Introduction
Non-alcoholic fatty liver disease (NAFLD) is the
most commonly occurring type of chronic liver disease in developed
and developing countries (1,2). The
prevalence of this disease has considerably increased in recent
years. NAFLD has been reported as the manifestation of metabolic
syndrome in the liver (3,4). Nonalcoholic steatohepatitis (NASH),
which is a pivotal stage in NAFLD, is more likely to progress to
fibrosis, cirrhosis and hepatocellular carcinoma (HCC) in certain
cases (5,6). Currently, NASH is the third most
common indicator of liver transplantation in the USA, although it
is predicted to become the most common one between 2020 and 2025.
Concerning the mechanism of NASH, the so-called ‘two hit theory’ is
the current hypothesis. The first hit is insulin resistance, which
causes accumulation of lipids in hepatocytes. The second hit
involves increased oxidative stress, lipid peroxidation and release
of proinflammatory cytokines, which lead to the occurrence and
development of NASH (7,8). However, the exact pathogenic
mechanism of NASH has not been fully elucidated.
Circular RNA (circRNA) is a recently discovered type
of endogenous noncoding RNA molecule with a closed loop structure
formed through covalent bonding, without the 5′-end cap and the
3′-end poly (A) tail, and is attracting a lot of attention in the
RNA family as the latest research ‘hot spot’, following microRNAs
(miRNAs) and long noncoding RNAs (lncRNAs) (9). CircRNAs are highly stable and
conservative across species (10).
Accumulating evidence has demonstrated that circRNAs may function
as ‘miRNAs sponges’, which bind with and inhibit the function of
miRNAs. CircRNAs are able to regulate gene expression at the
transcriptional and post-transcriptional levels (11–13).
An increasing number of studies have reported that circRNAs are
closely associated with disease development, and this class of
molecule is expected to become a novel target for disease diagnosis
and treatment.
Even though the functions of the majority of
circRNAs in various types of disease remain unclear (11), increasing evidence suggests that
the expression profile of circRNAs is altered in numerous diseases,
including neurological diseases, diabetes and cardiovascular
diseases (14–17). Recently, a growing number of
circRNAs have been reported to be dysregulated in liver diseases,
and their clinical significance has been investigated (18–23).
However, to date, very few studies have focused on the expression
profile of circRNAs in NASH. Jin et al (24) investigated the expression profile
of circRNAs in a methionine and choline-deficient (MCD)-induced
NASH mouse model, and identified four circRNA-miRNA-mRNA pathways;
furthermore, it was demonstrated that the circRNA profile in NASH
is able to provide potential candidates for future mechanistic
studies.
In the present study, changes in the circRNA
expression profile in a well-developed NASH mouse model were
investigated, with the aim of investigating the potential
pathogenic mechanisms of circRNAs during NASH development.
Combining circRNA microarray screening and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
validation with bioinformatics methods, a novel circRNA-miRNA
interaction pathway that is closely associated with the disease
processes of NASH was identified, enabling us to predict the
potential roles of circRNAs in NASH development.
Materials and methods
Animals
A total of 12 male C57BL/6 mice, aged 6 weeks,
weighing 20–25 g, were purchased from the Shanghai Xipuer-Bikai
Laboratory Animal Co., Ltd (Shanghai, China). All mice were housed
in cages under standard conditions with an ambient temperature of
24°C under a 12 h light/dark cycle. Mice had free access to food
and water (as detailed in the subsequent section). All experimental
protocols were approved by the Animal Ethics Committee of the
Eighth People's Hospital of Shanghai (Permit no. 2016-02-2).
NASH animal model construction
Animals were randomly divided into two groups:
Control group (n=6) and NASH group (n=6). All mice received food
and water ad libitum. Mice in the NASH group were provided
with an MCD diet. Mice in the control group were fed the same diet,
but with sufficient quantities of DL-methionine (3 g/kg) and
choline bitartrate (2 g/kg). At the end of 8 weeks of
administration of the diet, all the mice were anesthetized and
sacrificed, and their livers were removed. Serum and liver samples
were frozen in liquid nitrogen prior to biochemical analysis. A
small piece of the liver sample was fixed in 4% paraformaldehyde
(96 h at 4°C) for subsequent histological analysis.
The levels of serum triglycerides (TG) and total
cholesterol (TC), and the activities of the liver-associated
enzymes, alanine aminotransferase (ALT) and aspartate
aminotransferase (AST), were estimated with an automatic
biochemical analyzer (Roche/Hitachi P-800 Modular Analytics
Diagnostic System; Roche Diagnostics, Indianapolis, IN, USA).
Hepatic homogenates were used for determination of the TG and TC
content using a kit provided by Nanjing Jianchen Bioengineering
Institute (Nanning, China) according to the manufacturer's
protocol. Tissue lipids were extracted with methanol/chloroform
(1:2).
The liver samples removed from each mouse were fixed
in 4% paraformaldehyde for 96 h at 4°C. Following dehydration in
graded alcohol (once in 50, 70 and 95%, and twice in 100% for 2–3 h
each) and embedding in paraffin wax, the sections were cut to a
thickness of 4 µm, and stained with hematoxylin and eosin (H&E)
(25). The liver histology was
scored for ballooning (0–2), steatosis (0–3), and inflammation
(0–3), and the sum of these scores was used to create the NAFLD
Activity Score. Picrosirius red staining (0.1% Sirius Red in
saturated picric acid stained for 75 min at room temperature) was
used to assess collagen fibers in the liver tissues. Each section
was assessed under 10×20 light microscopic fields. For lipid
staining, 6 µm frozen liver sections were air-dried for 30 min,
followed by fixation in 4% formaldehyde. Oil red O staining was
performed with staining solution (0.5% Oil Red O in anhydrous
isopropanol, sections were stained for 30 min at 37°C)
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) according to the
manufacturer's protocol. Then, six slides from the control and NASH
groups were used for quantitative analysis. The stained area was
quantified in six randomly selected ×200 microscopic fields per
mouse section using ImageJ software (ImageJ2×; National Institutes
of Health, Bethesda, MD, USA).
Isolation and quality control of
RNA
Total RNA was isolated from the different groups
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and purified with an RNeasy
Mini kit (Qiagen GmbH, Hilden, Germany) according to the
manufacturer's protocol. The purity and concentration of RNA
samples were determined with the NanoDrop® ND-1000
spectrophotometer (Thermo Fisher Scientific, Inc.). The integrity
of the RNA was assessed by electrophoresis on a denaturing gel.
CircRNA microarray analysis
Three liver RNA samples from each group were
selected for microarray studies. Sample labeling and array
hybridization were performed according to the manufacturer's
protocol (Arraystar, Inc., Rockville, MD, USA). In brief, the
extracted total RNA was treated with RNase R
(Epicentre®; Illumina, Inc., Madison, WI, USA) to remove
linear RNAs and enrich the population of circRNAs. The enriched
circRNAs were amplified and transcribed into fluorescent
complementary RNA (cRNA) using random primers, according to the
instructions provided by the Arraystar Super RNA Labeling kit
(Arraystar, Inc.). Following purification of the labeled cRNAs with
the RNeasy Mini kit (Qiagen GmbH), the concentration and specific
activity of the labeled cRNAs were measured using the
NanoDrop® ND-1000 spectrophotometer. The labeled cRNA
was hybridized on to Arraystar mouse circRNA array v1.0. Following
washing of the slides, the arrays were scanned using the Agilent
G2505C scanner (Agilent Technologies, Inc., Santa Clara, CA,
USA).
RT-qPCR validations
RT-qPCR was used to confirm the circRNA expression
profiles obtained from the microarray data. Total RNA was isolated
from liver tissues using TRIzol® reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) and reverse transcribed into cDNA
using SuperScript™ III Reverse Transcriptase (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
The relative gene expression was determined using the Agilent
Mx3000P qPCR system. Amplifications were performed using a SYBR
Green PCR kit (Takara Biotechnology Co., Ltd., Dalian, China). The
qPCR program comprised an initial denaturation step of 3 min at
95°C, followed by 40 cycles of 15 sec at 95°C, 30 sec at 60°C, and
30 sec at 72°C. All samples were normalized to the signal generated
from GAPDH. Results were obtained from three independent wells and
shown as the fold change, according to the 2−ΔΔCq method
(26). Primers were designed to
amplify the circRNA-specific back splice junctions (Table I).
 | Table I.Primer sequences used for reverse
transcription-quantitative polymerase chain reaction
validation. |
Table I.
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction
validation.
| circRNA ID | Sequence |
|---|
| mGAPDH-Q-F |
TTCCTACCCCCAATGTGTCC |
| mGAPDH-Q-R |
GGTCCTCAGTGTAGCCCAAG |
|
mmu_circRNA_29981-Q-F |
ACCAAAACCTGCATTGGCAC |
|
mmu_circRNA_29981-Q-R |
ACAGAACATGGCGATCTGGG |
|
mmu_circRNA_004372-Q-F |
GCTGAGCTACACAACTGCATC |
|
mmu_circRNA_004372-Q-R |
TGGTCCATGAATCGCTCTGG |
|
mmu_circRNA_36096-Q-F |
CCCTGCATTGGAAGTTTGCT |
|
mmu_circRNA_36096-Q-R |
TGAAGGTCCCACCATTTCCTG |
|
mmu_circRNA_43021-Q-F |
AGGAACTACCGTGGGGATGT |
|
mmu_circRNA_43021-Q-R |
CACCATGGGCCAAGATAGGT |
|
mmu_circRNA_28028-Q-F |
AGCGAGATCCCTACTGGTGT |
|
mmu_circRNA_28028-Q-R |
GTGATCTTCTCGTGCAACGC |
|
mmu_circRNA_22646-Q-F |
GAAGTGCTACGGGCAACAGA |
|
mmu_circRNA_22646-Q-R |
CATCGAAACACGGAAACGCC |
|
mmu_circRNA_008234-Q-F |
CGTGAGATGTGTTGCTGAGG |
|
mmu_circRNA_008234-Q-R |
CAGCAGGTGCTAAAGCATGAC |
Microarray data and bioinformatics
analysis
Scanned images were imported into the Agilent
Feature Extraction software (version 11.0.1.1) for raw data
extraction. Quantile normalization of raw data and subsequent data
processing were performed using the R software package (R version
3.1.2). The circRNAs that exhibited fold changes (FC) ≥1.5 (P≤0.05)
between the two groups were categorized as significantly
differentially expressed. The selected differentially expressed
circRNAs were identified through volcano plotting and FC filtering.
Hierarchical clustering analysis was performed to show the
distinguishable circRNA expression pattern among samples.
The circRNA-miRNA interaction analysis was performed
using Arraystar's miRNA target prediction software, which includes
TargetScan (www.targetscan.org/) and miRanda (www.microrna.org/). Potential miRNA response elements
(MREs) for circRNA were predicted using the miRNA support vector
regression (mirSVR) which obtained from miRanda as previously
described (27). Gene Ontology
(GO) analysis was performed to explore the functional roles of host
linear transcripts in the terms biological processes (BPs),
cellular components (CCs) and molecular functions (MFs). Kyoto
Encyclopedia of Genes and Genomes (KEGG) was used to determine the
target genes in different biological pathways.
Statistical analysis
Data are presented as the mean ± standard deviation
for triplicate measurements. Statistically significant differences
between groups were estimated using the Student's t-test with SPSS
software, version 13.0 (SPSS, Inc., Chicago, IL, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
Successful construction of the NASH
mouse model
At the end of 8 weeks of feeding the mice on the
prescribed MCD diet, the body weight of the mice in the model group
was significantly decreased compared with that in the control group
(P<0.01; Table II). The
H&E-stained sections exhibited typical histopathological
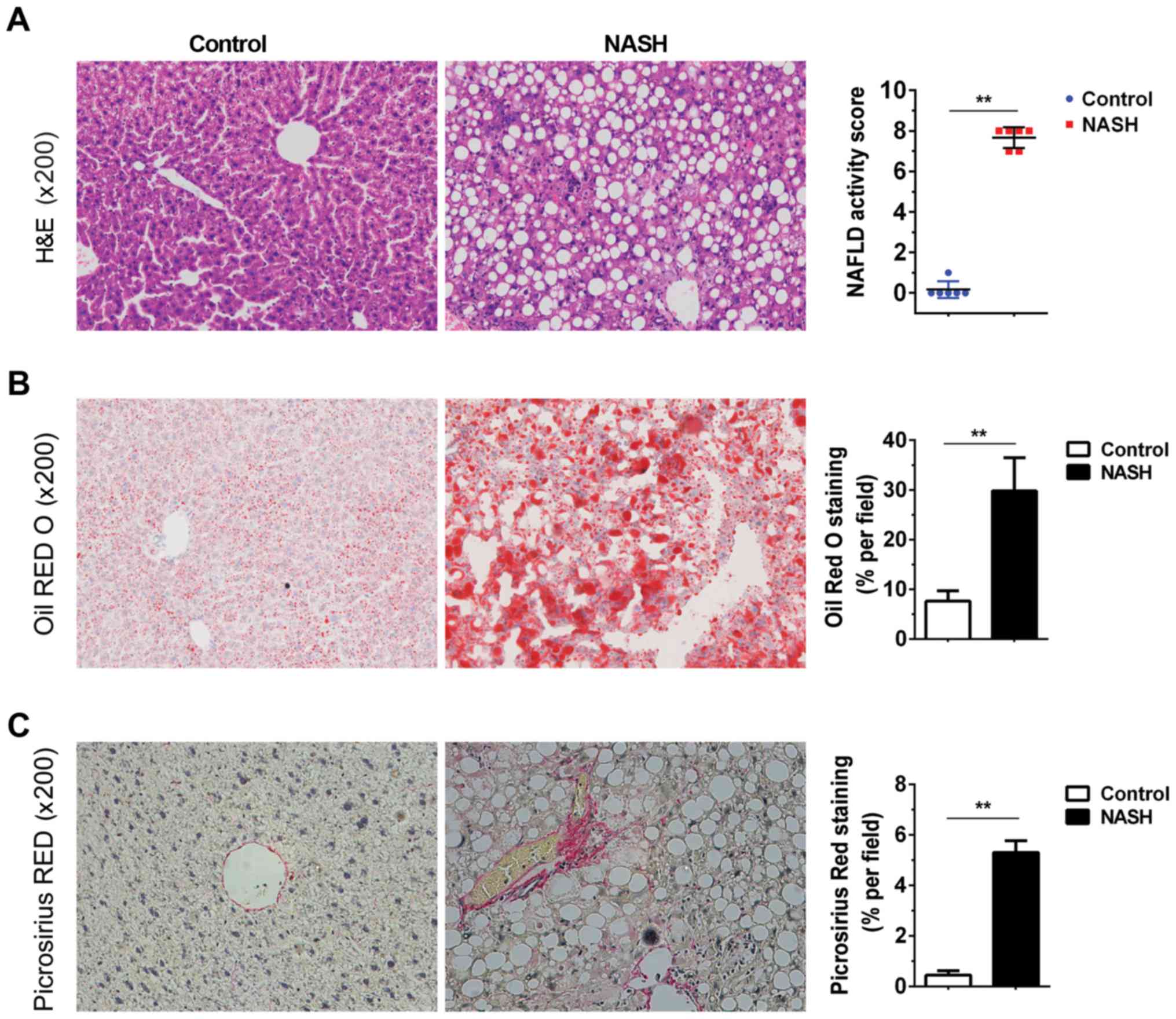
features of NASH. As presented in Fig.
1, sections from the NASH livers exhibited disordered hepatic
lobules, full fat vacuoles in lobule cells, infiltration of
inflammatory cells, and cell swelling (Fig. 1A). The livers of MCD diet-fed mice
exhibited marked lipid accumulation and liver fibrosis (Fig. 1B and C). Serum AST and ALT levels
indirectly reflect the failure of liver function. As indicated in
Table II, the serum ALT
(406.7±132.1 U/l) and AST (573.3±169.5 U/l) activities were
significantly increased following the administration of the MCD
diet, as compared with the normal group (25.0±7.5 and 175.3±29.5,
respectively; P<0.05). The TG and TC levels were lower in the
MCD diet-fed mice compared with that in control mice (P<0.05 and
P<0.01, respectively). The hepatic TG content (705±69.3 µmol/g
protein) was higher in the MCD diet-fed mice compared with the
control mice (179±17.6 µmol/g protein), whereas the hepatic TC
content did not exhibit any difference between the two groups
(Table II). These changes in
serum biomarkers and liver tissue indicated the successful
establishment of the NASH model in mice.
| Table II.Biochemical parameters in NASH
mice. |
Table II.
Biochemical parameters in NASH
mice.
| Variables | Control | NASH |
|---|
| Initial body weight
(g) | 22.9±0.1 | 22.3±0.4 |
| Final body weight
(g) | 30.1±2.3 |
13.8±0.1b |
| Serum alanine
transferase (U/l) | 25.0±7.5 |
406.7±132.1a |
| Serum aspartate
transferase (U/l) | 175.3±29.5 |
573.3±169.5a |
| Serum TG
(mmol/l) | 1.0±0.2 |
0.7±0.2a |
| Serum TC
(mmol/l) | 4.1±0.1 |
1.2±0.1b |
| Hepatic TG (µmol/g
protein) | 179±17.6 |
705±69.3b |
| Hepatic TC (µmol/g
protein) | 50.6±7.3 | 58.7±13.3 |
Expression profiles of circRNA in NASH
mice
To screen for circRNAs that were differentially
expressed between the NASH and control mice livers, circRNA
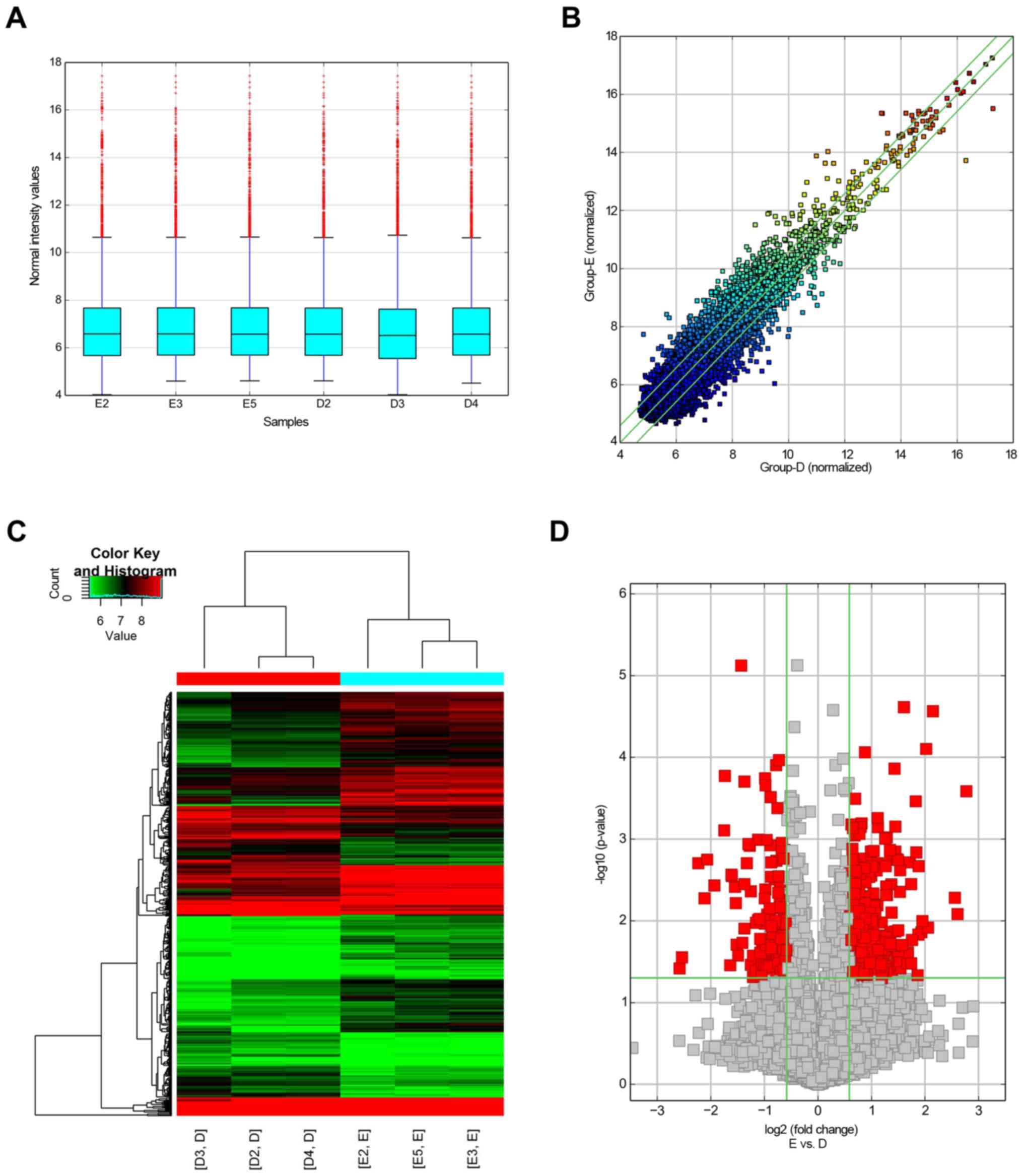
microarrays were performed in three samples from each group. A box
plot showed that the distributions of circRNA for the tested
samples were essentially identical following normalization
(Fig. 2A). Hierarchical
clustering, scatter plot visualization and volcano plot analyses
revealed the differences in circRNA expression levels in NASH and
control mice liver (Fig. 2B-D; FC
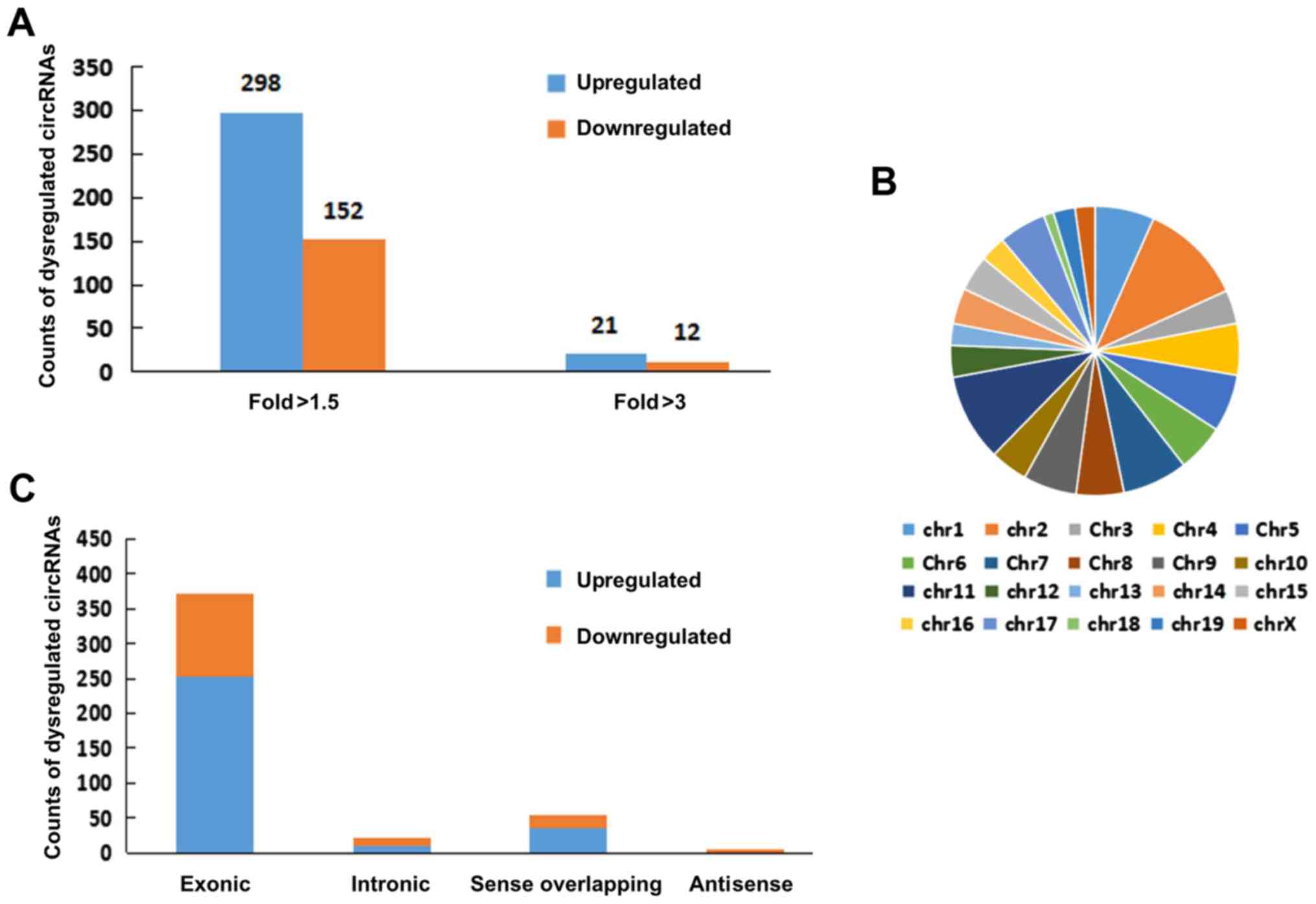
>2.0; P<0.05). Consequently, 450 circRNAs were identified as
dysregulated in the livers of NASH mice, with FC >1.5 and
P<0.05. Among these, 298 circRNAs were upregulated, and 152
circRNAs were downregulated, in the NASH group compared with those
in the control group (Fig. 3A).
Additionally, 21 circRNAs were upregulated, and 12 circRNAs were
downregulated, in the livers of NASH mice with FC >3.0 and
P<0.05 (Fig. 3A). CircRNAs on
mouse chromosomes are illustrated in the pie chart format in
Fig. 3B. Types and counts of
differentially regulated circRNAs are presented in Fig. 3C. The top 10 upregulated and
downregulated circRNAs sorted by their FC values are summarized in
Table III, along with their
potential miRNA targets. Taken together, these data indicated that
the circRNA expression patterns in NASH livers were different from
those in the control group.
| Table III.Annotation for differentially
expressed circRNAs/miR interaction. |
Table III.
Annotation for differentially
expressed circRNAs/miR interaction.
| circRNAs | Regulation | Fold-change | Type | MRE1 | MRE2 | MRE3 | MRE4 |
|---|
|
mmu_circRNA_28028 | Up | 6.8 | Exonic |
mmu-miR-9769-3p | mmu-miR-8097 |
mmu-miR-194-2-3p | mmu-miR-693-3p |
|
mmu_circRNA_29981 | Up | 6.1 | Exonic |
mmu-miR-7661-3p |
mmu-miR-181b-5p |
mmu-miR-6952-3p |
mmu-miR-3077-3p |
|
mmu_circRNA_39832 | Up | 5.9 | Exonic | mmu-miR-6344 |
mmu-miR-130a-5p |
mmu-miR-7091-3p |
mmu-miR-7214-5p |
|
mmu_circRNA_29980 | Up | 4.4 | Exonic |
mmu-miR-1249-5p |
mmu-miR-6923-3p |
mmu-miR-7058-3p |
mmu-miR-6976-3p |
|
mmu_circRNA_21454 | Up | 4.2 | Exonic | mmu-miR-1903 |
mmu-miR-7028-3p |
mmu-miR-7032-3p |
mmu-miR-3059-5p |
|
mmu_circRNA_34829 | Up | 4.0 | Exonic |
mmu-miR-6943-3p |
mmu-miR-7064-5p | mmu-miR-691 |
mmu-miR-7032-5p |
|
mmu_circRNA_40141 | Up | 3.9 | Exonic | mmu-miR-1903 | mmu-miR-223-3p | mmu-miR-881-3p |
mmu-miR-7646-3p |
|
mmu_circRNA_30398 | Up | 3.8 | Exonic |
mmu-miR-7061-3p |
mmu-miR-103-1-5p |
mmu-miR-103-2-5p | mmu-miR-136-5p |
|
mmu_circRNA_004372 | Up | 3.7 | Exonic |
mmu-miR-103-1-5p |
mmu-miR-103-2-5p | mmu-miR-107-5p | mmu-miR-143-5p |
|
mmu_circRNA_21453 | Up | 3.7 | Exonic | mmu-miR-1903 |
mmu-miR-7028-3p |
mmu-miR-6946-3p |
mmu-miR-6919-3p |
|
mmu_circRNA_28143 | Down | 6.0 | Intronic |
mmu-miR-7118-5p |
mmu-miR-1894-3p |
mmu-miR-7665-5p |
mmu-miR-1249-5p |
|
mmu_circRNA_28144 | Down | 5.8 | Intronic |
mmu-miR-7118-5p |
mmu-miR-1894-3p |
mmu-miR-7665-5p |
mmu-miR-1249-5p |
|
mmu_circRNA_22646 | Down | 4.7 | Exonic |
mmu-miR-196a-5p |
mmu-miR-6906-5p |
mmu-miR-6909-3p |
mmu-miR-196b-5p |
|
mmu_circRNA_31317 | Down | 4.3 | Exonic |
mmu-miR-3099-5p |
mmu-miR-6976-5p |
mmu-miR-5627-3p | mmu-miR-497b |
|
mmu_circRNA_008234 | Down | 4.2 | Antisense | mmu-miR-361-3p | mmu-miR-504-5p | mmu-miR-1291 |
mmu-miR-3090-3p |
|
mmu_circRNA_21487 | Down | 3.8 | Exonic |
mmu-miR-5621-5p |
mmu-miR-7665-5p |
mmu-miR-3069-5p |
mmu-miR-6907-5p |
|
mmu_circRNA_35127 | Down | 3.4 | Exonic |
mmu-miR-3618-5p |
mmu-miR-6973b-5p | mmu-miR-295-5p | mmu-miR-677-5p |
|
mmu_circRNA_40509 | Down | 3.3 | Exonic |
mmu-miR-6932-3p |
mmu-miR-26a-2-3p |
mmu-miR-7074-5p |
mmu-miR-3075-5p |
|
mmu_circRNA_29948 | Down | 3.1 | Intronic |
mmu-miR-6946-3p |
mmu-miR-7651-3p | mmu-miR-6399 |
mmu-miR-7116-3p |
|
mmu_circRNA_39830 | Down | 3.1 | Exonic |
mmu-miR-6908-3p |
mmu-miR-6975-3p | mmu-miR-149-5p | mmu-miR-500-5p |
Annotations for the circRNA-miRNA
interactions
To further validate the results of the present
study, Arraystar's miRNA target prediction software, based on
TargetScan and miRanda, was used to predict the circRNA-miRNA
interactions. A total of 20 differentially expressed exonic
circRNAs with the highest FC values were selected (FC >3;
P<0.05) to predict their miRNA response elements (MREs),
including 10 upregulated circRNAs and 10 downregulated circRNAs.
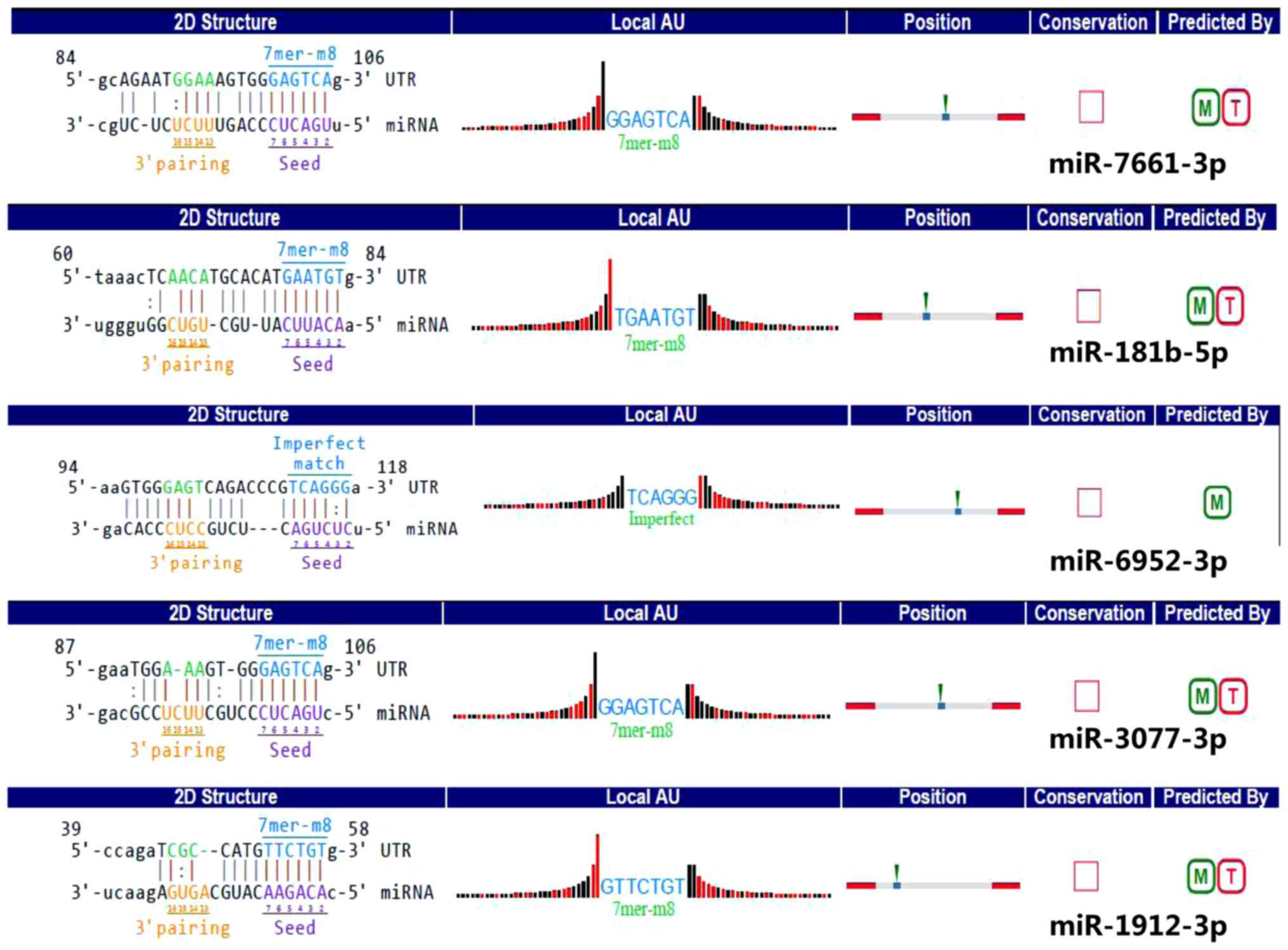
Four MREs that were identified to have ‘good’ mirSVR scores for
each circRNA are presented in Table
III.
Validation of selected circRNAs by
RT-qPCR
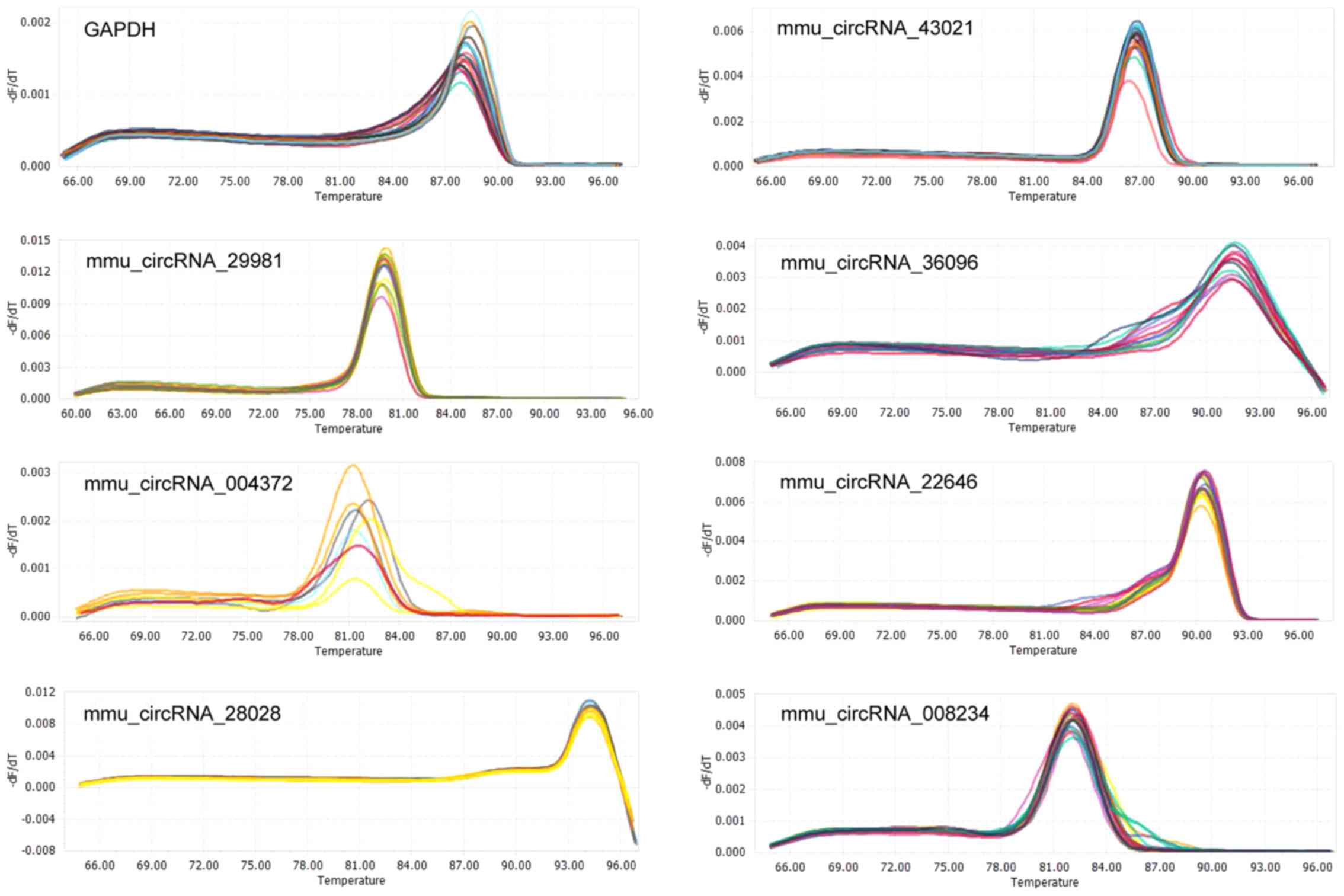
To validate the microarray results, seven
differentially expressed exonic circRNAs were randomly selected (FC
>3.5; P<0.05), including five upregulated circRNAs and two
downregulated circRNAs. Primers for circRNAs were designed (shown
in Table I), and the appearance of
a single peak in the melting curve confirmed the specificity of the
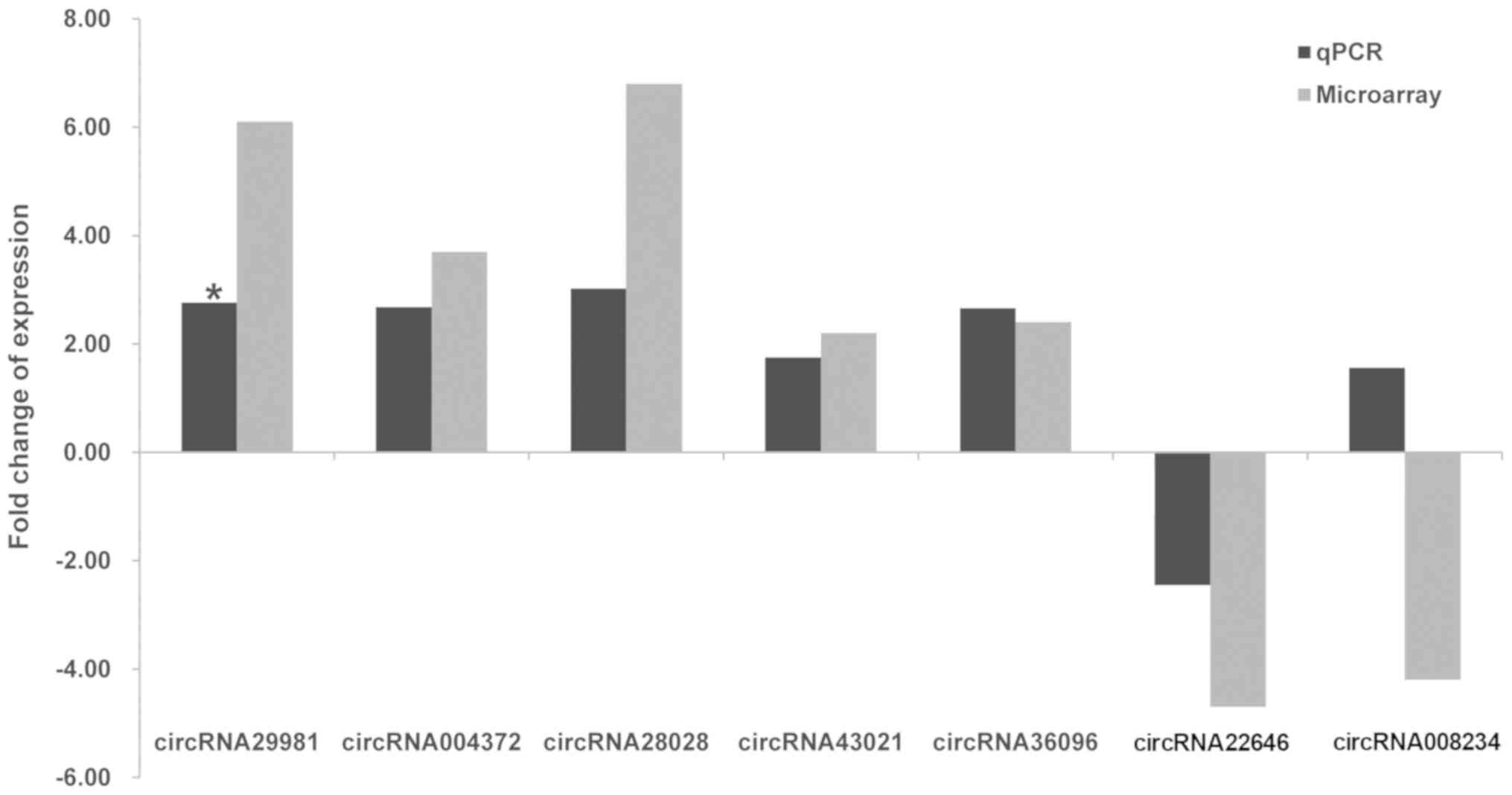
amplified products for each circRNA (Fig. 4). The results demonstrated that
five circRNAs (circRNA-28028, circRNA-29981, circRNA-004372,
circRNA-0043021 and circRNA-36096) were upregulated, and these
findings were broadly in accord with the data obtained from the
microarray analysis. However, only circRNA-29981 reached the
required level of statistical significance to become a candidate
for further study (Fig. 5). By
contrast, circRNA-008234 was observed to have the opposite
expression trend in microarray and RT-qPCR analysis, although the
difference was revealed not to be statistically significant.
Additionally, the expression levels of circRNA-22646 were not
significantly different between the NASH and control groups
(Fig. 5). Furthermore, the
detailed annotations for circRNA-29981 interaction with various
miRNAs (miR-7661-3p, miR-181b-5p, miR-6952-3p, miR-3077-3p, and
miR-1912-3p) are presented in Fig.
6. The effect of the circRNA-29981 regulation pathways in NASH
is worth further exploration.
Bioinformatics analysis predicting
biological function of host linear transcripts
CircRNAs are generally formed by alternative
splicing of pre-mRNA species, which are usually spliced to generate
a linear mRNA (11). A more recent
study has demonstrated that a class of circRNAs, termed exon-intron
circRNAs, enhances the expression of their pre-mRNAs (28). CircRNAs may also be potentially in
competition with their linear transcripts (24). To provide a comprehensive landscape
of the origination of circRNAs, GO and KEGG analyses were performed
to annotate the biological functions of host linear transcripts.
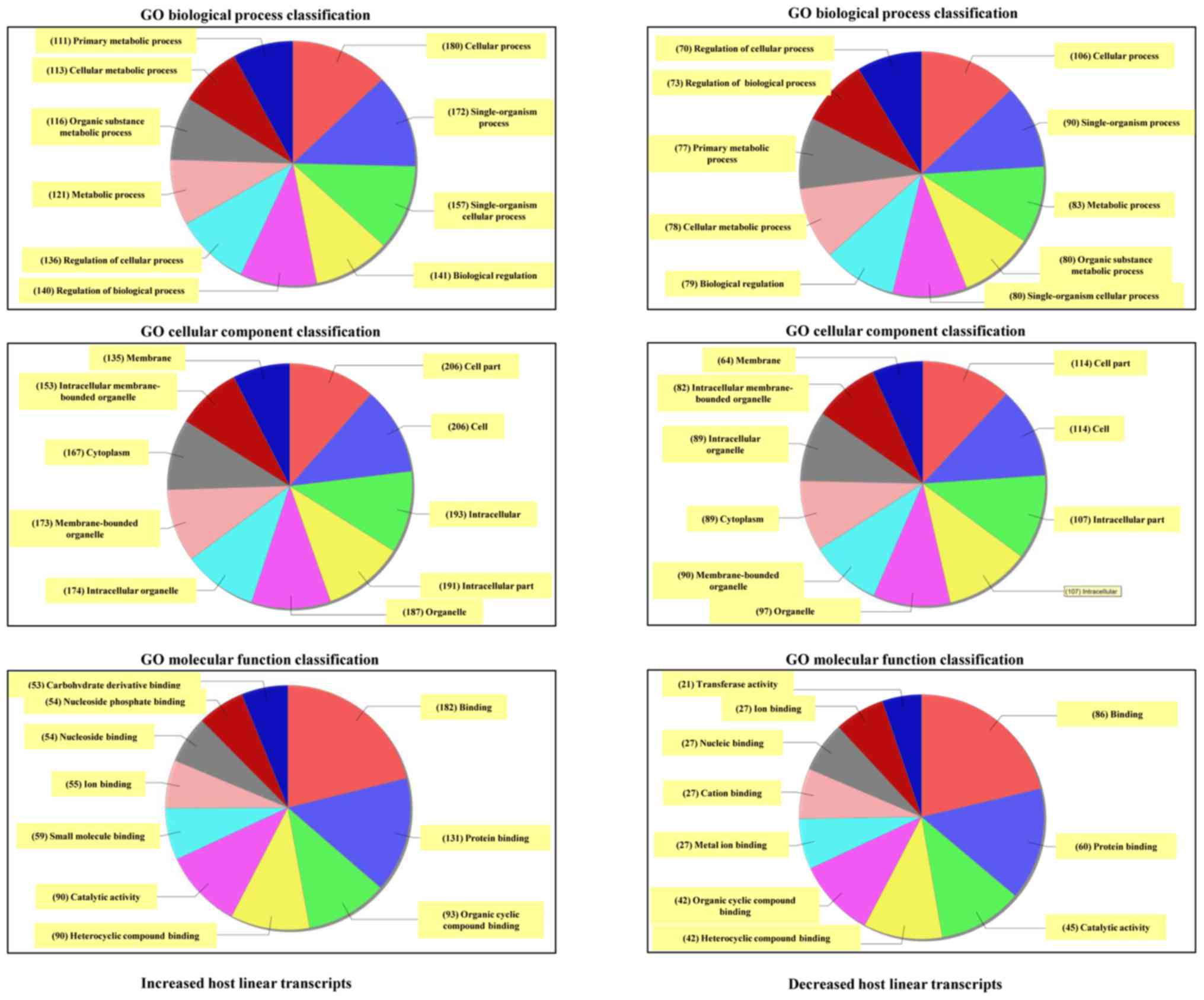
The top 10 generally altered GO terms between NASH and control
groups were classified according to the BP, CC and MF (Fig. 7). For increased mRNAs, the most
enriched and meaningful BP terms were ‘cellular process’,
‘single-organism process’ and ‘single-organism cellular process’.
The most enriched CC terms were ‘cell’, ‘cell part’,
‘intracellular’, and ‘intracellular part’. As for MF, the most
enriched terms were ‘binding’, ‘protein binding’, and ‘organic
cyclic compound binding’. In addition, the data from the GO
analysis on the downregulated transcripts revealed that the top two
GO processes were identical with those in the increased transcripts
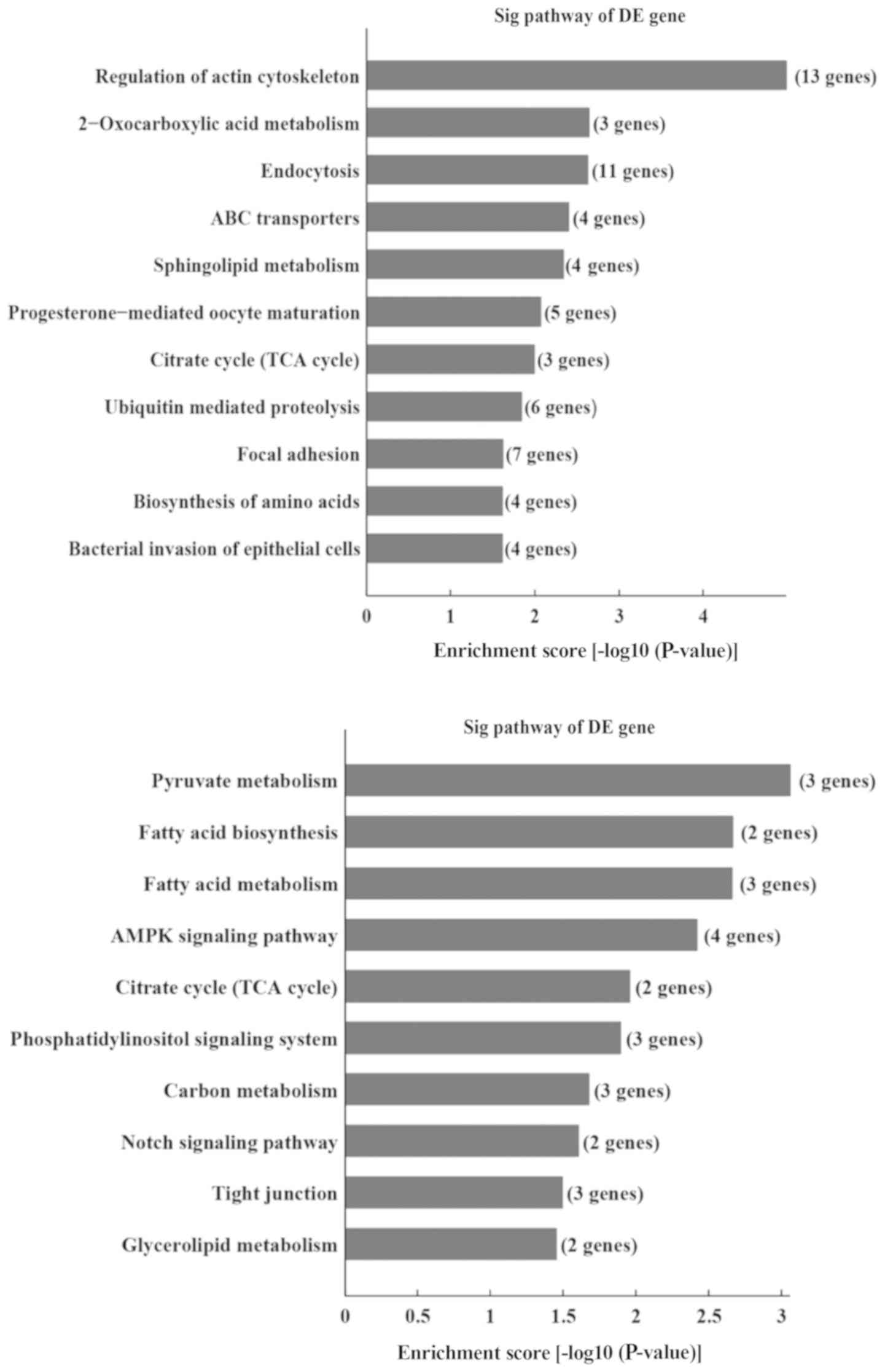
(Fig. 7). Furthermore, KEGG
analysis revealed that certain pathways, including regulation of
the actin cytoskeleton, 2-oxocarboxylic acid metabolism, and the
endocytosis signaling pathway, were associated with the upregulated
transcripts, whereas pathways such as the pyruvate metabolism,
fatty acid biosynthesis and fatty acid metabolism signaling
pathways were associated with the downregulated transcripts
(Fig. 8).
Discussion
The mechanism of NASH has yet to be fully
elucidated. Because of a lack of effective diagnosis and
therapeutic approaches, NASH has become the predominant cause of
cryptogenic cirrhosis. An in-depth analysis of the pathogenic
mechanism of NASH is essential for proper diagnosis and the
implementation of effective therapies. CircRNAs have been
associated with numerous diseases, cell processes and gene
expression studies (10). The
characteristics of circRNAs, such as tissue- and
development-specific expression, serving as a ‘miRNA sponge’ to
inhibit the activity of miRNAs, highlight the role of circRNAs in
disease development. Accumulating evidence has suggested that
circRNAs are associated with liver diseases, including HCC, liver
regeneration and hepatic steatosis (18–21,29).
Certain circRNAs have been revealed to have important roles in
regulating HCC growth, migration, and invasion (19,22).
Recently, Chen et al (30)
demonstrated that inhibiting the expression of has_circ_0071410
affected irradiation-induced hepatic stellate cell (HSC) activation
by regulating the expression of miR-9-5p. Guo et al
(29) demonstrated that the
dysregulation of circRNAs is associated with hepatic steatosis.
These studies suggested that circRNAs are closely associated with
liver diseases, and further in-depth analyses of circRNAs may
provide novel clues to obtain a deeper understanding of the
pathogenesis and processes of NASH.
Since it is difficult to obtain liver biopsy samples
from patients with NASH, either a NASH mouse model or a hepatic
steatosis cell model has been used to investigate the circRNA
profile (24,29). These studies demonstrated that
dysregulated circRNAs were associated with hepatic steatosis and
NASH development. However, currently there is no ideal NASH animal
model (31); therefore, a classic
NASH mouse model that is induced by an MCD diet was used in the
current study (32). To confirm
the success of the mouse model, H&E, Oil red O and picrosirius
red staining were performed. Additionally, the serum and liver
biochemistry also demonstrated the pathological features of NASH.
The aforementioned staining and biochemistry analysis confirmed a
successful simulation of the pathological characteristics of NASH.
In the present study, 450 significantly dysregulated circRNAs (FC
≥1.5, P<0.05) were identified, including 298 upregulated and 152
downregulated circRNAs in the NASH group. Furthermore, the most
significantly differentially expressed circRNAs comparing between
the NASH and control groups were annotated in detail, and the
circRNA-miRNA interaction was predicted. Following validation by
RT-qPCR, a novel circRNA, circRNA_29981, was confirmed to be
differentially expressed in NASH model mice, and the circRNA-miRNA
interaction information was predicted. With respect to the target
miRNAs of circRNA_29981, miR-181b has been validated as a key
regulator in steatosis via the targeting of sirtuin 1 (SIRT1)
(33). SIRT1 has been reported to
have an essential role in guiding the transition of the HSC
phenotype (34). miR-181b was also
reported to promote HSC proliferation, and has an important role in
liver fibrosis (23,35,36).
Another miRNA, mmu-miR-130a, has been demonstrated to enhance the
activation of HSCs by suppressing the expression of peroxisome
proliferator-activated receptor γ (37). Therefore, one may surmise that
circRNA_29981 could be involved in regulating the activation of
HSCs. Further research on the role of circRNA_29981 is required,
including experiments designed to validate the target miRNAs of
circRNA_29981, and investigate the molecular changes of a hepatic
cell line following circRNA knockdown or overexpression. Further
exploration of the circRNA-miRNA interaction networks will be the
subject of future investigations.
Jin et al (24) previously used BALB/c mice to create
a NASH model by providing an MCD diet for 2 weeks. In the present
study, C57BL/6 mice received an MCD diet for 8 weeks. No common
circRNAs were identified in the top 10 up- or downregulated
circRNAs when comparing between the two studies. This may be due to
the difference in the time for which the MCD diet was supplied, or
that different mouse strains were used. However, following the GO
pathway analysis, the results of the most enriched BP, CC, and MF
terms from the host linear transcripts in the present study were
identical with those of the study by Jin et al (24). This may potentially suggest that
there are certain associations between circRNAs and linear mRNAs in
NASH processes. The data obtained from the KEGG pathway analysis
revealed that host linear transcripts were associated with pathways
of energy metabolism, including ‘amino acid biosynthesis’ and
‘fatty acid biosynthesis and metabolism’, which have been credited
as being the important activities in NASH processes. In addition,
glycerolipid metabolism and inflammatory mediator regulation of
transient receptor potential channels were revealed to be important
in terms of NASH progression in the study by Jin et al
(24). Taken together, all these
findings reflect the complicated and critical mechanism underlying
circRNA-associated regulation.
Although circRNA_29981 in the mouse shares 89%
sequence homology with the sequence of circRNA_29981 in humans
(Fig. 9), the expression profile
of its homologous sequences in humans has yet to be determined.
Therefore, whether this result can be verified in human NASH
requires further confirmation. In addition, due to the low
validation rate of circRNA microarray data compared with the
RT-qPCR experiments, it is not possible to exclude that there may
be other dysregulated circRNAs that also participate in NASH
development. Further studies on these issues are required. As
circRNAs are stably expressed in blood, identifying the circulating
circRNAs in patients with NASH may provide a promising avenue for
research. This will also be an integral part of future studies.
Taken together, the present study has produced circRNA expression
profiles during the NASH process in mice, and a novel candidate
circRNA, circRNA_29981, that may be a potential regulator of HSC
activation was identified. The results of the present study have
corroborated the regulatory role of circRNAs in NASH, and provided
novel clues for understanding the mechanism of NASH
pathogenesis.
Acknowledgements
Not applicable.
Funding
The study was supported by the National Natural
Science Foundation of China (grant no. 81670129 to XW).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XW and QO participated in the research design. QO,
YZ and JZ performed the experiments. XW, QO and JZ performed the
data analysis; XW wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chitturi S, Farrell GC, Hashimoto E,
Saibara T, Lau GK and Sollano JD; Asia-Pacific Working Party on
NAFLD, : Non-alcoholic fatty liver disease in the Asia-Pacific
region: Definitions and overview of proposed guidelines. J
Gastroenterol Hepatol. 22:778–787. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Milić S and Stimac D: Nonalcoholic fatty
liver disease/steatohepatitis: Epidemiology, pathogenesis, clinical
presentation and treatment. Dig Dis. 30:158–162. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ma Z, Chu L, Liu H, Li J, Zhang Y, Liu W,
Dai J, Yi J and Gao Y: Paeoniflorin alleviates non-alcoholic
steatohepatitis in rats: Involvement with the ROCK/NF-κB pathway.
Int Immunopharmacol. 38:377–384. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Katsiki N, Perez-Martinez P, Anagnostis P,
Mikhailidis DP and Karagiannis A: Is nonalcoholic fatty liver
disease indeed the hepatic manifestation of metabolic syndrome?
Curr Vasc Pharmacol. 16:219–227. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Angulo P, Alba LM, Petrovic LM, Adams LA,
Lindor KD and Jensen MD: Leptin, insulin resistance, and liver
fibrosis in human nonalcoholic fatty liver disease. J Hepatol.
41:943–949. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bellentani S and Marino M: Epidemiology
and natural history of non-alcoholic fatty liver disease (NAFLD).
Ann Hepatol. 8 (Suppl 1):S4–S8. 2009.PubMed/NCBI
|
|
7
|
Day CP and James OF: Steatohepatitis: A
tale of two ‘hits’? Gastroenterology. 114:842–845. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Michelotti GA, Machado MV and Diehl AM:
NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol.
10:656–665. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen LL and Yang L: Regulation of circRNA
biogenesis. RNA Biol. 12:381–388. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Memczak S, Jens M, Elefsinioti A, Torti F,
Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer
M, et al: Circular RNAs are a large class of animal RNAs with
regulatory potency. Nature. 495:333–338. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ashwal-Fluss R, Meyer M, Pamudurti NR,
Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N and
Kadener S: circRNA biogenesis competes with pre-mRNA splicing. Mol
Cell. 56:55–66. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vicens Q and Westhof E: Biogenesis of
circular RNAs. Cell. 159:13–14. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hansen TB, Jensen TI, Clausen BH, Bramsen
JB, Finsen B, Damgaard CK and Kjems J: Natural RNA circles function
as efficient microRNA sponges. Nature. 495:384–388. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li TR, Jia YJ, Wang Q, Shao XQ and Lv RJ:
Circular RNA: A new star in neurological diseases. Int J Neurosci.
127:726–734. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu H, Guo S, Li W and Yu P: The circular
RNA Cdr1as, via miR-7 and its targets, regulates insulin
transcription and secretion in islet cells. Sci Rep. 5:124532015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fan X, Weng X, Zhao Y, Chen W, Gan T and
Xu D: Circular RNAs in cardiovascular disease: An overview. Biomed
Res Int. 2017:51357812017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao Z, Li X, Jian D, Hao P, Rao L and Li
M: Hsa_circ_0054633 in peripheral blood can be used as a diagnostic
biomarker of pre-diabetes and type 2 diabetes mellitus. Acta
Diabetol. 54:237–245. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yao T, Chen Q, Fu L and Guo J: Circular
RNAs: Biogenesis, properties, roles, and their relationships with
liver diseases. Hepatol Res. 47:497–504. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Han D, Li J, Wang H, Su X, Hou J, Gu Y,
Qian C, Lin Y, Liu X, Huang M, et al: Circular RNA circMTO1 acts as
the sponge of microRNA-9 to suppress hepatocellular carcinoma
progression. Hepatology. 66:1151–1164. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shang X, Li G, Liu H, Li T, Liu J, Zhao Q
and Wang C: Comprehensive circular RNA profiling reveals that
hsa_circ_0005075, a new circular RNA biomarker, is involved in
hepatocellular crcinoma development. Medicine (Baltimore).
95:e38112016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li L, Guo J, Chen Y, Chang C and Xu C:
Comprehensive CircRNA expression profile and selection of key
CircRNAs during priming phase of rat liver regeneration. BMC
Genomics. 18:802017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yao Z, Luo J, Hu K, Lin J, Huang H, Wang
Q, Zhang P, Xiong Z, He C, Huang Z, et al: ZKSCAN1 gene and its
related circular RNA (circZKSCAN1) both inhibit hepatocellular
carcinoma cell growth, migration, and invasion but through
different signaling pathways. Mol Oncol. 11:422–437. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen YJ, Zhu JM, Wu H, Fan J, Zhou J, Hu
J, Yu Q, Liu TT, Yang L, Wu CL, et al: Circulating microRNAs as a
fingerprint for liver cirrhosis. PLoS One. 8:e665772013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jin X, Feng CY, Xiang Z, Chen YP and Li
YM: CircRNA expression pattern and circRNA-miRNA-mRNA network in
the pathogenesis of nonalcoholic steatohepatitis. Oncotarget.
7:66455–66467. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cardiff RD, Miller CH and Munn RJ: Manual
hematoxylin and eosin staining of mouse tissue sections. Cold
Spring Harb Protoc. 2014:655–658. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Betel D, Koppal A, Agius P, Sander C and
Leslie C: Comprehensive modeling of microRNA targets predicts
functional non-conserved and non-canonical sites. Genome Biol.
11:R902010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li Z, Huang C, Bao C, Chen L, Lin M, Wang
X, Zhong G, Yu B, Hu W, Dai L, et al: Exon-intron circular RNAs
regulate transcription in the nucleus. Nat Struct Mol Biol.
22:256–264. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guo XY, He CX, Wang YQ, Sun C, Li GM, Su
Q, Pan Q and Fan JG: Circular RNA profiling and bioinformatic
modeling identify its regulatory role in hepatic steatosis. Biomed
Res Int. 2017:59361712017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen Y, Yuan B, Wu Z, Dong Y, Zhang L and
Zeng Z: Microarray profiling of circular RNAs and the potential
regulatory role of hsa_circ_0071410 in the activated human hepatic
stellate cell induced by irradiation. Gene. 629:35–42. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Haczeyni F, Yeh MM, Ioannou GN, Leclercq
IA, Goldin R, Dan YY, Yu J, Teoh NC and Farrell GC: Mouse models of
non-alcoholic steatohepatitis: A reflection on recent literature. J
Gastroenterol Hepatol. 33:1312–1320. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rinella ME, Elias MS, Smolak RR, Fu T,
Borensztajn J and Green RM: Mechanisms of hepatic steatosis in mice
fed a lipogenic methionine choline-deficient diet. J Lipid Res.
49:1068–1076. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang Y, Zhu K, Yu W, Wang H, Liu L, Wu Q,
Li S and Guo J: MiR-181b regulates steatosis in nonalcoholic fatty
liver disease via targeting SIRT1. Biochem Biophys Res Commun.
493:227–232. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li M, Hong W, Hao C, Li L, Wu D, Shen A,
Lu J, Zheng Y, Li P and Xu Y: SIRT1 antagonizes liver fibrosis by
blocking hepatic stellate cell activation in mice. FASEB J.
32:500–511. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang B, Li W, Guo K, Xiao Y, Wang Y and
Fan J: miR-181b promotes hepatic stellate cells proliferation by
targeting p27 and is elevated in the serum of cirrhosis patients.
Biochem Biophys Res Commun. 421:4–8. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yu F, Lu Z, Chen B, Dong P and Zheng J:
Identification of a novel lincRNA-p21-miR-181b-PTEN signaling
cascade in liver fibrosis. Mediators Inflamm. 2016:98565382016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lu L, Wang J, Lu H, Zhang G, Liu Y, Wang
J, Zhang Y, Shang H, Ji H, Chen X, et al: MicroRNA-130a and −130b
enhance activation of hepatic stellate cells by suppressing PPARγ
expression: A rat fibrosis model study. Biochem Biophys Res Commun.
465:387–393. 2015. View Article : Google Scholar : PubMed/NCBI
|