Introduction
An increasing trend has been observed in the
incidence of coronary atherosclerotic heart disease, which is seen
as one of the most common diseases that threatens human health
(1). In 2008, the World Health
Organization (WHO) released the ‘Global Health Statistics Report’
and reported that following malignant tumors, cardiovascular and
cerebrovascular diseases have become the principal diseases leading
to human mortality (2). Ischemic
heart disease was reported to be the leading cause of mortality
globally, resulting in >8 million deaths in 2013 (3). At present, the primary intervention
in the therapy of AMI is to promptly open the myocardial
infarction-associated artery and induce reperfusion by coronary
artery bypass graft. However, it was identified that these
treatments were accompanied by ischemia-reperfusion (I/R) injury,
which impacts the effects of the treatment (4). I/R injury is the focus of
cardiovascular research. The application of drug intervention
following reperfusion may effectively reduce myocardial I/R (MI/R)
injury (5–7). Therefore, the search for novel
therapeutic agents to reduce the MI/R injury in the clinical
setting is necessary.
Oxidative stress is a highly complex biological
process in MI/R injury. During MI/R injury, a variety of adverse
effects on the organism were observed, which further activated the
neuroendocrine regulatory network through the
hypothalamic-pituitary-adrenal axis and via sympathetic nervous
system pathways (8,9). Furthermore, the process of MI/R
injury leads to abnormal cell proliferation, hypertrophy,
differentiation and death, resulting in a series of
pathophysiological effects (10,11).
The transmission and regulation of stress signals depends on the
neuroendocrine network and cell-signaling network. Previous studies
have demonstrated that in the pathological process of
stress-induced cardiac disorders and cardiovascular diseases, MI/R
injury, including cardiomyocyte necrosis and apoptosis, occurs
(12–15). Overproduction of epinephrine and
GCs in stress not only activates the protein kinase A system and
other signaling pathways in cardiomyocytes (16); however, additionally enhances the
expression of fibroblast-associated FAS and promotes the activation
of caspase-3 and −9. These two phenomena may lead to an increased
mitochondrial membrane potential (MMP) in cardiomyocytes. These
processes further cause the release of mitochondrial cytochrome
c (cyto c), apoptosis-inducing factor (AIF) and
second mitochondria-derived activator of caspase (Smac), leading to
the apoptosis of cardiomyocytes (17–19).
However, the accurate mechanisms of stress and mitochondria in MI/R
injury remain unclear.
Uncaria rhynchophylla, a type of
Rubiaceae plant, has been used for treating cerebral
diseases (20). Uncaria
rhynchophylla functions by lowering blood pressure, reversing
cardiac hypertrophy, and reducing the force and frequency of
cardiac muscle contraction (20).
Rhynchophylline (RP), the primary active ingredient of Uncaria
rhynchophylla, possesses an antihypertensive effect and
protects against ischemia-induced neuronal damage (21,22).
Previous studies have demonstrated that RP is able to inhibit the
proliferation of vascular smooth muscle cells and reduced
angiotensin II-induced cardiomyocyte hypertrophy (23,24).
Other previous studies have additionally demonstrated that RP may
be used in cerebral ischemia protection, calcium channel blocking,
myocardial remodeling inhibition, anti-arrhythmia, anticonvulsant,
and anti-inflammatory and analgesic therapy (25–27).
However, little is known regarding the roles of RP in MI/R
injury.
The present study aimed to investigate whether RP
protects cardiomyocytes against MI/R injury. Furthermore, it was
noteworthy to investigate the exact roles and mechanisms of RP and
the mitochondrial mechanisms in MI/R injury.
Materials and methods
Establishment of cardiomyocyte
oxygen-glucose deprivation/reoxygenation (OGD/R) injury to simulate
MI/R damage
The rat H9c2 cardiomyocyte cell line was obtained
from the Cell Bank of Chinese Academy of Sciences (Shanghai,
China). To establish a model of MI/R in vitro, the
cardiomyocytes were first cultured in serum/glucose-free Dulbecco's
modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and an anoxic environment (95%
N2 and 5% CO2) at 37°C for 2 h. The gas
mixture of 95% N2 and 5% CO2 was replaced
with air at a flow velocity of 2 l/min. Subsequent to the hypoxia
process, the cardiomyocytes were transferred into fresh medium
(DMEM) and maintained in an oxygen-rich incubator (37°C; 95%
O2 and 5% CO2). Following incubation for 1 h,
the MI/R cell model was harvested.
Grouping
Five treatment groups were prepared, which were the
control group (cardiomyocytes with no treatment), MI/R group
(cardiomyocytes treated with OGD/R injury), 20 µM RP+MI/R group
(cardiomyocytes treated with OGD/R injury, and subsequently treated
with 20 µM RP at 37°C for 1 h), 40 µM RP+MI/R group (cardiomyocytes
treated with OGD/R injury, and subsequently treated with 40 µM RP
at 37°C for 1 h) and 80 µM RP+MI/R group (cardiomyocytes treated
with OGD/R injury, and subsequently treated with 80 µM RP at 37°C
for 1 h). RP was purchased from Beijing Solarbio Science &
Technology Co., Ltd. (Purity >98%; Beijing, China).
Cell viability analysis
A Cell Counting Kit-8 (CCK-8; Beyotime Institute of
Biotechnology, Haimen, China) was performed to assess the cell
viability of the cardiomyocytes. Cardiomyocytes in the logarithmic
phase were seeded into 96-well plates at a density of
~6×104 cells/ml/group, and maintained in a 5%
CO2 atmosphere at 37°C for 12 h. Subsequently, the
cardiomyocytes were maintained for 12, 24 and 48 h. A total of 10
µl CCK reagent was supplemented into the wells of the 96-well
plates. The cardiomyocytes were maintained for 3 h. A microplate
reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA) was used to
record the absorbance at 450 nm. Cell viability was evaluated as
the percentage of cell survival compared with the control.
Enzyme activity detection
The lactate dehydrogenase (LDH; cat. no. C0016)
detection kit, malondialdehyde (MDA; cat. no. S0131) detection kit,
superoxide dismutase (SOD; cat. no. S0101) assay kit and
glutathione peroxidase (GPx; cat. no. S0058) assay kit were
purchased from Beyotime Institute of Biotechnology. Detection of
MDA content, LDH activity, SOD activity and Gpx activity were
performed according to the manufacturer's protocols.
Reactive oxygen species (ROS) levels were detected
using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). A total of
2×106 cells/ml were seeded in a 6-well plate and 10 mM
DCFH-DA in PBS was added to the cells for 20 min at 37°C. DCF
fluorescence was measured using a BD LSR II flow cytometer (BD
Biosciences, Franklin Lakes, NJ, USA). The flow cytometry data were
analyzed using FlowJo software version 7.5.5 (FlowJo LLC, Ashland,
OR, USA).
Apoptosis assay
Flow cytometry (FCM) was conducted to assess the
apoptosis of cardiomyocytes. Following washing with PBS, cultured
cardiomyocytes were trypsinized with 0.25% trypsin (Beyotime
Institute of Biotechnology). The cells were harvested and
centrifuged at 1,000 × g for 5 min at 4°C. The supernatant was
removed and the cardiomyocytes were suspended in the incubation
buffer at a density of 1×106 cells/ml for assessment.
Apoptosis of cardiomyocytes was detected with the Annexin
V-fluorescein isothiocyanate and propidium iodide detection kit
(cat. no. V13242, Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's instructions. A flow cytometer
(FACSCalibur) running BD CellQuest™ software version 1.2 (both from
BD Biosciences) was used to assess cell apoptosis.
Evaluation of Ca2+ and MMP
in cardiomyocytes
PBS was added to the cultured cardiomyocytes until
the cell concentration reached 1×106 cells/ml. Rho 123
(5 µM; cat. no. R8004; Sigma-Aldrich; Merck KGaA) or Fluo-3AM [3
µM; cat. no. 70-F1243; Multisciences (Lianke) Biotech Co., Ltd.,
Hangzhou, China] were added to the cardiomyocytes. The
cardiomyocytes were incubated at room temperature and kept away
from light for 10 min. FCM was performed to assess the MMP and
Ca2+ level in cardiomyocytes, and the excitation
wavelength was 488 nm. In total, 10,000 cells were collected from
each sample. The data was analyzed using BD CellQuest™ software
version 1.2 (BD Biosciences).
Evaluation of mitochondrial
permeability transition pore (mPTP) in cardiomyocytes
Mitochondria were isolated from cells using a
Mitochondrial extraction kit (cat. no. SM0020; Beijing Solarbio
Science & Technology Co., Ltd.), according to the
manufacturer's protocols. Tumescent fluid (containing 120 mmol/l
KCl, 20 mmol/l MOPS, 10 mmol/l Tris-HCl and 5 mmol/l
KH2PO4; pH 7.4; Beyotime Institute of
Biotechnology) was added to the mitochondria and the mixture was
diluted to the point where the protein concentration reached 0.25
g/l at 25°C. Subsequently, 200 µmol/l CaCl2 was added to
the mitochondria and the absorbance at 520 nm was observed for 15
min. The alterations in absorbance suggested the degree of
mitochondrial swelling, which indicated the opening of mPTP.
Western blot analysis
Proteins were isolated using NP-40 lysis buffer
(Beyotime Institute of Biotechnology, China). The BCA Protein
quantification kit (cat. no. ab207002; Abcam, Cambridge, UK) was
used to determine protein concentration. A total of 25 µg/lane
protein was separated by 12% SDS-PAGE. The separated products were
transferred to a polyvinylidene difluoride membrane (EMD Millipore,
Billerica, MA, USA). The membranes were blocked with 5% skimmed
milk at room temperature for 2 h. Western blotting was performed
with specific primary antibodies at 4°C overnight:
anti-inactive-caspase (1:2,000; cat. no. ab184787; rabbit
anti-rat); anti-active-caspase-3 (1:500; cat. no. ab49822; rabbit
anti-rat); anti-inactive-caspase-9 (1:500; cat. no. ab138412;
rabbit anti-rat); anti-active-caspase-9 (1:1,000; cat. no. ab25758;
rabbit anti-rat); anti-mitochondrial cyto c (1:5,000; cat.
no. ab133504; rabbit anti-rat); anti-mitochondrial AIF (1:1,000;
cat. no. ab32516; rabbit anti-rat); anti-cyto c (1:1,000;
cat. no. ab110325; rabbit anti-rat); anti-AIF (1:1,000; cat. no.
ab1998; rabbit anti-rat); anti-cyclooxygenase (COX) IV (1:2,000;
cat. no. ab16056; rabbit anti-rat); and anti-actin (1:5,000; cat.
no. ab179467; rabbit anti-rat) (all from Abcam). Horseradish
peroxidase-conjugated secondary antibodies (1:5,000; Abcam; cat.
no. ab205718; goat anti-rabbit) were supplemented and incubated at
room temperature for 1 h. Enhanced chemiluminescent (ECL) reagents
(EMD Millipore) in combination with an ECL system (GE Healthcare,
Chicago, IL, USA) were used to assess the results. The density of
the protein bands was quantified using Quantity One®
software version 4.2.1 (Bio-Rad Laboratories, Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR) analysis
Total RNA was extracted from the cultured
cardiomyocytes using TRIzol® reagent (Thermo Fisher
Scientific, Inc.). RNA was reverse transcribed to cDNA using
BeyoRT™ II cDNA Synthesis kit (Beyotime Institute of
Biotechnology), according to the manufacturer's protocol. qPCR was
performed using a SYBR-Green PCR Master Mix kit (Applied
Biosystems; Thermo Fisher Scientific, Inc.) on a ABI 7500
Thermocycler (Applied Biosystems). PCR cycling conditions were as
follows: 10 min pretreatment at 94°C, at 95°C for 15 sec, at 68°C
for 45 sec (45 cycles), at 95°C for 15 sec, at 68°C for 1 min, at
94°C for 15 sec, a final extension at 75°C for 10 min and held at
4°C. The primers were purchased from Invitrogen (Thermo Fisher
Scientific, Inc.): Caspase-3, forward, 5′-TGTCGATGCAGCTAACCTCA-3′
and reverse, 5′-GCAGTAGTCGCCTCTGAAGA-3′ (product: 241 bp);
caspase-9, forward, 5′-CATTGGTTCTGGCAGAGCTC-3′ and reverse,
5′-AGCAGTCAGGTCGTTCTTCA-3′ (product: 238 bp); cyto c,
forward, 5′-CAACTGCACAAGACAACCCA-3′ and reverse,
5′-ATAGCACAATCCCCACCACA-3′ (product: 204 bp); AIF, forward,
5′-ACATGCGACCTCCTCTTTCA-3′ and reverse, 5′-TGCCTCTTACATCCAGGTGG-3′
(product: 213 bp); COX IV, forward, 5′-TGAAGGAGAAGGAGAAGGCC-3′ and
reverse, 5′-ACCCAGTCACGATCAAAGGT-3′ (product: 229 bp); actin,
forward, 5′-CAACATGGATGAGCGGAAGG-3′ and reverse,
5′-GCAGTGTAGCAGCATCGAAA-3′ (product: 233 bp). Actin was used as the
control of the input RNA level. The 2−ΔΔCq method was
used to analyze the relative gene expression (28).
Statistical analysis
Results in the present study are presented as the
mean ± standard error of the mean with at least three independent
experiments repeated. All experimental data were analyzed one-way
analysis of variance followed by the Dunnett's test. P<0.05 was
considered to indicate a statistically significant difference. Data
analysis was performed using GraphPad Prism version 6.0 (GraphPad
Software, Inc., La Jolla, CA, USA).
Results
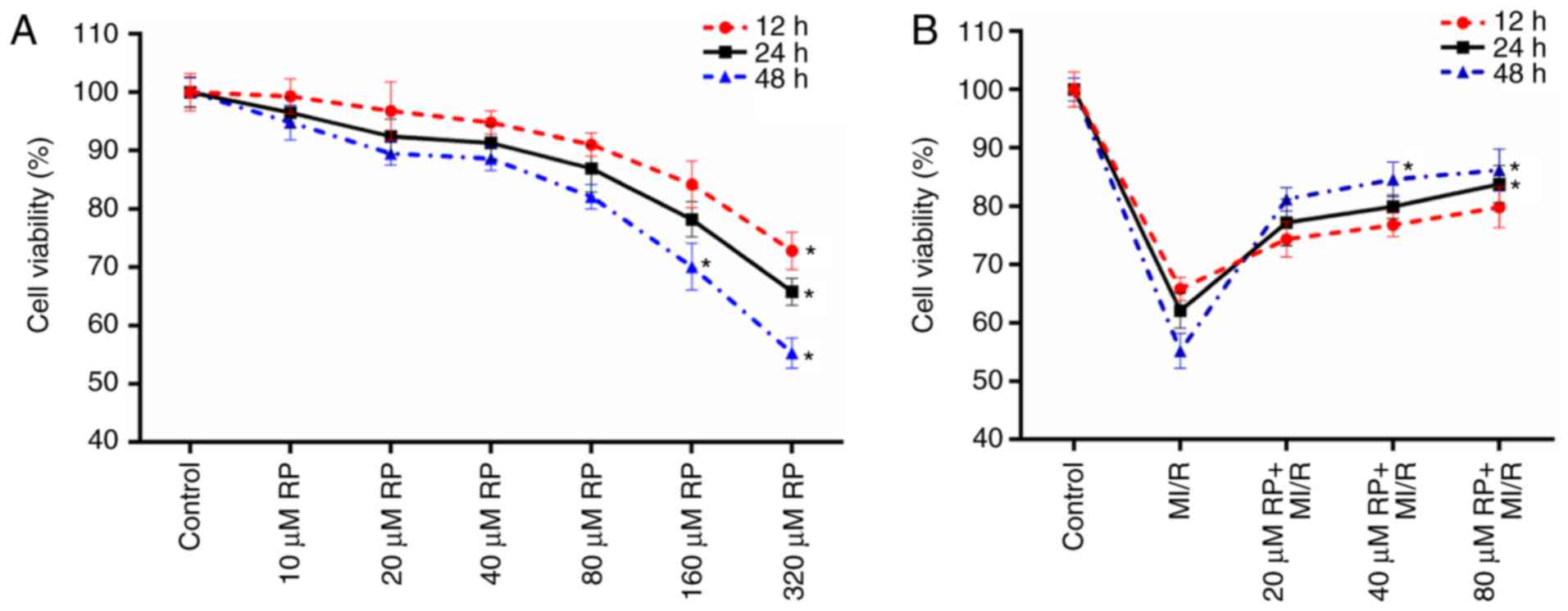
RP increases the cell viability of
MI/R-induced cardiomyocytes
The cell viabilities of cardiomyocytes treated with
different concentrations of RP were measured in the present study.
It was observed that compared with the control group, the cell
activities of cardiomyocytes treated with RP at concentrations of
10, 20, 40 and 80 µM exhibited no significant difference (Fig. 1A). However, it was identified that
with the treatments of 160 and 320 µM RP, the cell viabilities of
cardiomyocytes were significantly decreased, particularly at 48 h
treatment (P<0.05; Fig. 1A).
These results suggested that low concentrations of RP exhibited a
limited effect on the cell viability of cardiomyocytes. Therefore,
the cell viabilities of cardiomyocytes subjected to MI/R injury and
MI/R-induced cardiomyocytes treated with 20, 40 and 80 µM RP were
further evaluated. According to the results, it was observed that
MI/R injury decreased the cell viability of cardiomyocytes;
whereas, RP increased the cell viability of MI/R-induced
cardiomyocytes in a dose-dependent manner (Fig. 1B). The results indicated that RP
was able to enhance the cell viability of cardiomyocytes, which
were subjected to MI/R injury. Therefore, it was confirmed that
MI/R resulted in low cell viability of cardiomyocytes; whereas, RP
increased the cell viability of MI/R-induced cardiomyocytes within
a certain concentration range.
RP decreases the oxidative stress and
mPTP level of MI/R-induced cardiomyocytes
Oxidative stress has been proposed to contribute to
the development and progression of MI/R (5). Therefore, the levels of oxidative
stress markers, including ROS, MDA, LDH, SOD and GPx in
cardiomyocytes treated with MI/R and different concentrations of RP
were assessed. The ROS, MDA and LDH content in cardiomyocytes
treated with MI/R in advance were markedly higher compared with the
control, while treatment with ≥40 µM RP significantly decreased the
ROS, MDA and LDH content in MI/R-induced cardiomyocytes (P<0.05;
Fig. 2A-C). However, MI/R was
observed to be able to reduce the activities of SOD and GPx in
cardiomyocytes. The SOD and GPx activity in MI/R-induced
cardiomyocytes was significantly increased (P<0.05; Fig. 2D and E) following treatment with
≥40 µM RP. It was suggested that RP reduced oxidative stress in
MI/R-induced cardiomyocytes. The opening degree of mPTP in
cardiomyocytes was assessed by spectrophotometry. The results
revealed that MI/R increased the mPTP opening degree in
cardiomyocytes (P<0.05; Fig.
2F); whereas, RP decreased the opening degree of mPTP in
MI/R-induced cardiomyocytes.
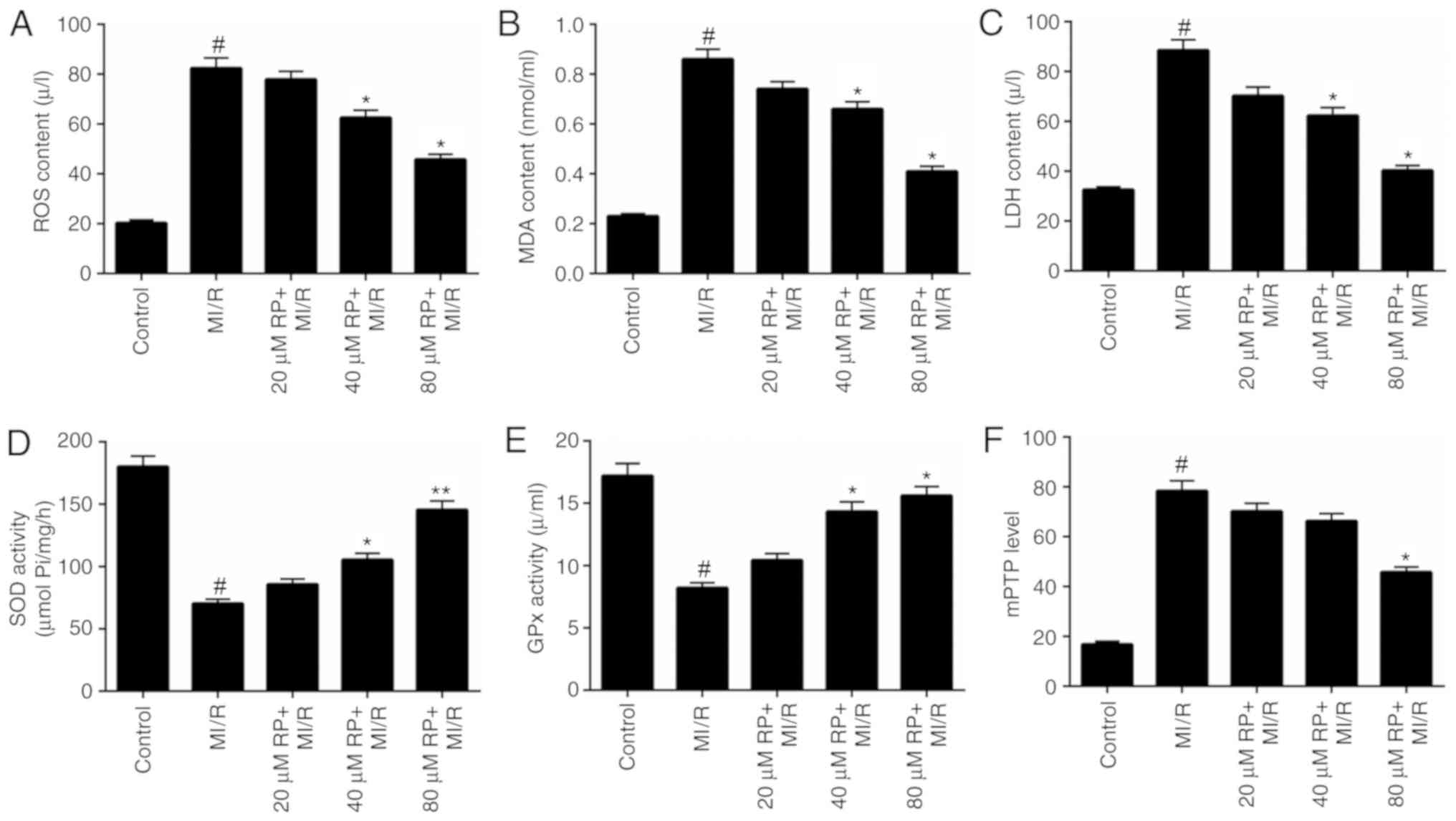 | Figure 2.RP decreases the oxidative stress and
mPTP level of MI/R-induced cardiomyocytes. Cardiomyocytes were
treated with MI/R, 20 µM RP+MI/R, 40 µM RP+MI/R and 80 µM RP+MI/R.
(A) ROS levels were detected by flow cytometric analysis. (B) MDA
content, (C) LDH activity, (D) SOD activity and (E) GPx activity in
cardiomyocytes were measured using commercial kits. (F)
Spectrophotometry was performed to detect the degree of mPTP
openness in cardiomyocytes. #P<0.05 vs. Control;
*P<0.05, **P<0.01 vs. MI/R. RP, rhynchophylline; mPTP,
mitochondrial permeability transition pore; ROS, reactive oxygen
species; MDA, malondialdehyde; LDH, lactate dehydrogenase; SOD,
superoxide dismutase; GPx, glutathione peroxidase; MI/R, myocardial
ischemia-reperfusion. |
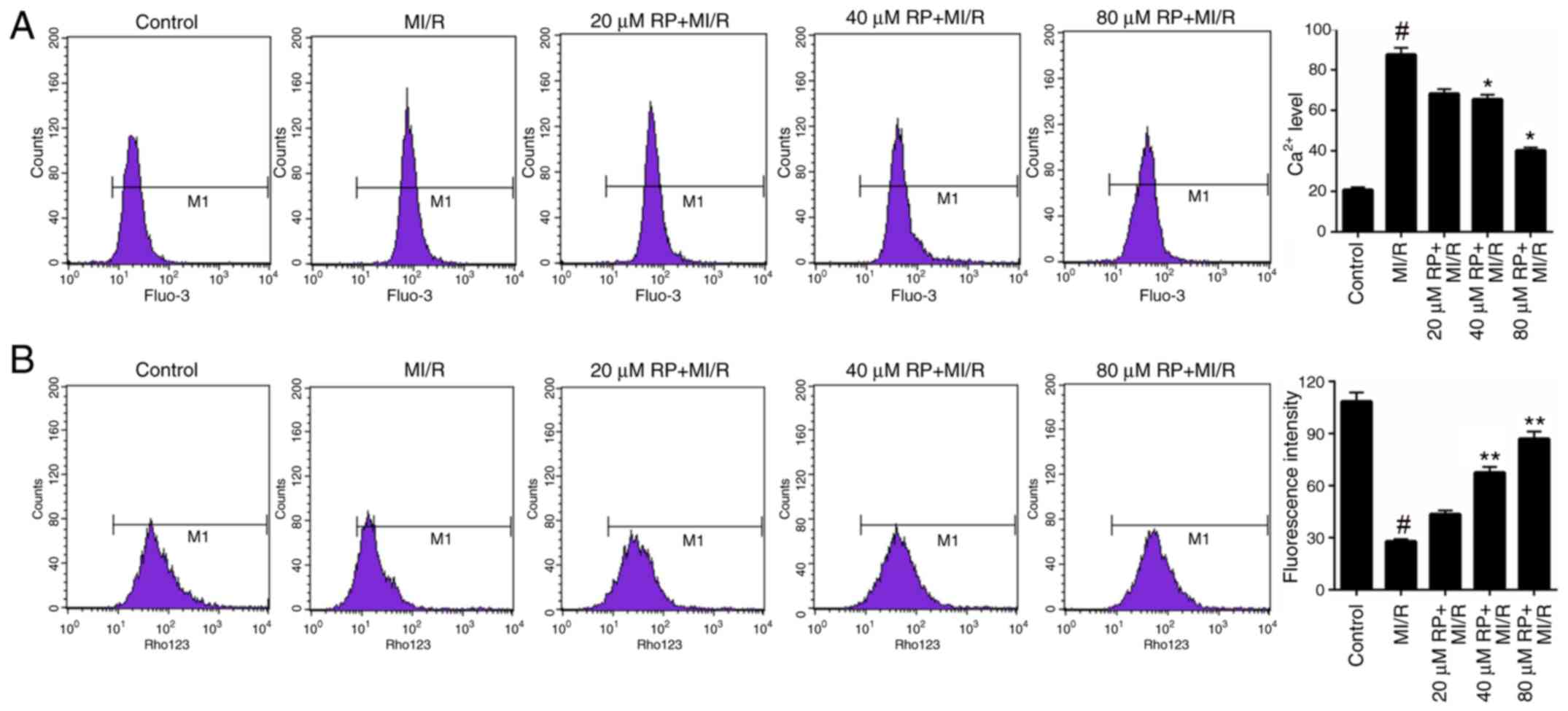
RP modulates the Ca2+ and
MMP levels in MI/R-induced cardiomyocytes
Furthermore, the Ca2+ and MMP levels in
cardiomyocytes treated with MI/R and different concentrations of RP
were measured. Based on the FCM data, it was identified that MI/R
markedly increased the Ca2+ level in cardiomyocytes.
Following treatment with 40 and 80 µM RP, the Ca2+ level
in MI/R-induced cardiomyocytes was significantly decreased compared
with the MI/R group (P<0.05; Fig.
3A), suggesting that RP was able to decrease the
Ca2+ level in MI/R-induced cardiomyocytes. Additionally,
the FCM results on MMP revealed that MI/R significantly decreased
the MMP level in cardiomyocytes; whereas, treatment with 40 and 80
µM RP was observed to significantly increase the MMP levels in
MI/R-induced cardiomyocytes (P<0.01; Fig. 3B). These results suggested that RP
was able to modulate the Ca2+ and MMP levels in
MI/R-induced cardiomyocytes.
RP regulates mitochondrial-associated
gene expression
The mitochondrial mechanisms of RP in protecting
cardiomyocytes against MI/R induced injury were examined. In this
analysis, the mitochondrial cyto c, mitochondrial AIF,
cytosolic cyto c and cytosolic AIF expression in
cardiomyocytes were measured. In the cardiomyocytes from the MI/R
groups, the mitochondrial cyto c expression was decreased
compared with the control, while the expression level of
mitochondrial AIF was markedly increased compared with the control.
Increases were observed in the mitochondrial cyto c
expression in MI/R-induced cardiomyocytes treated with 40 and 80 µM
RP. It was additionally identified that RP was able to downregulate
the expression level of mitochondrial AIF in MI/R-induced
cardiomyocytes, with significant decreases observed in the 40 and
80 µM RP+MI/R groups (P<0.01; Fig.
4A and B). Additionally, the expression level of cytosolic cyto
c in cardiomyocytes was upregulated upon MI/R injury, while
cytosol AIF expression was reduced by MI/R injury (P<0.05).
Following treatment with RP, the expression level of cytosolic cyto
c was decreased; whereas, the cytosolic AIF expression was
significantly increased in MI/R-induced cardiomyocytes (P<0.05;
Fig. 4C and D). Therefore, it was
demonstrated that RP upregulated the mitochondrial cyto c
and cytosolic AIF expression, and downregulated the expression
levels of mitochondrial AIF and cytosolic cyto c in
MI/R-induced cardiomyocytes.
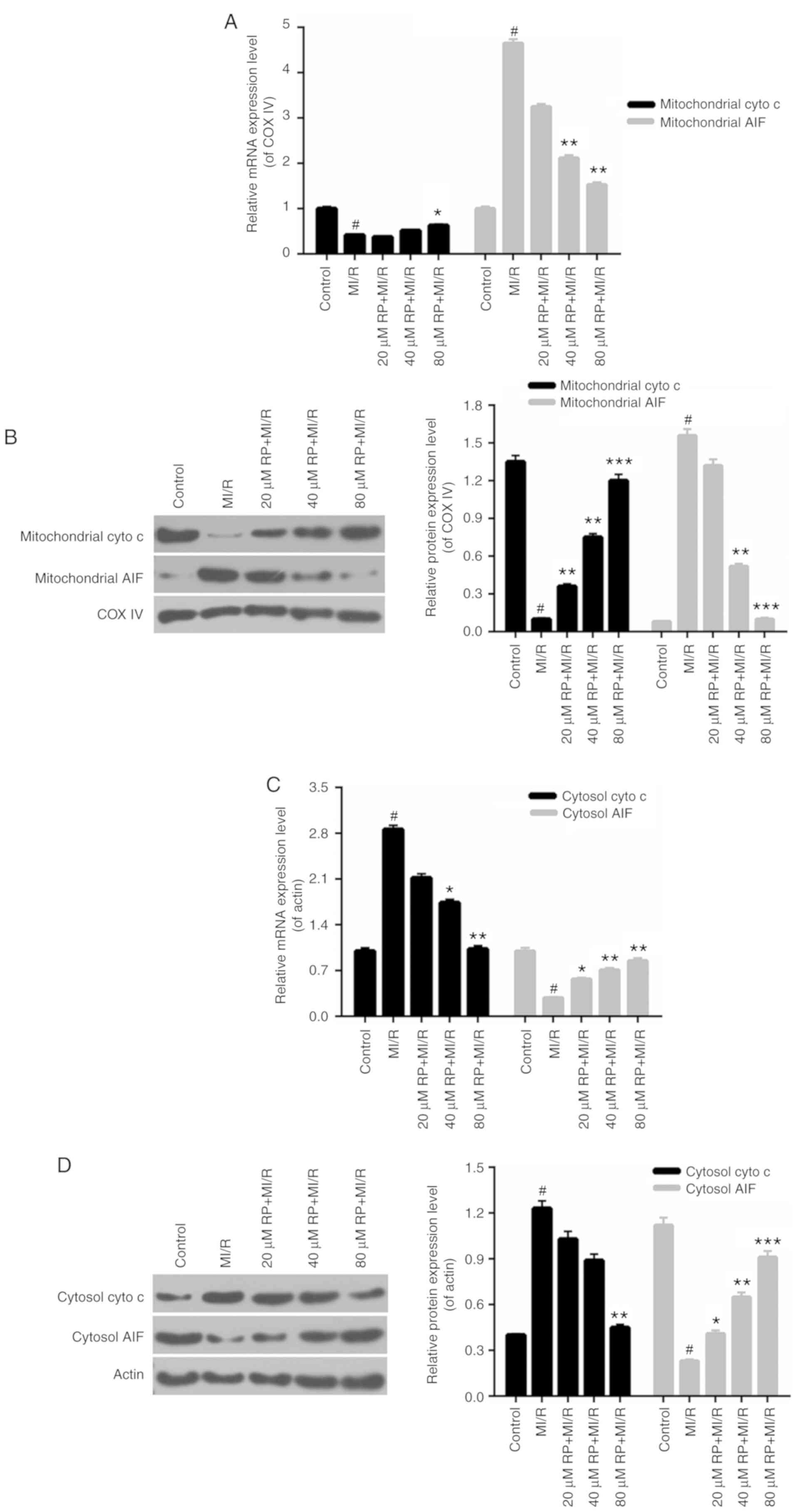 | Figure 4.RP regulates the
mitochondrial-associated gene expression. Cardiomyocytes were
treated with MI/R, 20 µM RP+MI/R, 40 µM RP+MI/R and 80 µM RP+MI/R.
(A) RT-qPCR and (B) western blot assays were performed on the
expression levels of mitochondrial cyto c and mitochondrial
AIF in cardiomyocytes. (C) RT-qPCR and (D) western blot assays were
conducted to determine the expression levels of cytosolic cyto
c and cytosolic AIF in cardiomyocytes. #P<0.05
vs. Control; *P<0.05, **P<0.01, ***P<0.001 vs. MI/R. Rp,
rhynchophylline; MI/R, myocardial ischemia-reperfusion; RT-qPCR,
reverse transcription-quantitative polymerase chain reaction; cyto
c, cytochrome c; AIF, apoptosis-inducing factor; COX,
cyclooxygenase. |
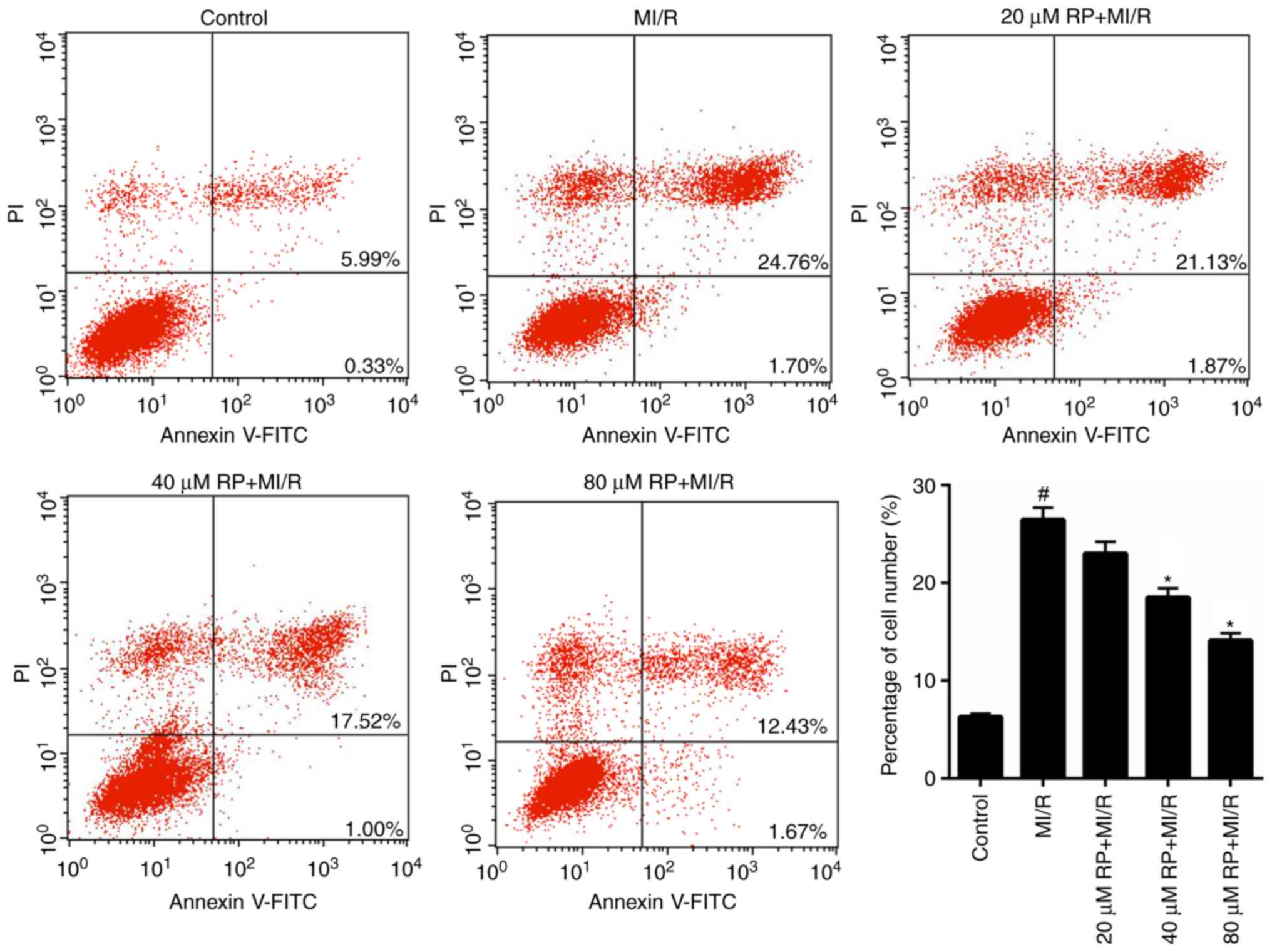
RP suppresses the MI/R-induced
apoptosis of cardiomyocytes
From the aforementioned results, it was determined
that RP was able to enhance the cell viability of MI/R-induced
cardiomyocytes; therefore, the apoptotic capacity of MI/R-induced
cardiomyocytes treated with RP was evaluated. It was demonstrated
that the proportion of apoptotic cardiomyocytes in MI/R group was
26.46%, which was significantly increased compared with the control
(6.32%). Furthermore, following treatment with 20, 40 and 80 µM RP,
the percentage of apoptotic MI/R-induced cardiomyocytes decreased
from 26.46 to 23, 18.52 and 14.1%, respectively (Fig. 5). In particular, significant
decreases in the percentage of apoptotic cells were observed in the
40 and 80 µM RP+MI/R groups (P<0.05). These data suggested that
RP markedly decreased the apoptotic capacity of MI/R-induced
cardiomyocytes in a dose-dependent manner.
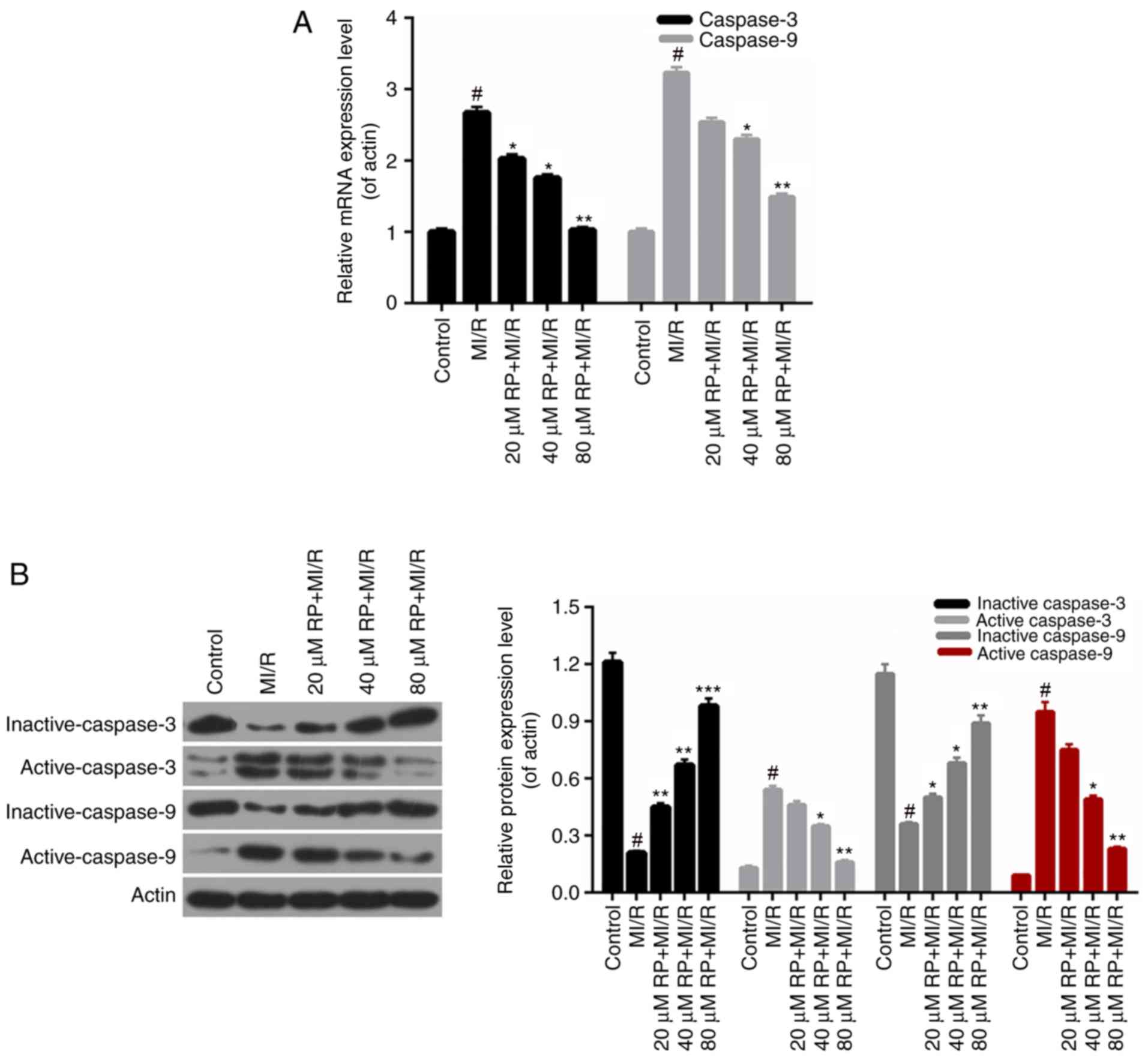
RP affects the apoptosis-associated
protein expression
According to the apoptosis results above, it was
demonstrated that RP inhibited the apoptosis of cardiomyocytes
induced by MI/R. Therefore, the apoptosis-associated proteins in
cardiomyocytes were further investigated. According to the RT-qPCR
data, it was observed that the caspase-3 and caspase-9 expression
in cardiomyocytes from MI/R group was significantly increased
compared with the control; whereas, the caspase-3 and caspase-9
expressions in MI/R-induced cardiomyocytes were decreased by RP in
a dose-dependent manner, with significant differences observed with
40 and 80 µM RP (P<0.05; Fig.
6A). Furthermore, the western blot analysis revealed that the
expression levels of inactive caspase-3 and −9 in cardiomyocytes
were markedly downregulated with MI/R injury; however, were
upregulated upon treatment with different concentrations of RP.
Additionally, it was identified that MI/R upregulated the active
caspase-3 and −9 expression in cardiomyocytes. However, RP was able
to decrease the expression levels of caspase-3 and −9 in
MI/R-induced cardiomyocytes compared with the control (Fig. 6B). Therefore, it was demonstrated
that RP modulated the apoptosis-associated protein expression in
MI/R-induced cardiomyocytes.
Discussion
Cardiomyocyte injury is a critical cytological basis
for stress-induced cardiovascular disease. Cell apoptosis, a
crucial cytological mechanism of cardiomyocyte injury, serves a
role in a number of cardiovascular diseases, including heart
failure, AMI, atherosclerosis and MI/R injury (29). RP represents a crucial active
substance isolated from Chinese herbal medicine. Previous studies
demonstrated that RP had neuroprotective functions (30,31).
Additionally, Cao et al (32) suggested that RP may prevent cardiac
dysfunction and improve survival in lipopolysaccharide-challenged
mice by suppressing macrophage inhibitor-κBα phosphorylation.
However, the roles of RP in MI/R injury remain unclear. Therefore,
an aim of the present study was to investigate the effect of RP in
the MI/R injury and the associated mechanisms. The cell viabilities
of cardiomyocytes treated with different concentrations of RP were
measured to assess the cytotoxicity of RP. It was identified that
the cell viability of cardiomyocytes began to decrease after the
cells were treated with 160 µM RP for 48 h. Therefore, the cell
viability of cardiomyocytes treated with MI/R injury in addition to
using 20, 40 and 80 µM RP, was further assessed. The results
demonstrated that MI/R injury inhibited the cell viability of
cardiomyocytes, while RP markedly increased the cell viability of
MI/R-induced cardiomyocytes, particularly with treatment duration
of 48 h. Therefore, it was demonstrated that RP increased the cell
viability of cardiomyocytes subjected to MI/R injury.
It was demonstrated that acute stress may lead to
spasmodic cardiac arrhythmia and sudden mortality (33). Long-term high-intensity oxidative
stress additionally causes a variety of serious cardiovascular
diseases, for instance, hypertension and atherosclerosis (10,34).
Additionally, it has been identified that MI/R injury stimulates
severe oxidative stress in cardiomyocytes (35–37).
Therefore, to identify whether RP was able to modulate the
oxidative stress in cardiomyocytes subjected to MI/R injury, the
levels of oxidative stress markers in cardiomyocytes were assessed.
From the present results, it was observed that RP markedly reduced
the levels of ROS, MDA and LDH, and increased the SOD and GPx
activities in MI/R-induced cardiomyocytes. These results suggested
that RP may function to reduce oxidative stress in the
cardiomyocytes with MI/R injury. Therefore, it was concluded that
RP is able to reduce the oxidative stress in cardiomyocytes
subjected to MI/R injury.
Previous studies suggested that mitochondria serve
as an important pathway of stress-induced cardiomyocyte injury
(38,39). Mitochondria not only provide ATP to
cells but additionally participate in multiple cytopathic processes
(40,41). Mitochondria act as important
pathways that mediate oxidative stress and cell damage in
organisms. Previous studies demonstrated that the mPTP opening
degree, Ca2+ disturbances and MMP levels serve crucial
roles in the modulation of oxidative stress (42–44).
Therefore, the mPTP, Ca2+ and MMP levels were evaluated
in cardiomyocytes with MI/R injury and treated with different
concentrations of RP to investigate the effect of RP on the
mitochondria. Based on the present results, it was observed that RP
reduced the opening degree of mPTP in MI/R-induced cardiomyocytes.
The FCM data revealed that RP decreased the Ca2+ level;
however, it increased the MMP level in MI/R-induced cardiomyocytes.
These results suggested that RP was able to affect the functions of
mitochondria in the cardiomyocytes with MI/R injury. It was
demonstrated that an increased MMP level may result in the release
of mitochondrial cyto c, AIF, Smac and other
apoptosis-inducing factor, resulting in cell apoptosis (17–19).
In order to further investigate the accurate mechanisms of RP in
the modulation of mitochondria function, the associated
mitochondrial-associated mechanisms in cardiomyocytes were
assessed. It was identified that in mitochondria of MI/R-induced
cardiomyocytes, RP markedly upregulated cyto c expression,
while it decreased the expression level of AIF. Nevertheless, the
cyto c and AIF expression modulations in the cytosol of
MI/R-induced cardiomyocytes by RP were reversed to the expression
of those in mitochondria. RP markedly downregulated the cyto
c expression, while it upregulated the AIF expression in the
cytosol of MI/R-induced cardiomyocytes. Therefore, it was
hypothesized that RP is able to reduce the release of cyto c
from the mitochondria into cytosol and stimulate the release of AIF
from mitochondria into the cytosol. Collectively, it was
demonstrated that RP affected the functions of mitochondria by
regulating the mitochondrial mechanisms in cardiomyocytes suffering
from MI/R injury.
Sinha et al (45) suggested that oxidative stress may
cause cellular apoptosis via the mitochondria-dependent pathway.
Based on the results described above, it was demonstrated that RP
affected the oxidative stress and mitochondria mechanisms in
cardiomyocytes suffering from MI/R injury. Therefore, it was
hypothesized that RP may affect the apoptotic capacity of
cardiomyocytes induced by MI/R injury. Therefore, the cell
apoptosis of cardiomyocytes treated with MI/R injury at 20, 40 and
80 µM of RP was evaluated. The FCM data indicated that MI/R
markedly accelerated the apoptosis ability of cardiomyocytes, while
RP evidently suppressed the apoptosis of MI/R-induced
cardiomyocytes in a dose-dependent manner. These results
demonstrated that RP was able to suppress the apoptosis of
cardiomyocytes induced by MI/R injury. In order to further examine
the accurate mechanisms of RP underlying the protection of
cardiomyocytes against MI/R injury, the apoptosis-associated
mechanisms were investigated. The RT-qPCR and western blotting data
confirmed that RP markedly downregulated the expression levels of
caspase-3 and caspase-9 in MI/R-induced cardiomyocytes. Therefore,
it was demonstrated that RP suppressed the apoptosis of
MI/R-induced cardiomyocytes by regulating the expression levels of
caspase-3 and caspase-9.
In conclusion, the present study demonstrated that
RP ameliorated MI/R injury through the modulation of mitochondrial
mechanisms. The present results have provided novel insight into
the mechanisms of RP and cardiomyocytes. The potential effects of
RP on the protection of MI/R-induced cardiomyocytes suggest that RP
may be an effective target for MI/R injury therapy.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated and/or analyzed during this study
are included in this published article.
Authors' contributions
QJQ, LQC and MLG designed the study. QJQ, LQC, PL,
YBW and XZZ performed the experiments. PL, YBW and XZZ performed
data analysis. QJQ wrote the main manuscript. QJQ, LQC, PL and MLG
contributed to manuscript revisions. All authors reviewed the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
McCullough PA: Coronary artery disease.
Clin J Am Soc Nephrol. 2:611–616. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roger VL, Go AS, Lloyd-Jones DM, Adams RJ,
Berry JD, Brown TM, Carnethon MR, Dai S, De Simone G, Ford ES, et
al: Heart disease and stroke statistics-2011 update: A report from
the american heart association. Circulation. 123:e18–e209. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
GBD 2013 Mortality and Causes of Death
Collaborators: Global, regional, and national age-sex specific
all-cause and cause-specific mortality for 240 causes of death,
1990–2013: A systematic analysis for the Global Burden of Disease
Study 2013. Lancet. 385:117–171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rodriguez-Sinovas A, Abdallah Y, Piper HM
and Garcia-Dorado D: Reperfusion injury as a therapeutic challenge
in patients with acute myocardial infarction. Heart Fail Rev.
12:207–216. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bolli R: Preconditioning: A paradigm shift
in the biology of myocardial ischemia. Am J Physiol Heart Circ
Physiol. 292:H19–H27. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kitakaze M: How to mediate
cardioprotection in ischemic hearts-accumulated evidence of basic
research should translate to clinical medicine. Cardiovasc Drugs
Ther. 24:217–223. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tissier R, Waintraub X, Couvreur N,
Gervais M, Bruneval P, Mandet C, Zini R, Enriquez B, Berdeaux A and
Ghaleh B: Pharmacological postconditioning with the phytoestrogen
genistein. J Mol Cell Cardiol. 42:79–87. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chida Y and Hamer M: Chronic psychosocial
factors and acute physiological responses to laboratory-induced
stress in healthy populations: A quantitative review of 30 years of
investigations. Psychol Bull. 134:829–885. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sinha R: Chronic stress, drug use, and
vulnerability to addiction. Ann N Y Acad Sci. 1141:105–130. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma XL, Kumar S, Gao F, Louden CS, Lopez
BL, Christopher TA, Wang C, Lee JC, Feuerstein GZ and Yue TL:
Inhibition of p38 mitogen-activated protein kinase decreases
cardiomyocyte apoptosis and improves cardiac function after
myocardial ischemia and reperfusion. Circulation. 99:1685–1691.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schwartz AR, Gerin W, Davidson KW,
Pickering TG, Brosschot JF, Thayer JF, Christenfeld N and Linden W:
Toward a causal model of cardiovascular responses to stress and the
development of cardiovascular disease. Psychosom Med. 65:22–35.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bozner A, Balaz V, Dostal J and Szigetiova
A: Electron microscopy morphometric analysis of the effect of
immobilization stress on the structure of the myocardium in rats
belonging to 2 age groups. Cesk Patol. 20:146–150. 1984.(In
Slovak). PubMed/NCBI
|
|
13
|
Jonsson L, Johansson G, Lannek N, Lindberg
P and Poupa O: Histochemical and electron microscopic studies of
acute cardiomyopathy induced by restraint stress in pigs. Recent
Adv Stud Cardiac Struct Metab. 6:461–470. 1975.PubMed/NCBI
|
|
14
|
Jonsson L and Johansson G: Cardiac muscle
cell damage induced by restraint stress. Virchows Arch B Cell
Pathol. 17:1–12. 1974.PubMed/NCBI
|
|
15
|
Kuder T: Electron microscopic studies of
neurocytes of the pterygopalatine ganglion in the rat after
immobilization. Arch Vet Pol. 32:93–102. 1992.PubMed/NCBI
|
|
16
|
Cai Q, Huang S, Zhu Z, Li H, Li Q, Jia N
and Liu J: The effects of prenatal stress on expression of p38 MAPK
in offspring hippocampus. Int J Dev Neurosci. 26:535–540. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Grinberg M, Sarig R, Zaltsman Y, Frumkin
D, Grammatikakis N, Reuveny E and Gross A: tBID Homooligomerizes in
the mitochondrial membrane to induce apoptosis. J Biol Chem.
277:12237–12245. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Komarov AP, Rokhlin OW, Yu CA and Gudkov
AV: Functional genetic screening reveals the role of mitochondrial
cytochrome b as a mediator of FAS-induced apoptosis. Proc Natl Acad
Sci USA. 105:14453–14458. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Perchellet EM, Wang Y, Weber RL,
Sperfslage BJ, Lou K, Crossland J, Hua DH and Perchellet JP:
Synthetic 1,4-anthracenedione analogs induce cytochrome c
release, caspase-9, −3, and −8 activities, poly(ADP-ribose)
polymerase-1 cleavage and internucleosomal DNA fragmentation in
HL-60 cells by a mechanism which involves caspase-2 activation but
not Fas signaling. Biochem Pharmacol. 67:523–537. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ndagijimana A, Wang X, Pan G, Zhang F,
Feng H and Olaleye O: A review on indole alkaloids isolated from
Uncaria rhynchophylla and their pharmacological studies.
Fitoterapia. 86:35–47. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kang TH, Murakami Y, Takayama H, Kitajima
M, Aimi N, Watanabe H and Matsumoto K: Protective effect of
rhynchophylline and isorhynchophylline on in vitro ischemia-induced
neuronal damage in the hippocampus: Putative neurotransmitter
receptors involved in their action. Life Sci. 76:331–343. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou J and Zhou S: Antihypertensive and
neuroprotective activities of rhynchophylline: The role of
rhynchophylline in neurotransmission and ion channel activity. J
Ethnopharmacol. 132:15–27. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang F, Sun AS, Yu LM, Wu Q and Gong QH:
Effects of isorhynchophylline on angiotensin II-induced
proliferation in rat vascular smooth muscle cells. J Pharm
Pharmacol. 60:1673–1678. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
He N, Sun A, Wu Q, Huang X and Shi J:
Inhibitory effect of rhynchophylline on cardiomyocyte hypertrophy
induced by angiotensin II. Chin J Pharmacol Toxicol. 24:255–260.
2010.
|
|
25
|
Doggrell SA and Brown L: Rat models of
hypertension, cardiac hypertrophy and failure. Cardiovasc Res.
39:89–105. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Haugen E, Chen J, Wikström J, Grönros J,
Gan LM and Fu LX: Parallel gene expressions of IL-6 and BNP during
cardiac hypertrophy complicated with diastolic dysfunction in
spontaneously hypertensive rats. Int J Cardiol. 115:24–28. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou JY and Zhou SW: Isorhynchophylline: A
plant alkaloid with therapeutic potential for cardiovascular and
central nervous system diseases. Fitoterapia. 83:617–626. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) methods. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Feuerstein GZ and Young PR: Apoptosis in
cardiac diseases: Stress- and mitogen-activated signaling pathways.
Cardiovasc Res. 45:560–569. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang H, Zhong R, Xia Z, Song J and Feng
L: Neuroprotective effects of rhynchophylline against ischemic
brain injury via regulation of the Akt/mTOR and TLRs signaling
pathways. Molecules. 19:11196–11210. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu DD, Hoeven R, Rong R and Cho WC:
Rhynchophylline protects cultured rat neurons against
methamphetamine cytotoxicity. Evid Based Complement Alternat Med.
2012:6360912012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cao W, Wang Y, Lv X, Yu X, Li X, Li H,
Wang Y, Lu D, Qi R and Wang H: Rhynchophylline prevents cardiac
dysfunction and improves survival in lipopolysaccharide-challenged
mice via suppressing macrophage I-κBα phosphorylation. Int
Immunopharmacol. 14:243–251. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ziegelstein RC: Acute emotional stress and
cardiac arrhythmias. JAMA. 298:324–329. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Esch T, Stefano GB, Fricchione GL and
Benson H: Stress-related diseases-a potential role for nitric
oxide. Med Sci Monit. 8:RA103–RA118. 2002.PubMed/NCBI
|
|
35
|
Frentzou GA, Drinkhill MJ, Turner NA, Ball
SG and Ainscough JF: A state of reversible compensated ventricular
dysfunction precedes pathological remodelling in response to
cardiomyocyte-specific activity of angiotensin II type-1 receptor
in mice. Dis Model Mech. 8:783–794. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ji L, Fu F, Zhang L, Liu W, Cai X, Zhang
L, Zheng Q, Zhang H and Gao F: Insulin attenuates myocardial
ischemia/reperfusion injury via reducing oxidative/nitrative
stress. Am J Physiol Endocrinol Metab. 298:E871–E880. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tao L, Gao E, Jiao X, Yuan Y, Li S,
Christopher TA, Lopez BL, Koch W, Chan L, Goldstein BJ and Ma XL:
Adiponectin cardioprotection after myocardial ischemia/reperfusion
involves the reduction of oxidative/nitrative stress. Circulation.
115:1408–1416. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Qian L, Song X, Ren H, Gong J and Cheng S:
Mitochondrial mechanism of heat stress-induced injury in rat
cardiomyocyte. Cell Stress Chaperones. 9:281–293. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xinxing W, Hong F, Rui Z, Yun Z, Jingbo G
and Lingjia Q: Phosphorylated nerve growth factor-induced clone B
(NGFI-B) translocates from the nucleus to mitochondria of stressed
rat cardiomyocytes and induces apoptosis. Stress. 15:545–553. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Loeffler M and Kroemer G: The
mitochondrion in cell death control: Certainties and incognita. Exp
Cell Res. 256:19–26. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lopez MF, Kristal BS, Chernokalskaya E,
Lazarev A, Shestopalov AI, Bogdanova A and Robinson M:
High-throughput profiling of the mitochondrial proteome using
affinity fractionation and automation. Electrophoresis.
21:3427–3440. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kim RH, Smith PD, Aleyasin H, Hayley S,
Mount MP, Pownall S, Wakeham A, You-Ten AJ, Kalia SK, Horne P, et
al: Hypersensitivity of DJ-1-deficient mice to
1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative
stress. Proc Natl Acad Sci USA. 102:5215–5220. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu H, Bowes RC III, van de Water B,
Sillence C, Nagelkerke JF and Stevens JL: Endoplasmic reticulum
chaperones GRP78 and calreticulin prevent oxidative stress, Ca2+
disturbances, and cell death in renal epithelial cells. J Biol
Chem. 272:21751–21759. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Satoh T, Enokido Y, Aoshima H, Uchiyama Y
and Hatanaka H: Changes in mitochondrial membrane potential during
oxidative stress-induced apoptosis in PC12 cells. J Neurosci Res.
50:413–420. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sinha K, Das J, Pal PB and Sil PC:
Oxidative stress: The mitochondria-dependent and
mitochondria-independent pathways of apoptosis. Arch Toxicol.
87:1157–1180. 2013. View Article : Google Scholar : PubMed/NCBI
|