Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic,
progressive, interstitial lung disease, with a poor prognosis and a
mechanism of pathogenesis that remains to be elucidated. The median
survival rate of patients with IPF is only 3–5 years, in which the
main pathological feature is the abnormal deposition of
extracellular matrix (ECM). The current therapeutic approaches for
IPF are ineffective (1,2), as the pathogenesis of IPF remains to
be fully elucidated. Multiple signaling pathways and various
factors have been reported to have meaningful roles in pulmonary
fibrosis (PF), among which transforming growth factor-β1 (TGF-β1)
is considered a key factor during the onset of PF. TGF-β1 has been
demonstrated to promote PF by enhancing the EMT of alveolar type II
epithelial cells (3,4). The transcription factor Smad3 is an
important downstream signaling factor of the TGF-β1 signaling
pathway, which is involved in various physiological and
pathological processes. TGF-β1 binds to its specific receptor,
stimulating Smad3-regulated and fibrosis-related gene transcription
and initiating fibrosis. Tribbles pseudokinase 3 (TRB3) is one
member of the mammalian kinase-like tribbles homologues belonging
to the pseudokinase family. It is involved in regulating various
signaling pathways, including mitogen-activated protein kinase,
Notch, and Wnt, and in modulating various processes, including cell
proliferation, differentiation, apoptosis, and carcinogenesis
(5,6). TRB3 can react with Smad3 by enhancing
the regulatory activity of Smad3-induced transcription and
targeting gene expression, leading to activation of the
TGF-β1/Smad3 signaling pathway. Studies have indicated that the
expression of TRB3 is upregulated in renal fibrosis, myocardial
fibrosis and tumor tissue when its expression level positively
correlated with the fibrosis; however, silencing TRB3 inhibits
fibrosis (7–9). The role of TRB3 in PF remains to be
fully elucidated. Therefore, in the present study, novel targets
for IPF treatment were investigated by demonstrating the role of
TRB3 in the pathogenesis of IPF.
Materials and methods
Adenovirus vector construction
Primers were designed based on the TRB3 sequence
published by NCBI GenBank (https://www.ncbi.nlm.nih.gov/nuccore?db=nucleotide;
ref. no. MGI:3562334). The target gene was amplified using TRB3
cDNA as a template, of which the product was constructed into a
vector for the TRB3 recombinant plasmid (Ad-TRB3). The
TRB3-specific short hairpin (sh)RNA was designed as follows:
mmu-TRB3-small interfering RNA1 (5′-GAAGAAACCGTTGGAGTTTG-3′) was
constructed for the recombinant plasmid Ad-TRB3-shRNA. The plasmids
were cultured and purified, and adenovirus packaging was performed.
The infectious titer of the recombinant adenovirus was evaluated
using a TCID50 assay, which was 1.1×1011 (PFU/ml).
Cell culturing
The MLE-12 murine alveolar type II epithelial cells,
from Fuxiang Biotechnology Co., Ltd. (Shanghai, China) were
cultured in a 5% CO2 atmosphere at 37°C with saturated
humidity.
Overexpression and downregulation of
TRB3
The cells were seeded (the density was
1.2×106 cells/ml) in a six-well plate and divided into
five groups randomly: i) control group (Group C); ii) TGF-β1 group
(Group T); iii) TGF-β1 + Ad-GFP group (Group T + G); iv) TGF-β1 +
Ad-TRB3 group (Group T + TRB3); and v) TGF-β1 + Ad-TRB3-shRNA group
(Group T + TRB3-shRNA). The cells were infected with the
corresponding adenovirus vectors when at 60% confluency, MLE-12
cells in logarithmic growth phase were infected with multiple types
of virus. Each virus was infected at three multiplicities of
infection: 50, 100 and 200 for Ad-GFP; 200, 400 and 800 for
Ad-TRB3; 100, 200 and 400 for Ad-TRB3-shRNA. The transduction time
was 48 h. Subsequently, the cells were harvested, and total protein
and RNA were extracted from the cells in each group.
Fluorogenic reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
Total RNA was extracted from the MLE-12 cells using
TRIzol (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Reverse transcription was performed using the PrimeScript RT
reagent kit (Takara Bio, Inc., Otsu, Japan) and incubated at 37°C
for 15 min and at 85°C for 5 sec. The target genes were then
amplified using the SYBR Premix Ex Taq kit (Takara Bio, Inc.).
Thermocycling conditions were as follows: Initial denaturation at
94°C for 4 min, followed by 40 cycles of 94°C for 30 sec, 58°C for
30 sec and 72°C for 30 sec. The results were analyzed using the
software of the ABI 7500 system (version 2.0.6; Applied Biosystems;
Thermo Fisher Scientific, Inc.). The primers for target genes are
listed in Table I. β-actin was
used as an internal control, and values for each target gene were
normalized using the expression level of β-actin (10).
 | Table I.Primers for each murine gene. |
Table I.
Primers for each murine gene.
| Gene name | Sequence (5′-3′) | Product length
(bp) |
|---|
| CollaI forward |
GAGACAGGCGAACAAGGTGA | 399 |
| CollaI reverse |
CTCAAGGTCACGGTCACGAA |
|
| α-SMA forward |
GTACCCAGGCATTGCTGACA | 271 |
| α-SMA reverse |
GAGGCGCTGATCCACAAAAC |
|
| TRB3 forward |
GGAACCTTCAGAGCGACTT | 341 |
| TRB3 reverse |
TGGCACTCAGGGAGCATC |
|
| β-actin forward |
CCACCATGTACCCAGGCATT | 189 |
| β-actin reverse |
CGGACTCATCGTACTCCTGC |
|
Western blot analysis
Total cell protein was extracted using cell lysis
buffer (Beyotime Institute of Biotechnology, Haimen, China),
according to the manufacturer's protocol. A total of 5 µg protein
was loaded in each lane. SDS-PAGE (10%) was performed for each
group, and the proteins were transferred from the gel onto PVDF
membranes. Following blocking for 2 h using 1X Bovine lacto
transfer technique optimizer (Thermo Fisher Scientific, Inc.), the
membranes were incubated with anti-TRB3 (cat. no. sc-390242; Santa
Cruz Biotechnology Inc., Dallas, TX, USA), anti-phosphorylated
(p-)Smad3 (cat. no. 9523S; Cell Signaling Technology, Inc.,
Danvers, MA, USA), anti-E-cadherin (cat. no. wl01482; Wanleibio
Co., Ltd., Shanghai, China), anti-Vimentin (cat. no. wl00742;
Wanleibio Co., Ltd.), anti-Fibronectin (cat. no. sc-69681; Santa
Cruz Biotechnology, Inc.) and anti-GAPDH (cat. no. sc-47724; Santa
Cruz Biotechnology, Inc.) antibodies at 4°C, overnight. The
concentration of each primary antibody was 1:1,000. Subsequently,
the membranes were incubated with secondary antibody labeled with
horseradish peroxidase (1:10,000; cat. no. sc-2004; Santa Cruz
Biotechnology, Inc.) at room temperature for 1.5 h. Following
incubation with the secondary antibody, all membranes were washed,
incubated in the Western Lightningä Chemiluminescence reagent
(PerkinElmer, Inc., Waltham, MA USA), and photographic films were
exposed to the membrane in a darkroom. Finally, images of the
membranes were captured using the gel imaging system from LabWorksä
4.5 (UVP, LLC, Phoenix, AZ, USA), and the band brightness of each
group was calculated.
Immunofluorescence (IF)
The cells were seeded onto coverslips and fixed
using paraformaldehyde. Following this, rabbit TRB3 antibody
(1:1,000; sc-390242; Santa Cruz Biotechnology, Inc.) was used as
the primary antibody and was incubated at 4°C for 24 h. Donkey
anti-rabbit immunoglobulin G labeled with FITC (1:1,000; cat. no.
sab4600003; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was used
as the secondary antibody and was incubated at 37° for 1.5 h. The
nucleus was stained with DAPI (Sigma; Merck KGaA). Finally, the
coverslip was placed onto the object slide, examined under a
confocal laser scanning microscope (CLSM, FV1000, Olympus
Corporation, Tokyo, Japan), and images were captured.
Flow cytometry
The MLE-12 cells were plated into a six-well plate
and infected according to the grouping. The cells were harvested
and resuspended 48 h after infection and stained following the
protocols of the ApoScreen Annexin V Apoptosis kit
(SouthernBiotech, Birmingham, AL, USA) and Propidium Iodide kit
(Roche Diagnostics, Basel, Switzerland). Following staining for 1
h, the cells were examined and analyzed using CellQuest software
(version 6.0; Becton, Dickinson and Company, Franklin Lakes, NJ,
USA).
Cell Counting Kit-8 (CCK-8)
The MLE-12 cells were cultured in 96-well plates and
infected according to the grouping. After 48 h, 10 µl CCK-8
solution was pipetted into each well, and the incubation was
continued for 2 h. The absorbance was measured at 450 nm using a
microplate reader. The OD value was in proportion to the living
cell number.
Statistical analysis
Using data from the present study, a database was
established using SPSS19.0 (IBM Corp., Armonk, NY, USA). All data
were consistent with normal distribution and are presented as the
mean ± standard deviation. Differences between two means were
compared using a t-test, while that among multiple groups using
analysis of variance. P<0.05 was considered to indicate a
statistically significant difference.
Results
mRNA and protein expression levels of
TRB3 increase in TGF-β1-stimulated MLE-12 cells
The mRNA and protein expression levels of TRB3 were
low in normal MLE-12 cells but were significantly higher in the
TGF-β1-stimulated cells, particularly in Group T + TRB3. When
TRB3-shRNA was added, the mRNA and protein levels of TRB3 were the
lowest (P<0.05; Table II).
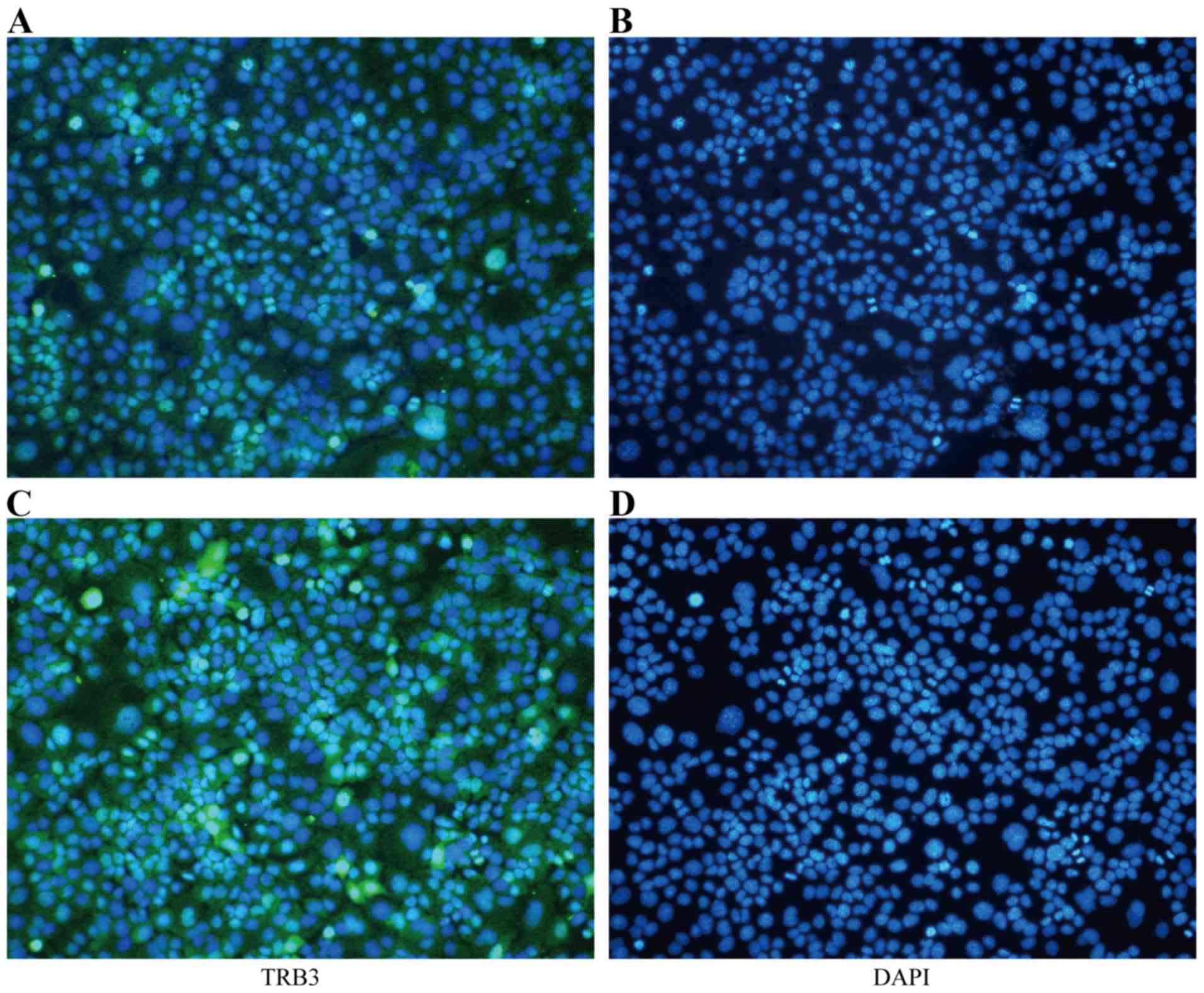
Furthermore, the IF assay suggested that the expression of TRB3 in
the nucleus was increased in the TGF-β1-stimulated MLE-12 cells
(Fig. 1).
| Table II.mRNA and protein expression levels of
TRB3 in each group. |
Table II.
mRNA and protein expression levels of
TRB3 in each group.
| Group |
TRB3mRNA/(2−ΔΔCq) | TRB3
protein/(TRB3/GAPDH) |
|---|
| C | 1.0000±0.0070 | 1.0000±0.0120 |
| T |
2.0250±0.0750a |
1.6820±0.2110a |
| T+G |
1.5400±0.1500b |
1.6520±0.0310b |
| T+TRB3 |
4.1250±0.3150c |
2.8520±0.2110c |
| T+TRB3-shRNA |
0.0190±0.0010c |
0.3220±0.0020c |
TRB3 stimulates the activation of the
TGF-β1/Smad3 signaling pathway
The protein expression level of p-Smad3 in the
MLE-12 cells was assessed when TRB3 was upregulated or
downregulated, respectively (P<0.05). The results demonstrated
that, when TRB3 was overexpressed, the protein expression of
p-Smad3 increased (P<0.05). By contrast, the downregulation of
TRB3 led to decreased protein expression of p-Smad3 (P<0.05;
Table III).
| Table III.Protein expression level of p-Smad3 in
each group. |
Table III.
Protein expression level of p-Smad3 in
each group.
| Group | p-Smad3/GAPDH | t |
|---|
| C | 1.0000±0.0112 |
|
| T | 1.7830±0.0085 | 55.81a |
| T+G | 1.7610±0.0048 |
2.239b |
| T+TRB3 | 3.8550±0.0293 | 71.24c |
| T+TRB3-shRNA | 0.5481±0.0289 | 41.49c |
Overexpression of TRB3 promotes the
EMT of TGF-β1-stimulated MLE-12 cells, which is inhibited by the
downregulation of TRB3
Further investigation found that, compared with the
TGF-β1 stimulation group, the overexpression of TRB3 enhanced the
mRNA expression levels of α-smooth muscle actin (α-SMA) and
collagen type I (CollaI), two fibroblast markers, in MLE-12 cells
(P<0.05; Table IV). In
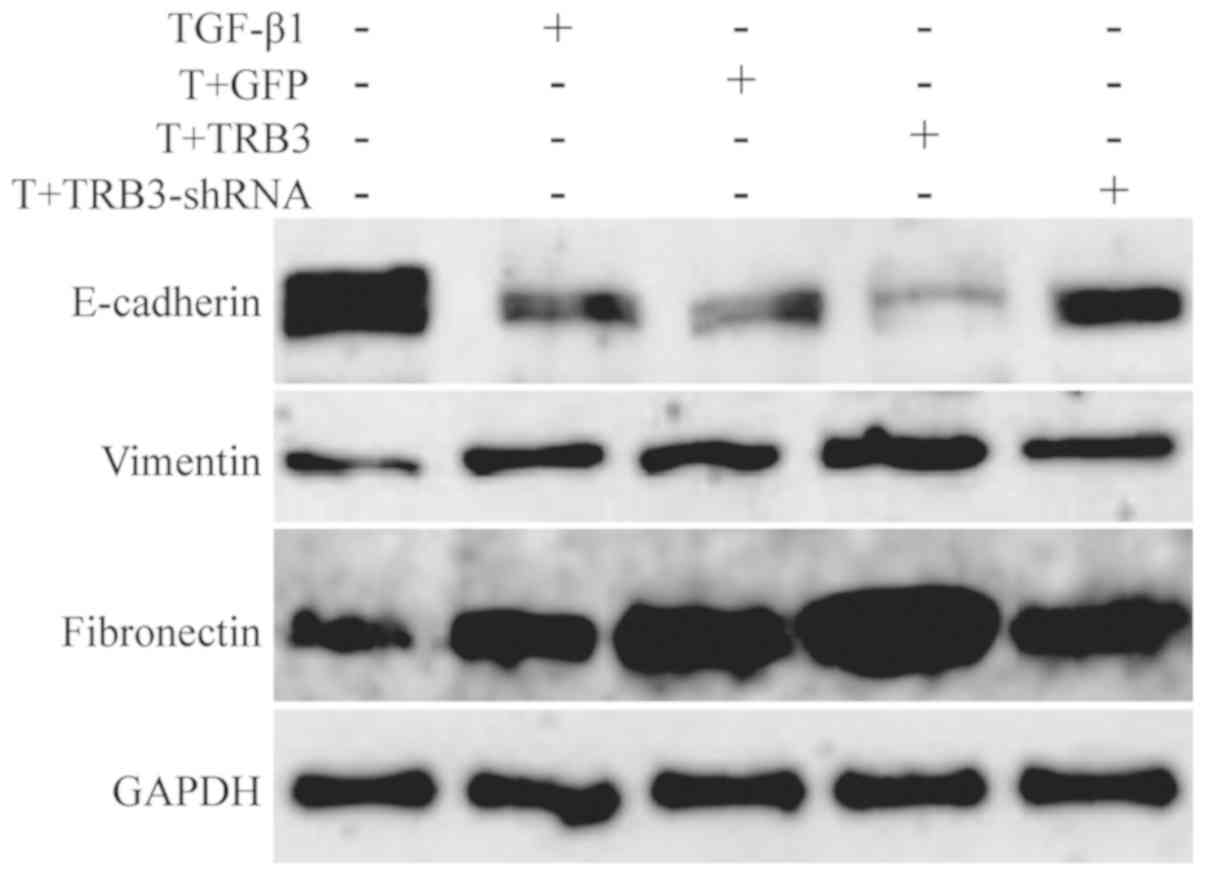
addition, the results of the western blot analysis indicated that
the expression of E-cadherin (epithelial marker) decreased, whereas
the expression levels of Vimentin and Fibronectin (mesenchymal
markers) increased (P<0.05; Table
V; Fig. 2). When the
expression of TRB3 was downregulated by RNA interference, the mRNA
expression levels of α-SMA and CollaI were also downregulated
(P<0.05; Table IV).
Furthermore, the expression of E-cadherin increased, and the
expression levels of Vimentin and Fibronectin decreased (P<0.05)
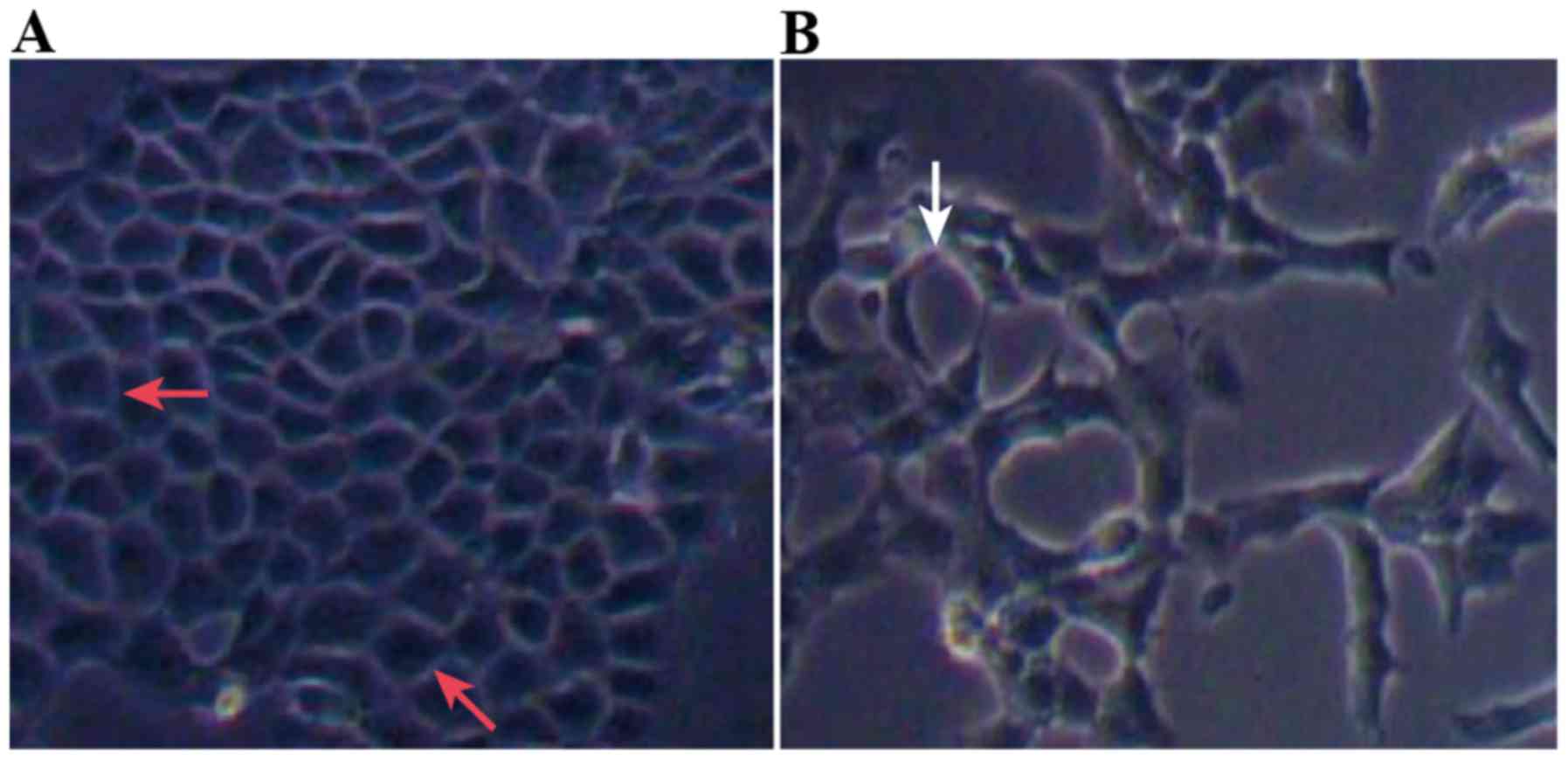
(Table V; Fig. 2). Compared with Group C, the
morphology of cells in Group T changed from polygonal or
rectangular to spindle-shaped (Fig. 3A
and B).
| Table IV.mRNA expression of α-SMA and CollaI in
MLE-12 cells in each group. |
Table IV.
mRNA expression of α-SMA and CollaI in
MLE-12 cells in each group.
| Group | CollaI | t | α-SMA | t |
|---|
| C | 1.0100±0.0130 |
| 1.0025±0.0007 |
|
| T | 2.2700±0.2300 | 5.522a | 2.4700±0.3900 | 17.911a |
| T+G | 1.2400±0.1700 | 3.601b | 2.3300±0.2500 |
0.302b |
| T+TRB3 | 6.2301±0.7300 | 6.657c | 6.2500±0.2500 | 11.092c |
| T+TRB3-shRNA | 0.2450±0.0450 | 5.658c | 0.4800±0.1200 |
6.671c |
| Table V.Protein expression levels of
E-cadherin, Vimentin and Fibronectin in MLE-12 cells in each group,
as measured by densitometry analysis. |
Table V.
Protein expression levels of
E-cadherin, Vimentin and Fibronectin in MLE-12 cells in each group,
as measured by densitometry analysis.
| Group |
E-cadherin/GAPDH | t | Vimentin/GAPDH | t |
Fibronectin/GAPDH | t |
|---|
| C | 1.0000±0.0560 |
| 1.0000±0.0577 |
| 1.0000±0.0231 |
|
| T | 0.5258±0.0145 |
7.967a | 1.3050±0.0579 | 3.728a | 1.4060±0.1156 |
3.443a |
| T+G | 0.5624±0.1179 |
1.958b | 1.3640±0.0878 | 0.565b | 1.6270±0.0145 |
1.892b |
| T+TRB3 | 0.2576±0.0043 | 24.272c | 1.9410±0.0231 | 6.353c | 3.2750±0.0380 | 40.572c |
| T+TRB3-shRNA | 0.8910±0.0059 | 24.973c | 0.8043±0.0579 | 5.326c | 0.4606±0.0580 | 74.581c |
Overexpression of TRB3 reduces MLE-12
cell proliferation by promoting TGF-β1-induced cellular
apoptosis
The results of the flow cytometry and CCK assays
demonstrated that, following TGF-β1 stimulation, MLE-12 cell
apoptosis increased (P<0.05) and proliferation decreased
(P<0.05). The overexpression of TRB3 promoted the cell apoptosis
induced by TGF-β1, leading to the significant inhibition of cell
proliferation. Furthermore, the downregulated expression of TRB3
reduced the cell apoptosis induced by TGF-β1 (P<0.05), thereby
attenuating the inhibitory effect of TGF-β1 on cell proliferation
(Fig. 4, Table VI).
| Table VI.Cell apoptosis and proliferation
assessment in each group. |
Table VI.
Cell apoptosis and proliferation
assessment in each group.
| Group | Apoptotic rate | t | OD value | t |
|---|
| C | 1.5730±0.0145 |
| 0.4387±0.0025 |
|
| T | 6.4872±0.1017 | 47.820a | 0.3537±0.0189 | 7.720a |
| T+G | 5.800±0.2930 |
2.214b | 0.3600±0.0240 | 0.359b |
| T+TRB3 | 10.3201±0.0727 | 14.963c | 0.2340±0.0102 | 8.369c |
| T+TRB3-shRNA | 3.8172±0.0035 |
6.721c | 0.4213±0.0114 | 3.997c |
Discussion
The main pathological feature of PF is the abnormal
deposition of ECM. The dynamic process of ECM deposition,
remodeling, and reabsorption regulates normal tissue growth,
development and wound healing. It also maintains internal
homeostasis, with malfunctioning processes giving rise to the
pathological remodeling of tissues and organs, including tissue and
organ fibrosis. Studies have indicated that during PF, alveolar
type II epithelial cells trans-differentiate into fibroblasts and
myofibroblasts through EMT (11–13)
and secrete excessive collagen and other proteins, resulting in
excessive ECM deposition (1). The
TGF-β1/Smad3 signaling pathway is an important regulatory pathway
of EMT. During PF, the expression of TGF-β1 increases, which
inhibits alveolar epithelial cell proliferation by transducing
signals through plasma membrane receptors, and it promotes EMT and
the differentiation of fibroblasts into myofibroblasts, finally
resulting in the stimulation of fibrosis. The expression of TRB3
has been found to be upregulated in tissues of various fibrotic
diseases; the overexpression of TRB3 has also been demonstrated to
stimulate the EMT of tumor cells and other types of epithelial
cells (5,6,8,9).
Silencing TRB3 alleviated collagen and fibronectin deposition in
tissues with pathological fibrosis; however, the role of TRB3 in
fibrotic lung tissue has remained unclear. In the present study, it
was hypothesized that TRB3 promotes the EMT of mouse alveolar type
II epithelial cells by interacting with the TGF-β1/Smad3 signaling
pathway.
The results of the present study demonstrated that,
following TGF-β1 stimulation, the mRNA and protein expression
levels of TRB3 in the MLE-12 cells increased significantly. The
expression levels of TRB3 were modulated using adenovirus vectors
in cells. When TRB3 was overexpressed, TGF-β1/Smad3 signaling
pathway activation was enhanced, p-Smad3 accumulation was induced,
fibrosis-related gene transcription was promoted, and finally the
EMT of MLE-12 cells and ECM deposition were stimulated. By
contrast, the downregulation of TRB3 was demonstrated to inhibit
TGF-β1/Smad3 signaling pathway activation, reduce the
phosphorylation of Smad3, suppress the EMT of MLE-12 cells, and
attenuate ECM deposition. In tumor tissue, a positive feedback loop
was found, which facilitated the onset of EMT of tumor cells. On
one hand, TRB3 was involved in maintaining the level of p-Smad3; on
the other hand, it interacted with Smad3 directly. In turn, the
overexpression of Smad3 promoted the transcription of TRB3
(5), which is consistent with the
present results.
Alveolar type II epithelial cells are the dominant
cells in lung tissue, which secrete multiple surface proteins and
other components of the ECM. Injury or apoptosis of cells influence
lung fibrosis, which may be the promoting factor of IPF onset
(14–17). The present study demonstrated that
the overexpression of TRB3 stimulated TGF-β1-induced cell
apoptosis, aggravating the inhibition of cell proliferation,
whereas the downregulation of TRB3 resulted in reduced apoptosis,
alleviating the inhibition of proliferation, which confirmed our
hypothesis.
TGF-β1 is a key factor that stimulates fibrosis not
only through the classic TGF-β1/Smad3 signaling pathway, but also
in the redox reaction to modulate epithelial cell apoptosis,
contributing to lung fibrosis (18,19).
Several studies have indicated that in numerous diseases, including
fibrosis, TRB3 stimulates the apoptosis of various cell types,
including myocardial and renal tubular cells, through the p53
pathway, oxidative stress and endoplasmic reticulum stress
(20–24), whereas silencing TRB3 led to
significantly reduced cell apoptosis. Until now, the effect of TRB3
on alveolar type II epithelial cell apoptosis has not been
evaluated. The present study demonstrated that the downregulation
of TRB3 attenuated the inhibitory effect of TGF-β1 on cell
proliferation, as TRB3 can stimulate TGF-β1-induced alveolar type
II epithelial cell apoptosis.
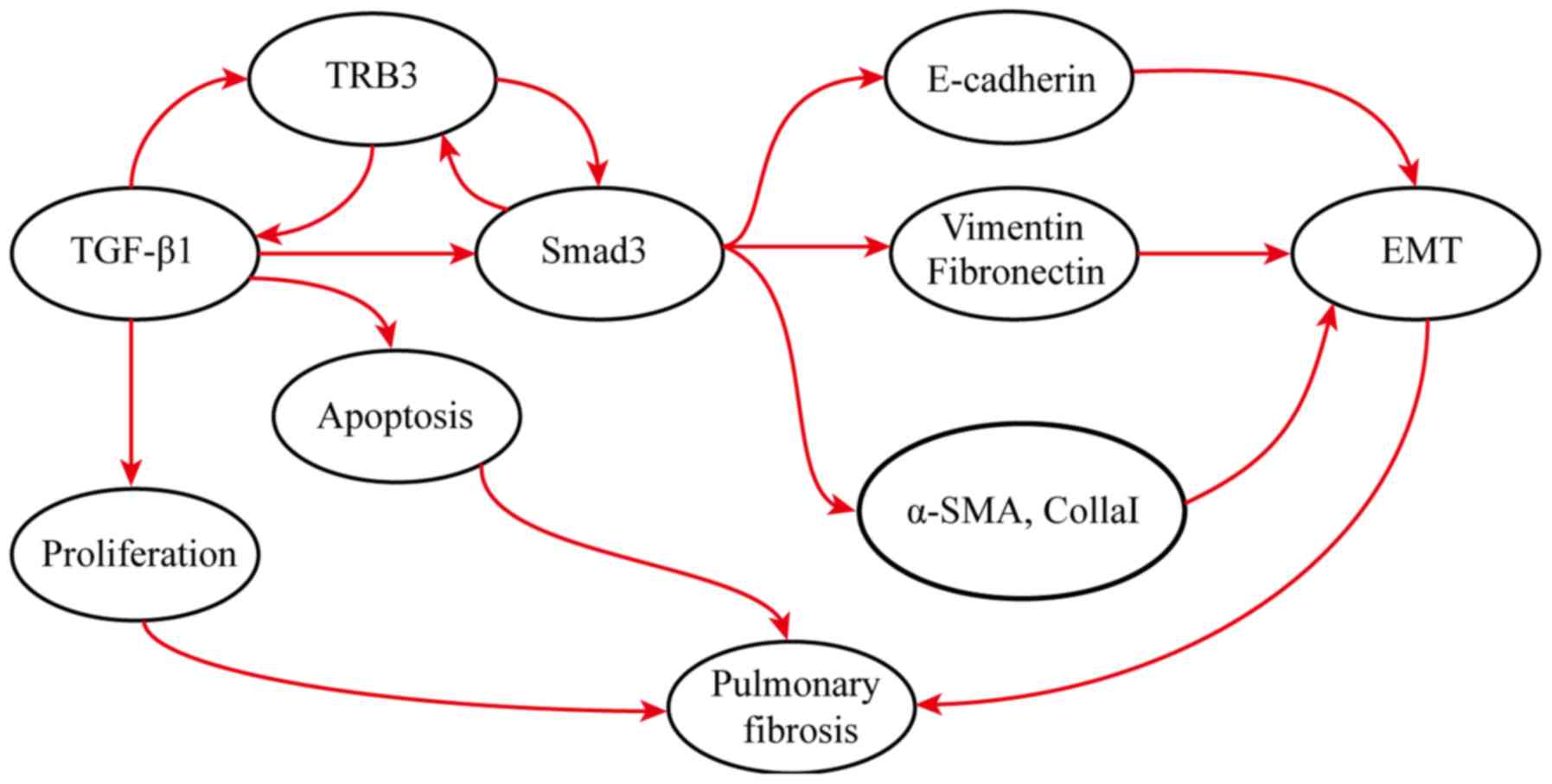
Therefore, it was hypothesized that during the early
stages of lung fibrosis, the expression of TRB3 is upregulated,
which interacts with the TGF-β1/Smad3 signaling pathway to promote
EMT, inhibit the proliferation of alveolar type II epithelial cells
by stimulating their apoptosis, and finally modulate the onset of
fibrosis. Interfering with the expression of TRB3 can regulate the
development of fibrosis by inhibiting alveolar type II epithelial
cell apoptosis. Additional investigation is required to answer
questions on additional components and elucidate more detailed
mechanisms within the regulatory network of the development of
fibrosis. Attention is also required on the difference between
in vitro and in vivo studies.
Acknowledgements
Not applicable.
Funding
This study was funded by the Natural Science Fund of
Shandong Province (grant no. ZR2014HM079).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
WY, WC and LM made substantial contributions to the
conception and design of the present study. WY, WC and JT
collected, analyzed and interpreted the data. WC and WY drafted the
manuscript. WY critically revised the manuscript. WY has given
final approval of the version to be published and agreed to be
accountable for all aspects of the work in ensuring that questions
related to the accuracy or integrity of any part of the work are
appropriately investigated and resolved. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Raghu G, Collard HR, Egan JJ, Martinez FJ,
Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, et
al: An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary
fibrosis: Evidence-based guidelines for diagnosis and management.
Am J Respir Crit Care Med. 183:788–824. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Raghu G, Rochwerg B, Zhang Y, Garcia CA,
Azuma A, Behr J, Brozek JL, Collard HR, Cunningham W, Homma S, et
al: An official ATS/ERS/JRS/ALAT clinical practice guideline:
Treatment of idiopathic pulmonary fibrosis. an update of the 2011
clinical practice guideline. Am J Respir Crit Care Med. 192:e3–19.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fernandez IE and Eickelberg O: The impact
of TGF-β on lung fibrosis: From targeting to biomarkers. Proc Am
Thorac Soc. 9:111–116. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shi Y, Gochuico BR, Yu G, Tang X, Osorio
JC, Fernandez IE, Risquez CF, Patel AS, Shi Y, Wathelet MG, et al:
Syndecan-2 exerts antifibrotic effects by promoting
caveolin-1-mediated transforming growth factor-β receptor I
internalization and inhibiting transforming growth factor-β1
signaling. Am J Respir Crit Care Med. 188:831–841. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hua F, Mu R, Liu J, Xue J, Wang Z, Lin H,
Yang H, Chen X and Hu Z: TRB3 interacts with SMAD3 promoting tumor
cell migration and invasion. J Cell Sci. 124:3235–3246. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Morse E, Schroth J, You YH, Pizzo DP,
Okada S, Ramachandrarao S, Vallon V, Sharma K and Cunard R: TRB3 is
stimulated in diabetic kidneys, regulated by the ER stress marker
CHOP, and is a suppressor of podocyte MCP-1. Am J Physiol Renal
Physiol. 299:F965–F972. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang L, Zhang J, Liu X, Liu S and Tian J:
Tribbles 3 regulates the fibrosis cytokine TGF-β1 through
ERK1/2-MAPK signaling pathway in diabetic nephropathy. J Immunol
Res. 2014:2403962014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang W, Sun A, Lv W, Cheng J, Lv S, Liu X,
Guan G and Liu G: TRB3, up-regulated in kidneys of rats with type1
diabetes, mediates extracellular matrix accumulation in vivo and in
vitro. Diabetes Res Clin Pract. 106:101–109. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tomcik M, Palumbo-Zerr K, Zerr P, Sumova
B, Avouac J, Dees C, Distler A, Becvar R, Distler O, Schett G, et
al: Tribbles homologue 3 stimulates canonical TGF-β signalling to
regulate fibroblast activation and tissue fibrosis. Ann Rheum Dis.
75:609–615. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tang M, Zhong M, Shang Y, Lin H, Deng J,
Jiang H, Lu H, Zhang Y and Zhang W: Differential regulation of
collagen types I and III expression in cardiac fibroblasts by AGEs
through TRB3/MAPK signaling pathway. Cell Mol Life Sci.
65:2924–2932. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang P, Wang Y, Nie X, Braïni C, Bai R and
Chen C: Multiwall carbon nanotubes directly promote
fibroblast-myofibroblast and epithelial-mesenchymal transitions
through the activation of the TGF-β/Smad signaling pathway. Small.
11:446–455. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tanjore H, Xu XC, Polosukhin VV, Degryse
AL, Li B, Han W, Sherrill TP, Plieth D, Neilson EG, Blackwell TS
and Lawson WE: Contribution of epithelial-derived fibroblasts to
bleomycin-induced lung fibrosis. Am J Respir Crit Care Med.
180:657–665. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim SJ, Cheresh P, Jablonski RP, Williams
DB and Kamp DW: The role of mitochondrial DNA in mediating alveolar
epithelial cell apoptosis and pulmonary fibrosis. Int J Mol Sci.
16:21486–21519. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Todd NW, Luzina IG and Atamas SP:
Molecular and cellular mechanisms of pulmonary fibrosis.
Fibrogenesis Tissue Repair. 5:112012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Matsuzawa Y, Kawashima T, Kuwabara R,
Hayakawa S, Irie T, Yoshida T, Rikitake H, Wakabayashi T, Okada N,
Kawashima K, et al: Change in serum marker of oxidative stress in
the progression of idiopathic pulmonary fibrosis. Pulm Pharmacol
Ther. 32:1–6. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sakai N and Tager AM: Fibrosis of two:
Epithelial cell-fibroblast interactions in pulmonary fibrosis.
Biochim Biophys Acta. 1832:911–921. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu RM and Desai LP: Reciprocal regulation
of TGF-β and reactive oxygen species: A perverse cycle for
fibrosis. Redox Biol. 6:565–577. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weidinger A and Kozlov AV: Biological
activities of reactive oxygen and nitrogen species: Oxidative
stress versus signal transduction. Biomolecules. 5:472–484. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang W, Cheng J, Sun A, Lv S, Liu H, Liu
X, Guan G and Liu G: TRB3 mediates renal tubular cell apoptosis
associated with proteinuria. Clin Exp Med. 15:167–177. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cheng WP, Wang BW, Lo HM and Shyu KG:
Mechanical stretch induces apoptosis regulator TRB3 in cultured
cardiomyocytes and volume-overloaded heart. PLoS One.
10:e01232352015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bromati CR, Lellis-Santos C, Yamanaka TS,
Nogueira TC, Leonelli M, Caperuto LC, Gorjão R, Leite AR, Anhê GF
and Bordin S: UPR induces transient burst of apoptosis in islets of
early lactating rats through reduced AKT phosphorylation via
ATF4/CHOP stimulation of TRB3 expression. Am J Physiol Regul Integr
Comp Physiol. 300:R92–R100. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Avery J, Etzion S, DeBosch BJ, Jin X, Lupu
TS, Beitinjaneh B, Grand J, Kovacs A, Sambandam N and Muslin AJ:
TRB3 function in cardiac endoplasmic reticulum stress. Circ Res.
106:1516–1523. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kage H and Borok Z: EMT and interstitial
lung disease: A mysterious relationship. Curr Opin Pulm Med.
18:517–523. 2012.PubMed/NCBI
|