Introduction
Type 1 diabetes mellitus (DM) is the most common
type of chronic metabolic disorder, and is characterized by insulin
deficiency, which leads to elevated blood glucose levels and
disordered protein metabolism (1).
Long-term hyperglycemia can result in various complications that
affect the eyes, kidneys, heart, blood vessels and nerves (2). Increasing evidence has demonstrated
that hyperglycemia leads to oxidative stress in the central nervous
system (CNS), and brain tissue is susceptible to oxidative damage
induced by diabetes (3). It has
been reported that neuronal death may be caused by
diabetes-mediated neurotoxicity (4). Furthermore, insulin resistance in DM
may cause β-amyloid deposition, which further exacerbates
neurotoxicity, causing damage to the brain and affecting cognitive
function (5). Previous studies
have indicated that hyperglycemia is associated with neurotoxicity
and neurotrophic alterations (6,7),
although the underling molecular mechanism remains to be fully
elucidated.
Regarding neurotrophic factors, brain-derived
neurotrophic factor (BDNF) is a member of the neurotrophin family,
which is widely expressed in the CNS. BDNF functions through
binding to its receptor, tyrosine receptor kinase B (TrKB). Through
its regulation of Trk receptors, BDNF affects the assembly of the
cytoskeleton, the pattern of innervation, synaptic strength,
plasticity and the expression of functionally important proteins
(8–10). Although numerous studies have
reported that BDNF and mitogen-activated protein kinase (MAPK)
signaling pathways enhance Ca2+-dependent
excitotoxicity, its specific mechanism remains to be fully
elucidated. In a number of neurological diseases, glutamate has
been reported to accumulate in the extracellular space. The
excessive synaptic release of glutamate and activation of glutamate
receptors subsequently leads to the dysregulation of
Ca2+ homeostasis and the induction of excitotoxic cell
death (11). In addition,
increased extracellular concentrations of glutamate or its analogs
may activate MAPK signaling pathways in primary neuronal cultures
(12). In a separate study, it was
suggested that BDNF increases glutamate release and
N-methyl-D-aspartate (NMDA) channel-gated Ca2+ influx
via TrkB, and thus regulates the frequency and amplitude of
Ca2+ oscillations (13). A study on diabetic rats reported
the downregulation of glutamate decarboxylase, subsequently leading
to the accumulation of glutamate (14). Impairment of glutamate transporters
is involved in various neurological diseases, including Alzheimer's
disease (15). A previous study on
DM revealed increased functional activity and augmented sensitivity
to the regulation of glutamate receptors, NMDA and
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) (14). These factors may contribute to
glutamate-mediated excitotoxicity in DM, as examined in the present
study.
In chronic diseases, the release of glutamate from
presynaptic membranes induces the influx of large quantities of
calcium through NMDA and AMPA receptors, subsequently activating
MAPK signaling pathways and BDNF. The extracellular
signal-regulated kinase (ERK) signaling pathway is a major branch
of the MAPK pathway. MAPK resides in the upstream region of the ERK
pathway and induces ERK1/2 by phosphorylating its threonine and
tyrosine residues (16). ERK
signaling is known to be essential in regulating cellular
proliferation, differentiation and survival, due to its effect on
translational and transcriptional events (17,18).
It has also been reported that the ERK signaling pathway has a
neuroprotective effect in the response of hippocampal neurons to
glutamate-induced excitotoxicity (19).
The striatum constitutes the largest component of
the basal ganglia, which integrates signals from the cerebral
cortex and is central to the appropriate selection of behavioral
action. As a key subcortical region, the role of the striatum has
been investigated in classical motor disorders, including
Parkinson's disease, Huntington's disease and Tourette syndrome, in
addition to depression, and learning and memory (20–24).
Approximately 95% of the striatal neurons are inhibitory projection
neurons, termed medium spiny neurons (MSNs) characterized by their
high spine density and GABAergic-inhibitory outputs (25,26).
Neurotoxicity and neurodegeneration induced by dopamine, glutamate
and environmental conditions, such as overexposure to manganese,
frequently occur in numerous neurological conditions, and the loss
of MSNs is a prominent feature (27,28).
Gastrodin is the primary chemical constituent of
Gastrodia elata Blume, a traditional Chinese medicine. In
view of the primary therapeutic effects of gastrodin in the CNS,
the pharmacokinetics of the herbal agent in the brain has attracted
increasing attention. Gastrodin, a phenolic glycoside chemically
known as 4-hydroxybenzyl alcohol-4-O-β-D-glucopyranoside, is
considered to be the main bioactive constituent of Rhizoma
gastrodiae. With the molecular formula,
C13H18O7, and a molecular weight
of 286 Da, gastrodin readily dissolves in methanol, ethanol and
water, but not in chloroform or ether (29). Gastrodin is able to pass though the
blood-brain barrier and readily gains access to brain tissue
following its entry into the systemic circulation. Lin et al
(30) reported that gastrodin was
detectable in the brain 5 min after intravenous administration (50
mg/kg), reaching a peak brain concentration after 15 min, in rats.
It is well-documented that gastrodin exhibits analgesic, sedative,
hypnagogic, anticonvulsant, antiepileptic and antineurodegenerative
properties (31). In addition,
gastrodin affects apoptosis and glutamate-induced intracellular
Ca2+ increases, indicating that the Ca2+
channel is a promising target of gastrodin. Gastrodin may exert its
neuroprotective effects by inhibiting excitotoxicity (32–34);
however, the specific molecular mechanism underlying the
association between gastrodin and neurotoxicity remains to be fully
elucidated.
In light of the aforementioned studies, the present
study aimed to investigate the association between BDNF, TrKB,
phosphorylated (p-)ERK1/2, p-MAPK kinase (MEK)1/2, excitotoxicity,
glutamate release and the effects of early intervention with
gastrodin in the striatum in DM. It is anticipated that the results
of the present study may provide a biochemical and molecular basis
for the neuroprotective effects of gastrodin in DM-induced
excitotoxicity in striatal neurons.
Materials and methods
Animals and induction of diabetes
All institutional and national guidelines for the
care and use of laboratory animals were followed. A total of 70
male Sprague-Dawley rats (age, 9 weeks; weight, 250–300 g) were
purchased from Chendu Dossy Experimental Animals Co., Ltd (Chendu,
China) and were provided with a standard rodent diet and water
ad libitum; rats were maintained at 23±1°C in a
specific-pathogen-free environment with a 12-hour light/dark cycle
and 60±10% relative humidity. Following adaptation for 2 weeks,
type 1 DM was induced using a single intraperitoneal injection of
streptozotocin (STZ) at 65 mg/kg. Rats with blood glucose levels
>16.7 mmol/l for 2 consecutive days were considered to be
diabetic. In the present study, the rats were randomly divided into
four groups: i) NC9W group, normal control rats gavaged with normal
saline (0.4 ml/100 g/day) and fed for 6 weeks; ii) DM9W + S group,
STZ-induced DM rats gavaged with normal saline (0.4 ml/100 g/day)
for 6 weeks at 3 weeks post-diabetes induction; iii) DM9W + G60
group, STZ-induced DM rats gavaged with gastrodin (60 mg/kg/day,
dissolved in saline, 0.4 ml/100 g/day) for 6 weeks at 3 weeks
post-diabetes induction; and iv) DM9W + G120 group, STZ-induced DM
rats gavaged with gastrodin (120 mg/kg/day, dissolved in saline,
0.4 ml/100 g/day) for 6 weeks at 3 weeks post-diabetes
induction.
Western blotting
The striatal tissue was freshly removed from the
brain of each rat and homogenized with radioimmunoprecipitation
assay buffer [cat. no. 9806; Cell Signaling Technology (CST), Inc.,
Danvers, MA, USA] containing a 10X protease inhibitor cocktail
(1:100; cat. no. 5871; CST, Inc.) and 10X phosphatase inhibitor
cocktail (1:100; cat. no. 5870; CST, Inc.). Following complete
lysis of the tissue, the homogenates were centrifuged at 3,028 × g
for 10 min at 4°C. The supernatants were collected, and the protein
concentration was determined using a bicinchoninic acid protein
assay kit. Equal quantities of sample protein (30 µg) were
separated on 10% SDS-PAGE gels and transferred onto a
polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA,
USA). Following blocking with 5% skim milk for 2 h at room
temperature, the membranes were incubated at 4°C overnight with the
following primary antibodies: Rabbit p-MEK1/2 monoclonal (1:1,000;
cat. no. 9154s; CST, Inc.), rabbit p-ERK1/2 monoclonal (1:1,000;
cat. no. 4370s; CST, Inc.), mouse TrKB monoclonal (1:2,000; cat.
no. sc-377218; Santa Cruz Biotechnology, Inc., Dallas, TX, USA),
mouse BDNF monoclonal (1:2,000; cat. no. ab203573; Abcam,
Cambridge, UK), rabbit MEK1/2 monoclonal (1:1,000; cat. no. 8727s;
CST, Inc.), rabbit ERK1/2 monoclonal (1:1,000; cat. no. 4695s; CST,
Inc.), and rabbit β-tubulin monoclonal (1:1,000; cat. no. 15115;
CST, Inc.). Following incubation for 2 h at room temperature with
goat-anti-rabbit (1:1,000; cat. no. 31460; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and goat-anti-mouse (1:1,000;
cat. no. 31430; Thermo Fisher Scientific, Inc.) immunoglobulin G
H&L secondary antibodies, the blots were developed with
enhanced chemiluminescence, and densitometric analysis of the film
was performed using ImageJ software (version 1.4.3.67; National
Institutes of Health, Bethesda, MD, USA).
Immunofluorescence
At designated time points, the rats were
anesthetized with ketamine (75 mg/kg) and xylazine (10 mg/kg),
administered intraperitoneally, and then perfused transcardially
with saline, followed by 4% paraformaldehyde. Following perfusion,
the brain was removed, fixed in 4% formaldehyde for 48 h at room
temperature, dehydrated in an ascending ethanol series, cleared
with xylene and embedded in paraffin blocks. The 3 µm thick
paraffin sections were deparaffinized and hydrated through a
descending series of gradient ethanol. The tissues were incubated
in citrate buffer for antigen retrieval. Subsequently, the tissue
sections were incubated with 5% normal goat serum (Beijing
Biosynthesis Biotechnology Co., Ltd, Beijing, China) for 2 h at
room temperature and then overnight at 4°C with the following
primary antibodies: Mouse dopamine- and cAMP-regulated neuronal
phosphoprotein (DARPP)-32 monoclonal (1:100; cat. no. sc-271111;
Santa Cruz Biotechnology, Inc.), rabbit p-ERK1/2 monoclonal
(1:1,000; cat. no. 4370s; Cell CST, Inc.), rabbit MEK1/2 monoclonal
(1:1,000; cat. no. 8727s; CST, Inc.) and rabbit ERK1/2 monoclonal
(1:1,000; cat. no. 4695s; CST, Inc.). This was followed by
incubation with goat-anti-rabbit immunoglobulin G H&L
Cross-Adsorbed Alexa Fluor 488 (1:100; cat. no. A11008; Thermo
Fisher Scientific, Inc.) or goat-anti-mouse immunoglobulin G
H&L Cross-Adsorbed Alexa Fluor 546 (1:100; cat. no. A11003;
Invitrogen; Thermo Fisher Scientific, Inc.) secondary antibodies
for 2 h at room temperature. Finally, the tissue sections were
washed with PBS. Images were captured at magnification, ×600 and
×1,000 under a confocal fluorescence microscope (FV1000; Olympus
Corporation, Tokyo, Japan;) with ChemiDoc XRS+ (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Hematoxylin and eosin (H&E)
staining
The aforementioned tissue sections were were first
incubated with hematoxylin for 5 min and subsequently washed with
1% ethanol hydrochloride for 3 sec at room temperature. Following
washing with water, the sections were stained with eosin. To assess
the abnormalities in neurons, the sections were examined at ×400
magnification under a light microscope in a blinded manner.
Amino acid analysis
The extracellular levels of glutamate were measured
using microdialysis followed by high-pressure-liquid-chromatography
(HPLC) in the striatum. HPLC uses a liquid as the mobile phase and
a high-pressure infusion system to pump a single solvent with
different polarities or a mixed solvent of different ratios, into a
column packed with a stationary phase; the components in the column
are subsequently separated. The derivative reagent was prepared by
dissolving 0.05 mg o-phthalaldehyde in 1 ml methanol to which 500
µl β-mercaptoethanol and 0.1 mol/l Na-tetraborate was added to make
10 ml. An HPLC device (Agilent 1200; Agilent Technologies, Inc.,
Santa Clara, CA, USA) with two mobile phases was used. Mobile phase
A consisted of 0.25% (v%) tetrahydrofuran in 0.03 mol/l sodium
acetate solution (solution pH, 7.2). Mobile phase B consisted of
80% (v%) acetonitrile and 20% (v%) 0.01 mol/l sodium acetate
solution. Glutamate standards were weighed and prepared into amino
acid standard solutions at different concentrations (1.00, 2.50,
5.00, 10.00, 25.00, 50.00 and 100.00 mg/l). A 20-µl sample solution
at each concentration was detected to plot a standard curve with
the concentrations represented on the X axis and the peak area
serving as the Y axis. The optimal fit for all standards was
0.9992. Glutamate concentrations in the striatum were measured in
each group based on this analytical method.
Statistical analysis
Data are expressed as the mean ± standard deviation
and were analyzed with SPSS 17.0 statistical software (SPSS, Inc.,
Chicago, IL, USA). Comparisons among groups were performed using
one-way analysis of variance and pairwise comparison was performed
using the least significant difference t-test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Early intervention with gastrodin
ameliorates neuronal injury in the striatum
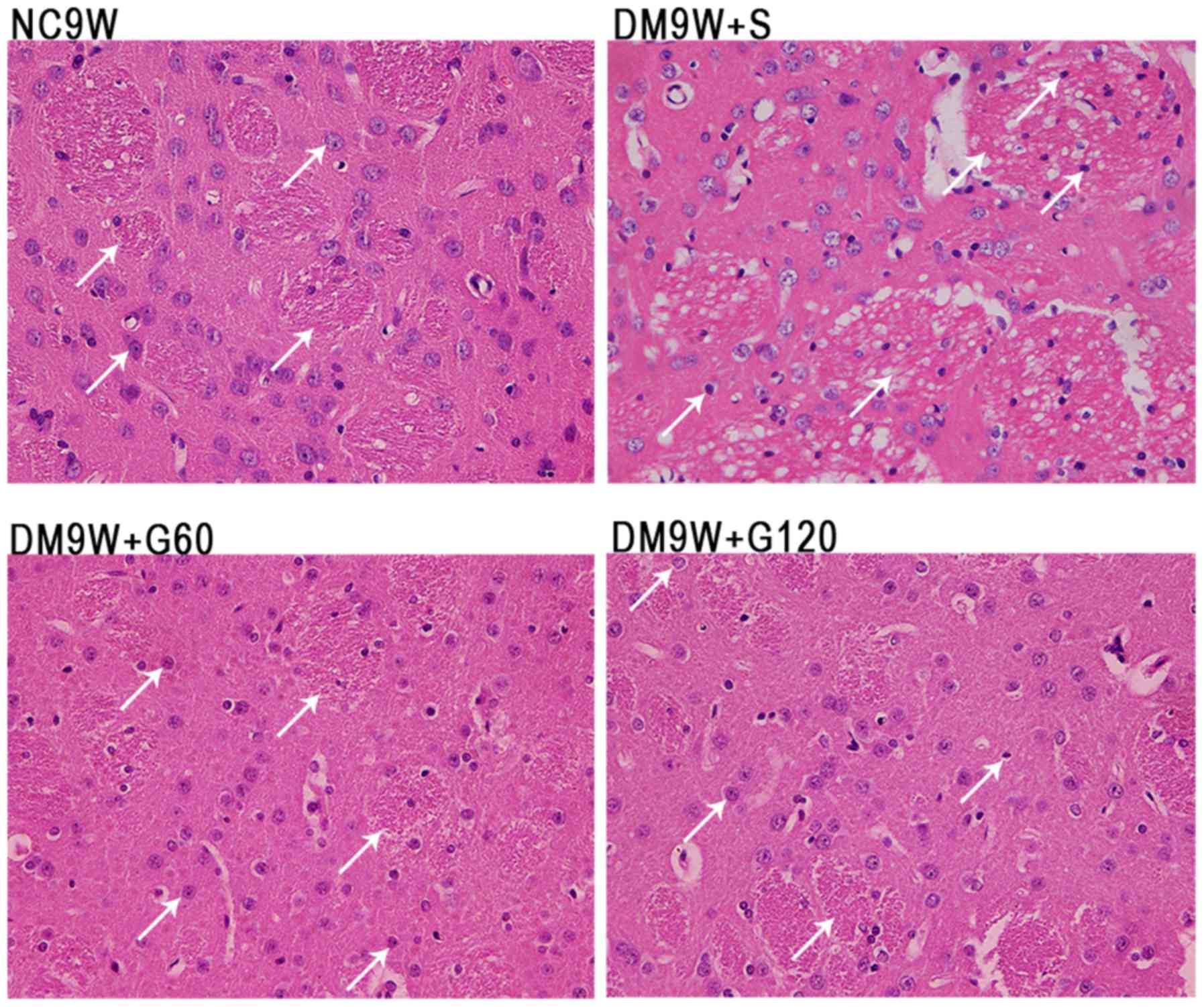
The neurons of the striatum were examined by H&E
staining (Fig. 1). In the normal
control (NC9W) group, the striatal neurons were arranged in an
orderly manner. The neurons were well-defined, exhibiting a
conspicuous round or oval nucleus with discrete chromatin
formations. By contrast, the striatal neurons in DM9W + S were
arranged in a disorderly manner with a pyknotic or shrunken
nucleus. Furthermore, the striatal tissue appeared vacuolated.
Compared with the NC9W group, neuronal damage was less evident in
the DM9W + G60 and DM9W + G120 groups.
Early intervention with gastrodin
decreases the expression of p-ERK1/2 in MSNs
MEK1/2, ERK1/2 and p-ERK1/2 (red, Alexa Fluor 546)
immunofluorescence was primarily localized to the MSNs (green,
Alexa 488) in the striatum. On closer scrutiny, ERK1/2, MEK1/2 and
p-ERK1/2 immunofluorescence was detected in the cytoplasm of the
MSNs; occasionally, the MSNs exhibited the ERK1/2, MEK1/2 and
p-ERK1/2 in the nucleus (Fig.
2A-D). Double immunofluorescence labeling of p-ERK1/2 revealed
significantly increased expression levels in the MSNs of the DM9W +
S group compared with that of the NC9W group (P<0.001). However,
expression of p-ERK1/2 in the MSNs of the DM9W + G60 and DM9W +
G120 groups was significantly attenuated compared with that in the
DM9W + S group (P<0.01; Fig.
2B).
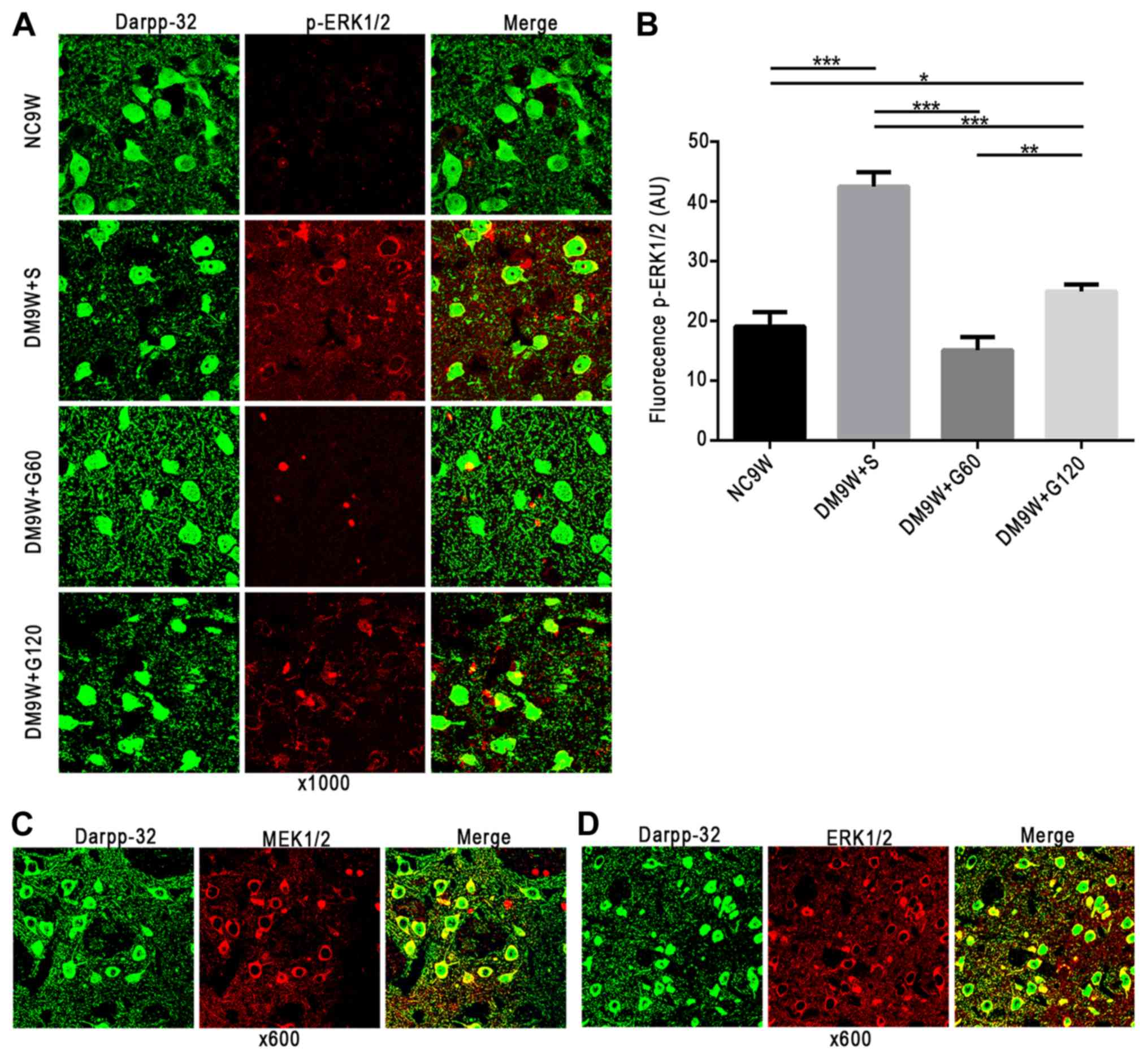 | Figure 2.Localization of MEK1/2, ERK1/2 and
p-ERK1/2 in MSNs of the striatum. MEK1/2, ERK1/2 and p-ERK1/2 are
shown in red (Alexa Fluor 546), and MSN marker DARPP-32 in green
(Alexa 488). (A) Gastrodin treatment decreased the expression
levels of p-ERK1/2 in MSNs; p-ERK1/2 was localized preferentially
in the cytoplasm. (B) Quantification of fluorescence intensity is
presented as the mean ± standard deviation. *P<0.05, **P<0.01
and ***P<0.001. (C) MEK1/2 and (D) ERK1/2 were also localized
preferentially in the cytoplasm. DARPP-23, dopamine- and
cAMP-regulated neuronal phosphoprotein-32; DM, diabetes mellitus;
ERK, extracellular signal regulated kinase; G60, gastrodin (60
mg/kg/day); Gl20, gastrodin (120 mg/kg/day); MEK, mitogen-activated
protein kinase kinase; MSNs, medium spiny neurons; NC, normal
control; S, normal saline; p-, phosphorylated. |
Gastrodin decreases the
phosphorylation of MEK1/2 and ERK1/2 and the expression of BDNF and
TrKB
To examine the contribution of MEK1/2, ERK1/2, BDNF
and TrKB to the neuroprotective effect of gastrodin, the expression
levels of these biomarkers were determined by western blotting
(Fig. 3). The results demonstrated
that the expression of p-MEK1/2 was significantly increased in the
DM9W + S group compared with that in the NC9W group (P<0.001).
In the DM9W + G60 group, the expression of p-MEK1/2 was
significantly lower compared with that in the DM9W + S group
(P<0.001); and the expression of p-MEK1/2 in the DM9W + G120
group was significantly higher compared with that in the GM9W + G60
group (P<0.001; Fig. 3Aa-c).
Additionally, the expression of p-ERK1/2 was significantly higher
in the DM9W + S group compared with that in the NC9W group
(P<0.001), and was significantly lower in the DM9W + G60 group
when compared with that in the DM9W + S group (P<0.001; Fig. 3Ba-c). Alterations in the expression
levels of BDNF and TrKB followed the same trend as those for
p-ERK1/2 (Fig. 3Ca-c).
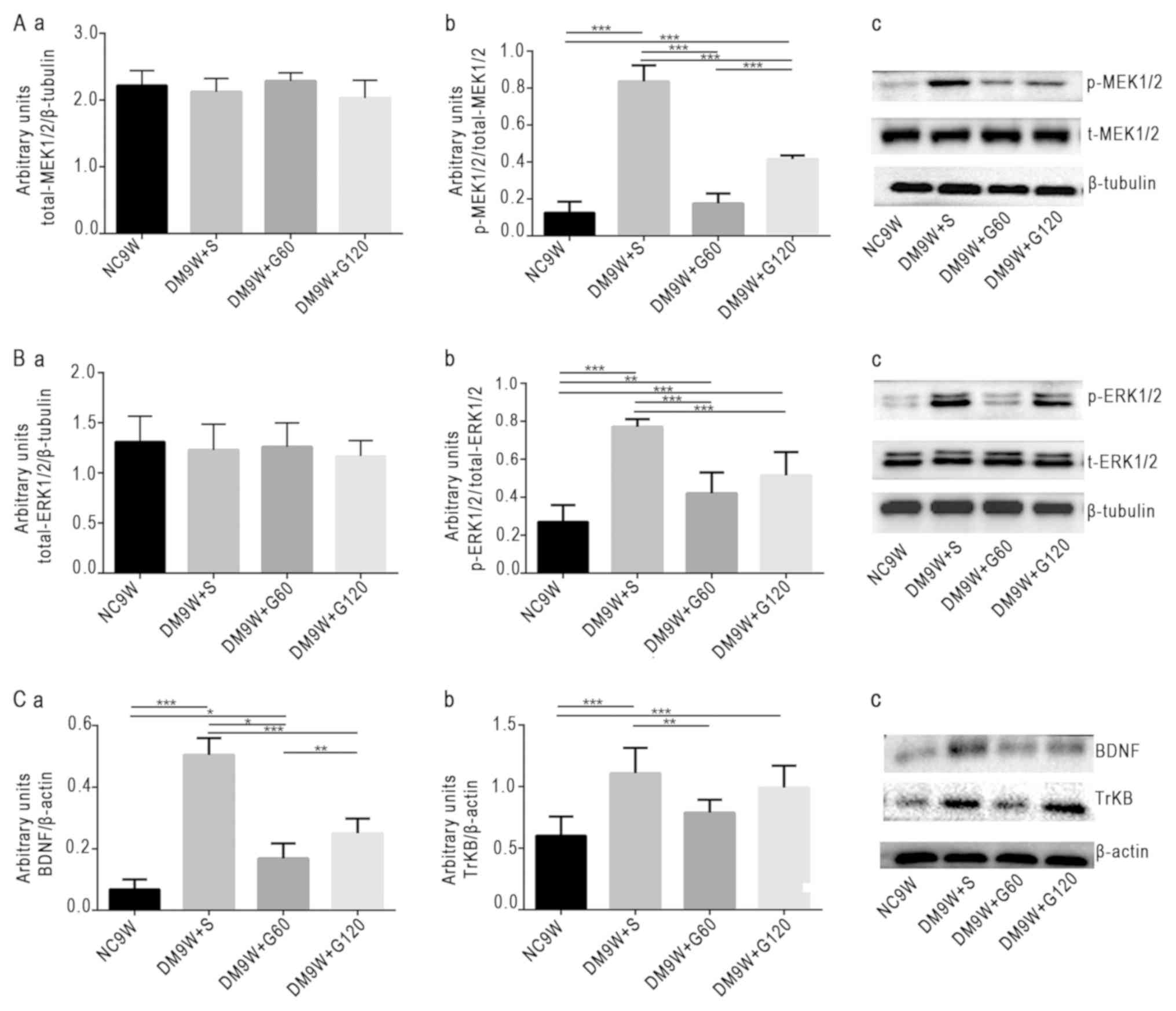 | Figure 3.Western blot analysis of protein
expression levels of p-ERK1/2, p-MEK1/2, BDNF and TrKB. Gastrodin
decreased the expression levels of p-ERK1/2, p-MEK1/2, BDNF and
TrKB in the striatum of type 1 DM rats. (A) Densitometric
quantification of (a) t-MEK1/2 and (b) p-MEK1/2:t-MEK1/2 from (c)
western blots. (B) Densitometric quantification of (a) t-ERK1/2 and
(b) p-ERK1/2:t-ERK1/2 from (c) western blots. (C) Densitometric
quantification of (a) BDNF:β-actin and (b) TrKB:β-actin from (c)
western blots. The bars represent total gray values (mean ±
standard deviation). *P<0.05, **P<0.01 and ***P<0.001.
BDNF, brain-derived neurotrophic factor; DM, diabetes mellitus;
ERK, extracellular signal regulated kinase; G60, gastrodin (60
mg/kg/day); Gl20, gastrodin (120 mg/kg/day); MEK, mitogen-activated
protein kinase kinase; NC, normal control; p-, phosphorylated; S,
normal saline; t-, total; TrKB, tyrosine receptor kinase B. |
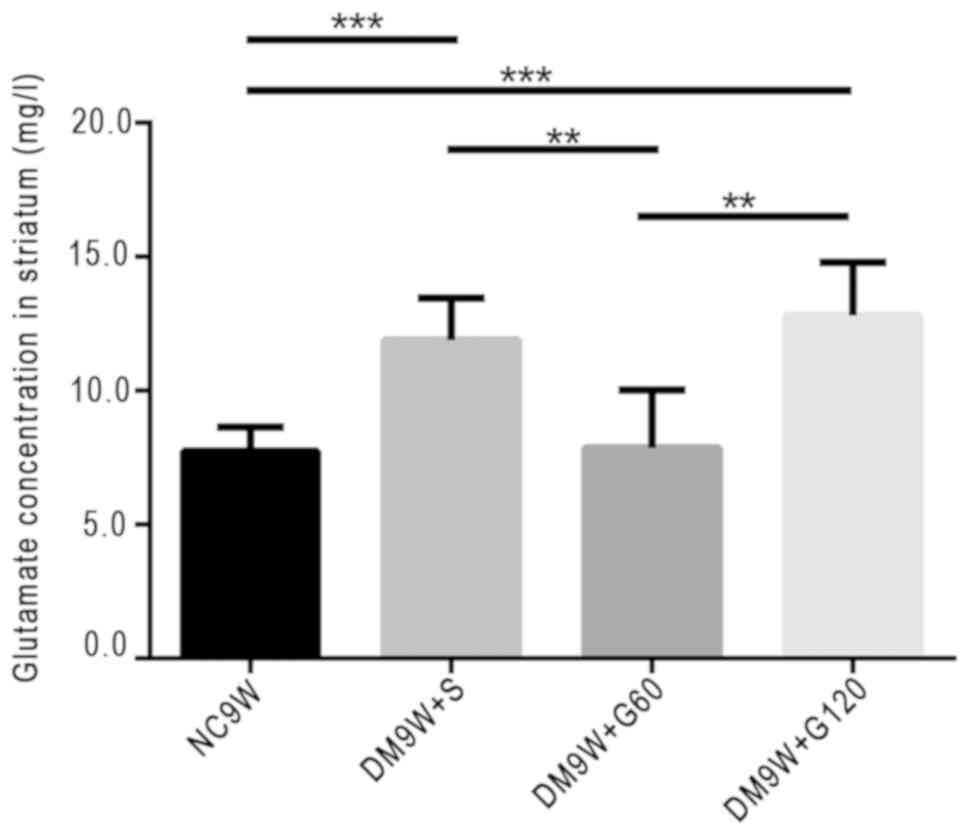
Gastrodin decreases glutamate
concentration
The extracellular levels of glutamate were measured
by HPLC. Compared with the NC9W group, glutamate levels were
significantly increased in the DM9W + S (P<0.001) and DM9W +
G120 (P<0.001) groups. However, the glutamate level in the DM9W
+ G60 group was significantly lower compared with that in the DM9W
+ S group (P<0.01); no significant differences were detected
between the DM9W + S and DM9W + G120 groups (Fig. 4).
Discussion
Previous studies have reported that
experimentally-induced diabetes may reduce the proliferation of
granular cells and enhance neuronal death (necrosis/apoptosis) in
the CA3 and dentate gyrus regions (35,36).
Duration-associated apoptosis is likely to account for the loss of
neurons and the concomitant emergence of cognitive impairments
observed in diabetic animals (37). In the present study, it was
demonstrated that striatal neurons underwent evident pathological
alterations 9 weeks subsequent to diabetic induction in rats.
Notably, these pathological alterations in the striatal neurons
were ameliorated following early intervention with gastrodin.
Glutamate is the primary excitatory neurotransmitter
in the mammalian brain. It has been demonstrated to be important in
numerous processes in the brain, including neurodevelopmental
processes, such as differentiation, migration and survival. By
contrast, glutamate has also been implicated in acute
neurodegeneration, chronic neurodegeneration, the stress response
and anxiety disorders. Furthermore, the ERK/MAPK signaling cascade
is essential in neuronal cell survival and death, generally
depending on its magnitude and duration of activation. However,
evidence suggests that the activation of ERK1/2 also contributes to
cell death in certain cell types and organs under specific
conditions (38). In addition, the
excessive release of glutamate and the subsequent influx of calcium
are associated with a number of neurological insults that result in
neuronal death (39). Glutamate
may lead to the persistent activation of ERK1/2, which is
associated with neuronal cell death (40). This is due to the fact that the
stimulation of glutamate receptors and influx of Ca2+
are associated with excitotoxic injury, leading to the
phosphorylation of p44/42 MAPK in neurons. Extracellular glutamate
may be released from neurons and astrocytes, and according to other
studies, activated microglia also release certain amounts of
glutamate under various pathological processes, including
inflammation (15,41). Therefore, the possibility that
glutamate may be released from glial cells and neurons in the
striatal tissue in DM should be considered.
The activation of ERKs has been revealed to
contribute to neuronal cell death in certain in vitro models
of neurotoxicity. Persistent activation of ERK1/2 contributes to
glutamate-induced oxidative toxicity (42–45),
which is consistent with the results of the present study. ERK also
contributes to cell death through the suppression of anti-apoptotic
signaling molecule RAC-α serine/threonine-protein kinase (34). The results of the present study
indicated that, 9 weeks following the induction of diabetes, the
expression levels of p-ERK1/2 and glutamate were significantly
elevated. Notably, following early intervention with gastrodin,
glutamate levels and the expression of ERK1/2 were reduced. As a
corollary, it may be that gastrodin reduces glutamate-induced
excitotoxicity by reducing the content of glutamate and suppressing
the expression level of ERK.
It is widely known that BDNF has a neuroprotective
effect by preventing the neuronal death induced by metabolic and
oxidative stress and excitotoxicity, and modulating calcium
responses to NMDA and AMPA receptors, which may be associated with
the MAPK signaling pathway. Its specific mechanism remains to be
fully elucidated, but may involve the enhancement of antioxidant
systems (46–48). However, certain neurotrophins may
have opposing effects on different types of cell death within the
same neuron (49,50). Previous reports have indicated that
certain trophic factors have the capacity to exacerbate
excitotoxicity (13,51) which is consistent with the results
of the present study on diabetes; however, the specific mechanisms
involved remain to be elucidated. Previous studies have reported
that excitotoxicity caused by glutamate and the deregulation of
intracellular Ca2+ homeostasis may contribute to
numerous forms of pathological neuronal death, including acute
injuries, such as hypoxia, hypoglycemia and seizures, and chronic
neurodegenerative disorders, such as Parkinson's, Alzheimer's and
Huntington's disease (51).
Furthermore, it has been reported that glutamate-induced
intracellular Ca2+ accumulation is time- and
dose-dependent (52). Previous
studies have demonstrated that AMPA and NMDA receptors are involved
in the regulation of BDNF (41).
The upregulation of AMPA and NMDA receptor subunits, mediated by
the binding of BDNF to Trk receptors, enhances calcium responses to
NMDA, increasing neuronal vulnerability to excitotoxic necrosis
(51).
The results of the present study demonstrated marked
upregulation in the levels of glutamate, BDNF and TrKB in the DM
rats, and this may enhance neurotoxicity resulting in neuronal
injury. These results suggested that gastrodin was able to decrease
neuronal injury by reducing glutamate, and the overexpressed BDNF
and TrKB. Therefore, damage to striatal neurons, as observed in the
present DM rat model, may be attributed to excitotoxicity. An
increase in glutamate in striatal tissue may be the primary
contributing factor to neuronal damage. Notably, an increase in
extracellular glutamate levels was coupled with the overactivation
of p-ERK1/2, p-MEK1/2, BDNF and TrKB. It is possible that these
factors and signaling pathways collectively lead to neuronal
dysfunction and structural alterations (Fig. 5). Furthermore, the results revealed
that gastrodin protected neurons through inhibiting the activation
of these factors and thus reducing the levels of neurotoxicity in
experimentally-induced diabetes. Gastrodin has been described as a
‘top grade medicine’, capable of improving health and extending
life without toxicity, and may be used long term without causing
harm (29). A previous study
showed that pretreatment with gastrodin at 60 and 120 mg/kg had
neuroprotective effects (53),
which is consistent with the results of the present study. Of note,
in the present experimentally-induced DM model, gastrodin at a
lower dose (60 mg/kg) was demonstrated to be more potent in
reducing neurotoxicity levels compared with a higher dose (120
mg/kg). This suggested that gastrodin at 60 mg/kg is the optimal
dose for the attenuation of diabetes-induced neurotoxicity in
striatal neurons.
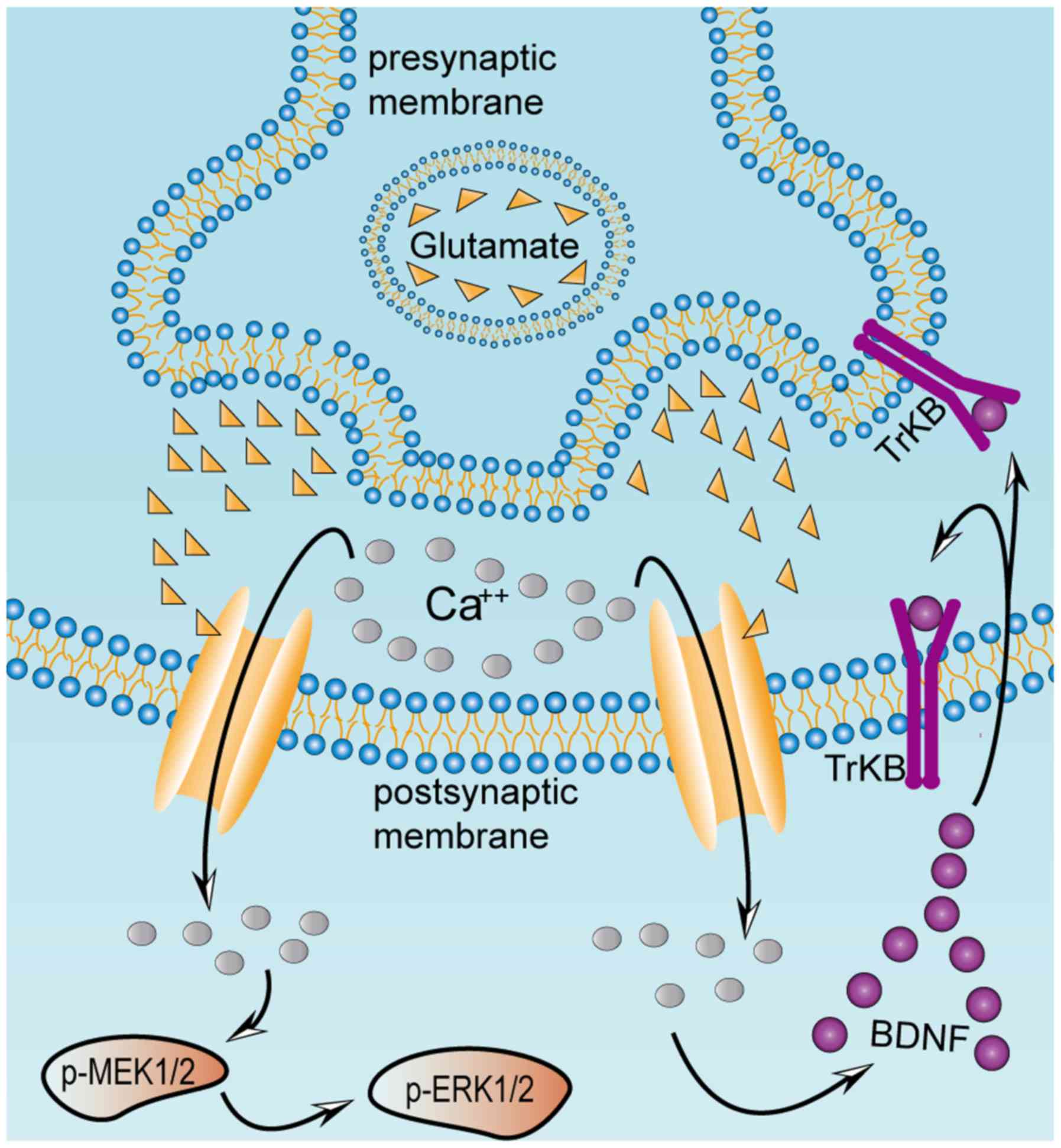 | Figure 5.Excessive release of glutamate from
presynaptic terminals activates BDNF, TrkB, p-MEK1/2 and p-ERK1/2
in postsynaptic neurons. Excitatory damage may be caused by chronic
metabolic diseases, including diabetes mellitus. This may stimulate
excessive release of glutamate, which induces a massive influx of
calcium mediated by NMDA and
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors in the
postsynaptic striatal neurons, thereby activating the MAPK pathway
and BDNF. Once BDNF secretion is activated, it binds to its
receptor TrkB, localized on the presynaptic and postsynaptic
membranes. Presynaptic TrkB receptors are known to mediate the
enhancement of glutamate release, whereas postsynaptic TrkB
receptors enhance the response of NMDA receptors to calcium ions.
Excessive influx of calcium ions into the postsynaptic striatal
neurons results in continuous activation of the MAPK and BDNF
pathways, which ultimately causes excitatory damage to the
striatum. BDNF, brain-derived neurotrophic factor; ERK,
extracellular signal regulated kinase; MEK, mitogen-activated
protein kinase kinase; NMDA, N-methyl-D-aspartate; p-,
phosphorylated; TrKB, tyrosine receptor kinase B. |
Acknowledgements
Not applicable.
Funding
The present study was supported in part by The
National Natural Sciences Foundation of China (grant nos. 81760149,
81460210, 81360176, 31760292 and 81200840), The National University
Students Innovation and Entrepreneurship Training Program (grant
no. 201710678004), The Department of Science and Technology of
Yunnan Province (grant no. 2013HB078), The Joint Special Funds for
the Department of Science and Technology of Yunnan Province-Kunming
Medical University [grant nos. 2017FE468 (−171), 2017FE468 and
2014FZ007] and The Yunnan provincial Education Commission (grant
nos. 2018JS156 and 2018Y040).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YYZ and DL designed the project, and contributed to
the analysis of data and finalization of the manuscript. YHQ and RZ
performed the majority of the experiments, participated in
discussion and analysis of data, and prepared the first draft of
the manuscript. QW and QL conducted part of experiments, and
participated in discussion and analysis of data. YDL designed and
guided the use of gastrodin. ZYQ and ZHY performed paraffin
embedding, sectioning and H&E staining. ZHM guided the
molecular biology experiment and analysis of data. XJL, MYZ, XW and
XYL helped with removal of tissue samples and took care of the
experimental rats. QW revised the manuscript.
Ethics approval and consent to
participate
This study was approved by the Medical Ethics
Committee of Kunming Medical University. All institutional and
national guidelines for the care and use of laboratory animals were
followed.
Patients consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing
interests.
References
|
1
|
Ding Y, Sun X and Shan PF: MicroRNAs and
cardiovascular disease in diabetes mellitus. Biomed Res Int.
2017:40803642017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gispen WH and Biessels GJ: Cognition and
synaptic plasticity in diabetes mellitus. Trends Neurosci.
23:542–549. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ashafaq M, Varshney L, Khan MH, Salman M,
Naseem M, Wajid S and Parvez S: Neuromodulatory effects of
hesperidin in mitigating oxidative stress in streptozotocin induced
diabetes. Biomed Res Int. 2014:2490312014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kong FJ, Ma LL, Guo JJ, Xu LH, Li Y and Qu
S: Endoplasmic reticulum stress/autophagy pathway is involved in
diabetes-induced neuronal apoptosis and cognitive decline in mice.
Clin Sci (Lond). 132:111–125. 2017. View Article : Google Scholar
|
|
5
|
Zhang Y, Huang NQ, Yan F, Jin H, Zhou SY,
Shi JS and Jin F: Diabetes mellitus and Alzheimer's disease: GSK-3β
as a potential link. Behav Brain Res. 339:57–65. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li Y, Zhang Y, Wang L, Wang P, Xue Y, Li
X, Qiao X, Zhang X, Xu T, Liu G, et al: Autophagy impairment
mediated by S-nitrosation of ATG4B leads to neurotoxicity in
response to hyperglycemia. Autophagy. 13:1145–1160. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhong Y, Zhu Y, He T, Li W, Li Q and Miao
Y: Brain-derived neurotrophic factor inhibits hyperglycemia-induced
apoptosis and downregulation of synaptic plasticity-related
proteins in hippocampal neurons via the PI3K/Akt pathway. Int J Mol
Med. 43:294–304. 2019.PubMed/NCBI
|
|
8
|
Huang EJ and Reichardt LF: Trk receptors:
Roles in neuronal signal transduction. Annu Rev Biochem.
72:609–642. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yamada K and Nabeshima T: Brain-derived
neurotrophic factor/TrkB signaling in memory processes. J Pharmacol
Sci. 91:267–270. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huang EJ and Reichardt LF: Neurotrophins:
Roles in neuronal development and function. Annu Rev Neurosci.
24:677–736. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Arundine M and Tymianski M: Molecular
mechanisms of calcium-dependent neurodegeneration in
excitotoxicity. Cell Calcium. 34:325–337. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rivera-Cervantes MC, Castañeda-Arellano R,
Castro-Torres RD, Gudino-Cabrera G, Feria y Velasco AI, Camins A
and Beas-Zarate C: P38 MAPK inhibition protects against glutamate
neurotoxicity and modifies NMDA and AMPA receptor subunit
expression. J Mol Neurosci. 55:596–608. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sakai N, Yamada M, Numakawa T, Ogura A and
Hatanaka H: BDNF potentiates spontaneous Ca2+ oscillations in
cultured hippocampal neurons. Brain Res. 778:318–328. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jayanarayanan S, Smijin S, Peeyush KT,
Anju TR and Paulose CS: NMDA and AMPA receptor mediated
excitotoxicity in cerebral cortex of streptozotocin induced
diabetic rat: Ameliorating effects of curcumin. Chem Biol Interact.
201:39–48. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Junpei T, Koki F, Marie M, Takeshi S, Yuko
S and Kaoru S: L-glutamate released from activated microglia
downregulates astrocytic L-glutamate transporter expression in
neuroinflammation: The ‘collusion’ hypothesis for increased
extracellular L-glutamate concentration in neuroinflammation. J
Neuroinflammation. 9:2752012.PubMed/NCBI
|
|
16
|
Li ZY, Huang Y, Yang YT, Zhang D, Zhao Y,
Hong J, Liu J, Wu LJ, Zhang CH, Wu HG, et al: Moxibustion eases
chronic inflammatory visceral pain through regulating MEK, ERK and
CREB in rats. World J Gastroenterol. 23:6220–6230. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Birkner K, Wasser B, Loos J, Plotnikov A,
Seger R, Zipp F, Witsch E and Bittner S: The role of ERK signaling
in experimental autoimmune encephalomyelitis. Int J Mol Sci.
18:E19902017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Plotnikov A, Chuderland D, Karamansha Y,
Livnah O and Seger R: Nuclear extracellular signal-regulated kinase
1 and 2 translocation is mediated by casein kinase 2 and
accelerated by autophosphorylation. Mol Cell Biol. 31:3515–3530.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ortuño-Sahagún D, González RM, Verdaguer
E, Huerta VC, Torres-Mendoza BM, Lemus L, Rivera-Cervantes MC,
Camins A and Zarate CB: Glutamate excitotoxicity activates the
MAPK/ERK signaling pathway and induces the survival of rat
hippocampal neurons in vivo. J Mol Neurosci. 52:366–377. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shepherd GM: Corticostriatal connectivity
and its role in disease. Nat Rev Neurosci. 14:278–291. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Caligiore D, Mannella F, Arbib MA and
Baldassarre G: Dysfunctions of the basal
ganglia-cerebellar-thalamo-cortical system produce motor tics in
Tourette syndrome. PLoS Comput Biol. 13:e10053952017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chang C, Crottaz-Herbette S and Menon V:
Temporal dynamics of basal ganglia response and connectivity during
verbal working memory. Neuroimage. 34:1253–1269. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Murty VP, DuBrow S and Davachi L: The
simple act of choosing influences declarative memory. J Neurosci.
35:6255–6264. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
van Duinkerken E, Schoonheim MM, Steenwijk
MD, Klein M, Ijzerman RG, Moll AC, Heymans MW, Snoek FJ, Barkhof F
and Diamant M: Ventral striatum, but not cortical volume loss, is
related to cognitive dysfunction in type 1 diabetic patients with
and without microangiopathy. Diabetes Care. 37:2483–2490. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gerfen CR and Surmeier DJ: Modulation of
striatal projection systems by dopamine. Annu Rev Neurosci.
34:441–466. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Reinius B, Blunder M, Brett FM, Eriksson
A, Patra K, Jonsson J, Jazin E and Kullander K: Conditional
targeting of medium spiny neurons in the striatal matrix. Front
Behav Neurosci. 9:712015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Stansfield KH, Bichell TJ, Bowman AB and
Guilarte TR: BDNF and Huntingtin protein modifications by
manganese: Implications for striatal medium spiny neuron pathology
in manganese neurotoxicity. J Neurochem. 131:655–666. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang J, Saur T, Duke AN, Grant SG, Platt
DM, Rowlett JK, Isacson O and Yao WD: Motor impairments, striatal
degeneration, and altered dopamine-glutamate interplay in mice
lacking PSD-95. J Neurogenet. 28:98–111. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu Y, Gao J, Peng M, Meng H, Ma H, Cai P,
Xu Y, Zhao Q and Si G: A review on central nervous system effects
of gastrodin. Front Pharmacol. 9:242018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin LC, Chen YF, Tsai TR and Tsai TH:
Analysis of brain distribution and biliary excretion of a nutrient
supplement, gastrodin, in rat. Anal Chim Acta. 590:173–179. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhan HD, Zhou HY, Sui YP, Du XL, Wang WH,
Dai L, Sui F, Huo HR and Jiang TL: The rhizome of Gastrodia elata
Blume-an ethnopharmacological review. J Ethnopharmacol.
189:361–385. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee YS, Ha JH, Yong CS, Lee DU, Huh K,
Kang YS, Lee SH, Jung MW and Kim JA: Inhibitory effects of
constituents of Gastrodia elata Bl. on glutamate-induced apoptosis
in IMR-32 human neuroblastoma cells. Arch Pharm Res. 22:404–409.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao X, Zou Y, Xu H, Fan L, Guo H, Li X,
Li G, Zhang X and Dong M: Gastrodin protect primary cultured rat
hippocampal neurons against amyloid-beta peptide-induced
neurotoxicity via ERK1/2-Nrf2 pathway. Brain Res. 1482:13–21. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jang JH, Son Y, Kang SS, Bae CS, Kim JC,
Kim SH, Shin T and Moon C: Neuropharmacological potential of
Gastrodia elata Blume and its components. Evid Based Complement
Alternat Med. 2015:3092612015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang WJ, Tan YF, Yue JT, Vranic M and
Wojtowicz JM: Impairment of hippocampal neurogenesis in
streptozotocin-treated diabetic rats. Acta Neurol Scand.
117:205–210. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li ZG, Zhang W, Grunberger G and Sima AA:
Hippocampal neuronal apoptosis in type 1 diabetes. Brain Res.
946:221–231. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sadeghi A, Hami J, Razavi S, Esfandiary E
and Hejazi Z: The effect of diabetes mellitus on apoptosis in
hippocampus: Cellular and molecular aspects. Int J Prev Med.
7:572016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhuang S and Schnellmann RG: A
death-promoting role for extracellular signal-regulated kinase. J
Pharmacol Exp Ther. 319:991–997. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Murray B, Alessandrini A, Cole AJ, Yee AG
and Furshpan EJ: Inhibition of the p44/42 MAP kinase pathway
protects hippocampal neurons in a cell-culture model of seizure
activity. Proc Natl Acad Sci USA. 95:11975–11980. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Choi BH, Hur EM, Lee JH, Jun DJ and Kim
KT: Protein kinase Cdelta-mediated proteasomal degradation of MAP
kinase phosphatase-1 contributes to glutamate-induced neuronal cell
death. J Cell Sci. 119:1329–1340. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Poitry-Yamate CL, Vutskits L and Rauen T:
Neuronal-induced and glutamate-dependent activation of glial
glutamate transporter function. J Neurochem. 82:987–997. 2010.
View Article : Google Scholar
|
|
42
|
Stanciu M, Wang Y, Kentor R, Burke N,
Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW
and DeFranco DB: Persistent activation of ERK contributes to
glutamate-induced oxidative toxicity in a neuronal cell line and
primary cortical neuron cultures. J Biol Chem. 275:12200–12206.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cagnol S, Van Obberghen-Schilling E and
Chambard JC: Prolonged activation of ERK1,2 induces
FADD-independent caspase 8 activation and cell death. Apoptosis.
11:337–346. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
de Bernardo S, Canals S, Casarejos MJ,
Solano RM, Menendez J and Mena MA: Role of extracellular
signal-regulated protein kinase in neuronal cell death induced by
glutathione depletion in neuron/glia mesencephalic cultures. J
Neurochem. 91:667–682. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shen GN, Liu L, Feng L, Jin Y, Jin MH, Han
YH, Jin CH, Jin YZ, Lee DS, Kwon TH, et al: Knockdown of
peroxiredoxin V increases glutamateinduced apoptosis in HT22
hippocampal neuron cells. Mol Med Rep. 17:7827–7834.
2018.PubMed/NCBI
|
|
46
|
Cheng B and Mattson MP: NT-3 and BDNF
protect CNS neurons against metabolic/excitotoxic insults. Brain
Res. 640:56–67. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Xiong H, Futamura T, Jourdi H, Zhou H,
Takei N, Diverse-Pierluissi M, Plevy S and Nawa H: Neurotrophins
induce BDNF expression through the glutamate receptor pathway in
neocortical neurons. Neuropharmacology. 42:903–912. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Almeida RD, Manadas BJ, Melo CV, Gomes JR,
Mendes CS, Graos MM, Carvalho RF, Carvalho AP and Duarte CB:
Neuroprotection by BDNF against glutamate-induced apoptotic cell
death is mediated by ERK and PI3-kinase pathways. Cell Death
Differ. 12:1329–1343. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hu P and Kalb RG: BDNF heightens the
sensitivity of motor neurons to excitotoxic insults through
activation of TrkB. J Neurochem. 84:1421–1430. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Koh JY, Gwag BJ, Lobner D and Choi DW:
Potentiated necrosis of cultured cortical neurons by neurotrophins.
Science. 268:573–575. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Glazner GW and Mattson MP: Differential
effects of BDNF, ADNF9, and TNFalpha on levels of NMDA receptor
subunits, calcium homeostasis, and neuronal vulnerability to
excitotoxicity. Exp Neurol. 161:442–452. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
El IA and Trenkner E: Growth factors and
taurine protect against excitotoxicity by stabilizing calcium
homeostasis and energy metabolism. J Neurosci. 19:9459–9468. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bian L, Bi X, Ai Q, Guo J, Dong S, Xu J,
Zhong L and Lu D: Effects of gastrodin on apoptotic factors of
cerebral cortex neuron in epileptic rats. Chin J Neuroanat.
32:37–43. 2016.
|