the localization of NF-κB p65 and western
blot analysis of cytoplasmic and nuclear NF-κB p65 expression
levels to establish whether nuclear translocation activation
occurs. The results demonstrated that activation of NF-κB was
largely reduced in the CCN3 knockdown group, whereas it was greatly
stimulated after LPS exposure (Fig.
5A). As shown in Fig. 5B,
pretreatment of the cells with PDTC, which inhibits the NF-κB
signaling pathway, significantly attenuated the overexpression of
CCN3 induced by LPS treatment. Meanwhile, the protein expression
levels of NF-κB p65 in the nucleus following pretreatment with PDTC
or CCN3 knockdown, were significantly decreased compared to those
without pretreatment, namely the LPS-treated only group
(P<0.001, Fig. 5C). Therefore,
we suggest that CCN3 caused activation of NF-κB in A549 cells by
the promotion of nuclear translocation of NF-κB p65.
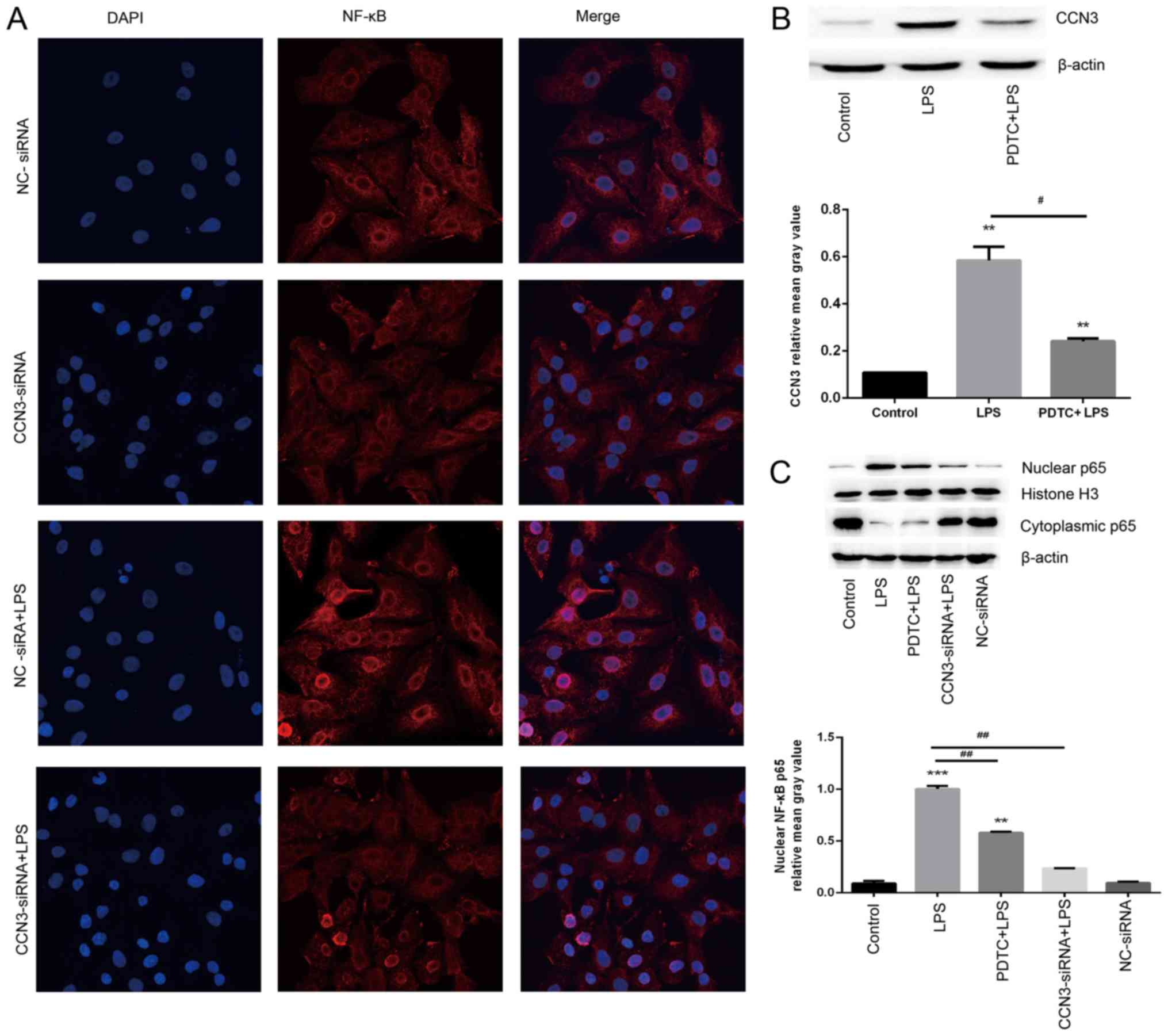 | Figure 5.CCN3-siRNA knockdown inhibits the
LPS-induced NF-κB signaling pathway. (A) Confocal microscopy of
NF-κB nuclear translocation in cultured A549 cells. Magnification
×600. DAPI (blue), NF-κB p65 (red), in A549 cells challenged with
LPS. (B and C) After 36 h of CCN3-siRNA transfection or not, cells
were pretreated with PDTC (10 µM) for 30 min, followed by
stimulation with LPS (0.1 µg/ml) for 12 h. The protein levels of
(B) CCN3 and (C) NF-κB p65 in the cytoplasm and nucleus were
assessed by western blot analysis. **P<0.01, ***P<0.001 vs.
control group; #P<0.05, ##P<0.01 vs.
LPS group. NC-siRNA, siRNA-negative control group; CCN3-siRNA, CCN3
siRNA-transfected group; CCN3, nephroblastoma overexpressed (also
known as NOV); LPS, lipopolysaccharide; NF-κB, nuclear
factor-κB. |
Discussion
Acute lung injury (ALI)/acute respiratory distress
syndrome (ARDS) is a highly refractory disease, and its complex
pathology in lung epithelial cells is not fully understood. In the
present study, we aimed to identify a potential signaling pathway
to target in order to prevent or treat the disease. This study
demonstrated that nephroblastoma overexpressed (NOV; also known as
CCN3) is critical for LPS-induced lung alveolar epithelial cell
injury and apoptosis. Firstly, we investigated the presence of CCN3
using an in vitro cell culture ALI model. We found that CCN3
mRNA and protein expression were significantly increased after LPS
treatment. These observations confirmed that LPS treatment strongly
induced the expression of CCN3 in lung alveolar epithelial cells.
This was consistent with a previous study (16), revealing elevated CCN3/NOV levels
as a potential indicator for lung injury severity.
However, the precise biological role, mechanism of
action and physiological function of CCN3 proteins in ARDS has
remained elusive until recently. Kular et al (7) indicated that CCN3 expression in
vitro is finely regulated by diverse inflammatory mediators and
cytokines, such as TNF-α, IL-1β and TGF-β. In this study, an in
vitro siRNA approach showed that inhibition of CCN3
significantly attenuated the expression levels of pro-inflammatory
cytokines (IL-1β and TGF-β1), which have been linked to the
initiation and amplification of the inflammatory response in
ALI/ARDS (2,34). Surprisingly, we found no
significant changes in the expression level of TNF-α after
transfection with CCN3-siRNA. This could be explained by the
insensitivity of TNF-α production by the alveolar epithelial cells
in our study. TNF-α was possibly not involved in this process. In
addition, recent studies have revealed that CCNs and TNF-α are
co-expressed at sites of inflammation (35). It is therefore tempting to
speculate that CCNs may help to counterbalance the inflammatory
effects of TNF-α. A previous study has shown that A549 cells
release IL-1β and TNF-α after LPS stimulation though autocrine
modes (36). This exerts a strong
and synergic induction signal for IL-6 (37), since a high increase of IL-6 in
ALI/ARDS accelerates the development of the local inflammatory
microenvironment (38). Moreover,
high levels of IL-1β, IL-6 and TNF-α have been considered as the
most promising biomarkers for predicting morbidity and mortality in
patients with ALI/ARDS (39).
Taken together, we demonstrated that CCN3 plays an important
pro-inflammatory role in lung epithelial cell injury by promoting
the activities of specific cytokines, such as IL-1β and TGF-β1.
TGF-β1 is a secretory cytokine that binds to the
Type II and Type I TGF β receptor (TGF-β RII and TGF-β RI,
respectively), which initiates TGF-β signaling via Smad
phosphorylation and nuclear translocation (40,41).
Many studies have demonstrated that the TGF-β1 pathway plays a
critical role in the development of ALI (18,40–42).
One study has demonstrated that early activation of TGF-β1/Smad2
signaling might contribute to acute pancreatitis-associated ALI,
through regulation of lung permeability, epithelial ion transport,
fibrinolysis and the extracellular matrix (31). In the present study, we observed a
reduction in the levels of TGF-βR II and p-Smad2/3 following CCN3
silencing. Notably, overexpression of CCN3 in A549 cells might
contribute to the activation of the TGF-β signaling pathway,
leading to the destruction of epithelial integrity and aggregation
of lung injury. Therefore, to further explore the direct roles of
the TGF-β and p-Smad2/3 signaling pathways in the effect of CCN3,
TP0427736, which inhibits the phosphorylation of Smad2/3 (43), was chosen to treat the A549 cells.
We observed a significant decrease in CCN3 expression induced by
LPS after TP0427736 treatment in the A549 cells, suggesting that
CCN3 may promote the release of inflammatory mediators by the
TGF-β/p-Smad2/3 signaling pathway in A549 cells. This reveals an
alternative pathway that could aid in future studies of the
pathogenesis of ALI.
Apoptosis of AEC II cells plays an essential role in
the pathogenesis of ARDS (44).
Our study showed that 0.1 µg/ml LPS could promote apoptosis in A549
cells. CCN3 knockdown greatly reduced the apoptotic rate of AEC II
cells, while overexpression of CCN3 promoted AEC II apoptosis.
LPS-triggered alveolar epithelial type II cell apoptosis is thought
to primarily depend on the mitochondrial apoptosis signaling
pathway (45). Bcl-2 regulates the
mitochondrial apoptotic pathway by preventing the release of
cytochrome c from mitochondria to cytosol, and activating
caspase-3 (46). Doghman et
al (47) showed that CCN3
induced human adrenocortical cell apoptosis through the activation
of caspase 3. Consistent with these results, the changes in Bcl-2
and caspase-3 levels highlighted that the inhibition of CCN3
expression prevented AEC II apoptosis at the cellular level.
Therefore, the activation of CCN3 induced by LPS may be involved in
promoting AEC II cell apoptosis through the Bcl-2/caspase-3
pathway.
Moreover, this study showed that CCN3 activated
NF-κB in A549 cells by promoting nuclear translocation of NF-κB
p65. As a central mediator of the human immune response, NF-κB is a
critical transcription factor in the pathogenesis of ALI (48). It directly activates downstream
signaling pathways by interacting with MyD88, promoting the
degradation of IκBα and the phosphorylation of NF-κB (49), leading to epithelial cell apoptosis
by the Fas/Fas ligand (Fas L) signaling pathway (50–52).
In addition, previous studies have demonstrated that the NF-κB
pathway promoted lung inflammation and injury in response to local
and systemic stresses in airway epithelial cells, by triggering the
transcription of inflammatory cytokines and chemokines (TNF-α,
IL-1β and IL-6) (53–55). Because this study did not
illuminate the relationship between IL-1β and NF-κB, it is not
clear whether the same response to stimuli could be observed in
A549 cells. Additionally, overexpression of CCN3 has been observed
to have anti-inflammatory effects on endothelial cells by
inhibiting the activation of NF-κB (9), which is in contrast with our results.
These differences may be due to differences in the cell types
analyzed. Our results demonstrated that pretreatment with PDTC (a
specific NF-κB inhibitor) significantly suppressed LPS-induced CCN3
and nuclear NF-κB p65 expression, indicating that CCN3 plays a role
in human lung alveolar epithelial cells through the NF-κB signaling
pathway. It has been shown that excessive epithelial cell apoptosis
could lead to the damage of the pulmonary alveolar-capillary
barrier and aggravate the inflammatory responses in lung diseases
(52,56), suggesting a close association
between inflammation and apoptosis. In summary, our study revealed
that CCN3 may have potential clinical value in the occurrence and
development of ALI via the TGF-β/p-Smad or NF-κB signaling
pathways. Further research is needed to validate our findings using
in vivo models, yet our data suggest a novel potential
target for future clinical studies.
In conclusion, the present study demonstrated that
CCN3 expression in alveolar epithelial cells was significantly
increased under inflammatory conditions and/or in response to
stimuli such as LPS. The overexpression of CCN3 can be perturbed by
a TGF-β/p-Smad or NF-κB inhibitor, which explains how CCN3-siRNA
led to the inhibition of the release of inflammatory cytokines and
apoptosis in human alveolar epithelial cells.
Acknowledgements
Not applicable.
Funding
This research study was supported by the Wenzhou
Municipal Science and Technology Plan Project (grant no.
2017Y0877), National Key R&D Program of China (grant nos.
2017YFC0908700 and 2017YFC0908703), Key R&D Program of Shandong
Province (grant no. 2016ZDJS07A14), Natural Science Foundation of
Zhejiang Province (grant no. LY18H010006) and National Nature
Science Foundation of China (grant no. 81600062).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HPZ, CSC and YGC conceived and designed the
experiments, analyzed the data and wrote the manuscript. HYH, DMW,
CLC, LD and ND carried out the experiments, prepared and analyzed
the figures and tables. LD, CSC, CLC and YGC obtained the study
materials and reagents in preparation for the experiments. All
authors reviewed drafts of the paper. All authors read and approved
the manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
The protocol of the present study was approved by
the Use Committee of Wenzhou Medical University (Wenzhou,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Matthay MA, Ware LB and Zimmerman GA: The
acute respiratory distress syndrome. J Clin Invest. 122:2731–2740.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ware LB and Matthay MA: The acute
respiratory distress syndrome. N Engl J Med. 342:1334–1349. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rooney SA: Regulation of surfactant
secretion. Comp Biochem Physiol A Mol Integr Physiol. 129:233–243.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Galani V, Tatsaki E, Bai M, Kitsoulis P,
Lekka M, Nakos G and Kanavaros P: The role of apoptosis in the
pathophysiology of Acute Respiratory Distress Syndrome (ARDS): An
up-to-date cell-specific review. Pathol Res Pract. 206:145–150.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Imazu Y, Yanagi S, Miyoshi K, Tsubouchi H,
Yamashita SI, Matsumoto N, Ashitani JI, Kangawa K and Nakazato M:
Ghrelin ameliorates bleomycin-induced acute lung injury by
protecting alveolar epithelial cells and suppressing lung
inflammation. Eur J Pharmacol. 672:153–158. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kutsukake M, Matsutani T, Tamura K,
Matsuda A, Kobayashi M, Tachikawa E and Uchida E: Pioglitazone
attenuates lung injury by modulating adipose inflammation. J Surg
Res. 189:295–303. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kular L, Pakradouni J, Kitabgi P, Laurent
M and Martinerie C: The CCN family: A new class of inflammation
modulators. Biochimie. 93:377–388. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Perbal B: NOV (nephroblastoma
overexpressed) and the CCN family of genes: Structural and
functional issues. Mol Pathol. 54:57–79. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin Z, Natesan V, Shi H, Hamik A, Kawanami
D, Hao C, Mahabaleshwar GH, Wang W, Jin ZG, Atkins GB, et al: A
novel role of CCN3 in regulating endothelial inflammation. J Cell
Commun Signal. 4:141–153. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu W, Hu X, Zhou X, Klenotic PA, Zhou Q
and Lin Z: Myeloid deficiency of CCN3 exacerbates liver injury in a
mouse model of nonalcoholic fatty liver disease. J Cell Commun
Signal. 12:389–399. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen PP, Li WJ, Wang Y, Zhao S, Li DY,
Feng LY, Shi XL, Koeffler HP, Tong XJ and Xie D: Expression of
Cyr61, CTGF, and WISP-1 correlates with clinical features of lung
cancer. PLoS One. 2:e5342007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dolinay T, Kaminski N, Felgendreher M, Kim
HP, Reynolds P, Watkins SC, Karp D, Uhlig S and Choi AM: Gene
expression profiling of target genes in ventilator-induced lung
injury. Physiol Genomics. 26:68–75. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Llinàs L, Peinado VI, Ramon Goñi J,
Rabinovich R, Pizarro S, Rodriguez-Roisin R, Barberà JA and Bastos
R: Similar gene expression profiles in smokers and patients with
moderate COPD. Pulm Pharmacol Ther. 24:32–41. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Moon HG, Kim SH, Gao J, Quan T, Qin Z,
Osorio JC, Rosas IO, Wu M, Tesfaigzi Y and Jin Y: CCN1 secretion
and cleavage regulate the lung epithelial cell functions after
cigarette smoke. Am J Physiol Lung Cell Mol Physiol. 307:L326–L337.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shahzeidi S, Mulier BD, de Crombrugghe B,
Jeffery PK, McAnulty RJ and Laurent GJ: Enhanced type III collagen
gene expression during bleomycin induced lung fibrosis. Thorax.
48:622–628. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen C, Shi L, Li Y, Wang X and Yang S:
Disease-specific dynamic biomarkers selected by integrating
inflammatory mediators with clinical informatics in ARDS patients
with severe pneumonia. Cell Biol Toxicol. 32:169–184. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cui X, Zeni F, Vodovitz Y,
Correa-de-Araujo R, Quezado M, Roberts A, Wahl S, Danner RL, Banks
SM, Gerstenberger E, et al: TGF-beta1 increases microbial clearance
but worsens lung injury during Escherichia coli pneumonia in
rats. Cytokine. 24:115–127. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dhainaut JF, Charpentier J and Chiche JD:
Transforming growth factor-beta: A mediator of cell regulation in
acute respiratory distress syndrome. Crit Care Med. 31 (Suppl
4):S258–S264. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fahy RJ, Lichtenberger F, McKeegan CB,
Nuovo GJ, Marsh CB and Wewers MD: The acute respiratory distress
syndrome: A role for transforming growth factor-beta 1. Am J Respir
Cell Mol Biol. 28:499–503. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lin J, Tian J, Wang L, Wu W, Li H, Wang X,
Zeng X and Zhang W: Apoptosis and surfactant protein-C expression
inhibition induced by lipopolysaccharide in AEC II cell may
associate with NF-κB pathway. J Toxicol Sci. 42:53–61. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang KY, Arcaroli JJ and Abraham E: Early
alterations in neutrophil activation are associated with outcome in
acute lung injury. Am J Respir Crit Care Med. 167:1567–1574. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Boots AW, Gerloff K, Bartholomé R, van
Berlo D, Ledermann K, Haenen GR, Bast A, van Schooten FJ, Albrecht
C and Schins RP: Neutrophils augment LPS-mediated pro-inflammatory
signaling in human lung epithelial cells. Biochim Biophys Acta.
1823:1151–1162. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cabrera-Benítez NE, Pérez-Roth E,
Ramos-Nuez Á, Sologuren I, Padrón JM, Slutsky AS and Villar J:
Inhibition of endotoxin-induced airway epithelial cell injury by a
novel family of pyrrol derivates. Lab Invest. 96:632–640. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chuang CY, Chen TL, Cherng YG, Tai YT,
Chen TG and Chen RM: Lipopolysaccharide induces apoptotic insults
to human alveolar epithelial A549 cells through reactive oxygen
species-mediated activation of an intrinsic mitochondrion-dependent
pathway. Arch Toxicol. 85:209–218. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
MacRedmond R, Singhera GK and Dorscheid
DR: Erythropoietin inhibits respiratory epithelial cell apoptosis
in a model of acute lung injury. Eur Respir J. 33:1403–1414. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Muroya M, Chang K, Uchida K, Bougaki M and
Yamada Y: Analysis of cytotoxicity induced by proinflammatory
cytokines in the human alveolar epithelial cell line A549. Biosci
Trends. 6:70–80. 2012.PubMed/NCBI
|
|
27
|
Rodríguez-González R, Ramos-Nuez Á,
Martín-Barrasa JL, López-Aguilar J, Baluja A, Álvarez J, Rocco PR,
Pelosi P and Villar J: Endotoxin-induced lung alveolar cell injury
causes brain cell damage. Exp Biol Med (Maywood). 240:135–142.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shao L, Meng D, Yang F, Song H and Tang D:
Irisin-mediated protective effect on LPS-induced acute lung injury
via suppressing inflammation and apoptosis of alveolar epithelial
cells. Biochem Biophys Res Commun. 487:194–200. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fong YC, Maa MC, Tsai FJ, Chen WC, Lin JG,
Jeng LB, Yang RS, Fu WM and Tang CH: Osteoblast-derived TGF-beta1
stimulates IL-8 release through AP-1 and NF-kappaB in human cancer
cells. J Bone Miner Res. 23:961–970. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Akbarshahi H, Sam A, Chen C, Rosendahl AH
and Andersson R: Early activation of pulmonary TGF-β1/Smad2
signaling in mice with acute pancreatitis-associated acute lung
injury. Mediators Inflamm. 2014:1480292014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Scorrano L and Korsmeyer SJ: Mechanisms of
cytochrome c release by proapoptotic BCL-2 family members. Biochem
Biophys Res Commun. 304:437–444. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin WC, Chen CW, Huang YW, Chao L, Chao J,
Lin YS and Lin CF: Kallistatin protects against sepsis-related
acute lung injury via inhibiting inflammation and apoptosis. Sci
Rep. 5:124632015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dolinay T, Kim YS, Howrylak J, Hunninghake
GM, An CH, Fredenburgh L, Massaro AF, Rogers A, Gazourian L,
Nakahira K, et al: Inflammasome-regulated cytokines are critical
mediators of acute lung injury. Am J Respir Crit Care Med.
185:1225–1234. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen CC and Lau LF: Deadly liaisons: Fatal
attraction between CCN matricellular proteins and the tumor
necrosis factor family of cytokines. J Cell Commun Signal. 4:63–69.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Feng T, Yunfeng N, Jinbo Z, Zhipei Z,
Huizhong Z, Li L, Tao J and Yunjie W: Single immunoglobulin IL-1
receptor-related protein attenuates the lipopolysaccharide-induced
inflammatory response in A549 cells. Chem Biol Interact.
183:442–449. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Aloisi F, Carè A, Borsellino G, Gallo P,
Rosa S, Bassani A, Cabibbo A, Testa U, Levi G and Peschle C:
Production of hemolymphopoietic cytokine s (IL-6, IL-8,
colony-stimulating factors) by normal human astrocytes in response
to IL-1 beta and tumor necrosis factor-alpha. J Immunol.
149:2358–2366. 1992.PubMed/NCBI
|
|
38
|
Shi L, Dong N, Ji D, Huang X, Ying Z, Wang
X and Chen C: Lipopolysaccharide-induced CCN1 production enhances
interleukin-6 secretion in bronchial epithelial cells. Cell Biol
Toxicol. 34:39–49. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Butt Y, Kurdowska A and Allen TC: Acute
lung injury: A clinical and molecular review. Arch Pathol Lab Med.
140:345–350. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hurst V VI, Goldberg PL, Minnear FL,
Heimark RL and Vincent PA: Rearrangement of adherens junctions by
transforming growth factor-beta1: Role of contraction. Am J
Physiol. 276:L582–L595. 1999.PubMed/NCBI
|
|
41
|
Sureshbabu A, Syed MA, Boddupalli CS,
Dhodapkar MV, Homer RJ, Minoo P and Bhandari V: Conditional
overexpression of TGFβ1 promotes pulmonary inflammation, apoptosis
and mortality via TGFβR2 in the developing mouse lung. Respir Res.
16:42015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pittet JF, Griffiths MJ, Geiser T,
Kaminski N, Dalton SL, Huang X, Brown LA, Gotwals PJ, Koteliansky
VE, Matthay MA and Sheppard D: TGF-beta is a critical mediator of
acute lung injury. J Clin Invest. 107:1537–1544. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Naruse T, Aoki M, Fujimoto N, Arase S,
Oura H, Ueda Y and Ikeda A: Novel ALK5 inhibitor TP0427736 reduces
TGF-β induced growth inhibition in human outer root sheath cells
and elongates anagen phase in mouse hair follicles. Pharmacol Rep.
69:485–491. 2017. View Article : Google Scholar
|
|
44
|
Bardales RH, Xie SS, Schaefer RF and Hsu
SM: Apoptosis is a major pathway responsible for the resolution of
type II pneumocytes in acute lung injury. Am J Pathol. 149:845–852.
1996.PubMed/NCBI
|
|
45
|
Aggarwal S, Dimitropoulou C, Lu Q, Black
SM and Sharma S: Glutathione supplementation attenuates
lipopolysaccharide-induced mitochondrial dysfunction and apoptosis
in a mouse model of acute lung injury. Front Physiol. 3:1612012.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Borner C: The Bcl-2 protein family:
Sensors and checkpoints for life-or-death decisions. Mol Immunol.
39:615–647. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Doghman M, Arhatte M, Thibout H, Rodrigues
G, De Moura J, Grosso S, West AN, Laurent M, Mas JC, Bongain A, et
al: Nephroblastoma overexpressed/cysteine-rich protein
61/connective tissue growth factor/nephroblastoma overexpressed
gene-3 (NOV/CCN3), a selective adrenocortical cell proapoptotic
factor, is down-regulated in childhood adrenocortical tumors. J
Clin Endocrinol Metab. 92:3253–3260. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Pahl HL: Activators and target genes of
Rel/NF-kappaB transcription factors. Oncogene. 18:6853–6866. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Nighot M, Rawat M, Al-Sadi R, Castillo EF,
Nighot P and Ma TY: Lipopolysaccharide-induced increase in
intestinal permeability is mediated by TAK-1 activation of IKK and
MLCK/MYLK gene. Am J Pathol. 189:797–812. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lopez AD, Avasarala S, Grewal S, Murali AK
and London L: Differential role of the Fas/Fas ligand apoptotic
pathway in inflammation and lung fibrosis associated with reovirus
1/L-induced bronchiolitis obliterans organizing pneumonia and acute
respiratory distress syndrome. J Immunol. 183:8244–8257. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Matsui K, Fine A, Zhu B, Marshak-Rothstein
A and Ju ST: Identification of two NF-kappa B sites in mouse CD95
ligand (Fas ligand) promoter: Functional analysis in T cell
hybridoma. J Immunol. 161:3469–3473. 1998.PubMed/NCBI
|
|
52
|
White MK, Baireddy V and Strayer DS:
Natural protection from apoptosis by surfactant protein A in type
II pneumocytes. Exp Cell Res. 263:183–192. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cheng DS, Han W, Chen SM, Sherrill TP,
Chont M, Park GY, Sheller JR, Polosukhin VV, Christman JW, Yull FE,
et al: Airway epithelium controls lung inflammation and injury
through the NF-kappa B pathway. J Immunol. 178:6504–6513. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gu LZ, Sun H and Chen JH: Histone
deacetylases 3 deletion restrains PM2.5-induced mice lung injury by
regulating NF-κB and TGF-β/Smad2/3 signaling pathways. Biomed
Pharmacother. 85:756–762. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Rosendahl A, Checchin D, Fehniger TE, ten
Dijke P, Heldin CH and Sideras P: Activation of the
TGF-beta/activin-Smad2 pathway during allergic airway inflammation.
Am J Respir Cell Mol Biol. 25:60–68. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sharp C, Millar AB and Medford AR:
Advances in understanding of the pathogenesis of acute respiratory
distress syndrome. Respiration. 89:420–434. 2015. View Article : Google Scholar : PubMed/NCBI
|