Introduction
Lung cancer remains the chief cause of cancer death
worldwide (1). Most lung cancer is
non-small cell lung cancer (NSCLC) corresponding to more than 85%
of lung cancers (2).
Platinum-based chemotherapy has been prescribed as a first-line
treatment for NSCLC patients but yielded limited outcomes in terms
of overall survival and 5-year survival rate (3). In the past decade, there has been
dramatic paradigm shift in the treatment of NSCLC, since
constitutive activation of epidermal growth factor receptor (EGFR)
have been discovered and verified to be closely correlated with the
initiation, progression and poor prognosis of NSCLC (4–6).
EGFR is a member of the HER/ErbB family of receptor
tyrosine kinase (RTK) composed of four closely related RTKs: EGFR
(ErbB1), HER2/neu (ErbB2), HER3 (ErbB3) and HER4 (ErbB4) (7,8).
These receptors are located in the plasma membrane and have similar
structural features, that are extracellular ligand-binding domain,
a single transmembrane region, and a cytoplasmic tyrosine kinase
domain (9). A number of ligands
including epidermal growth factor (EGF) and transforming growth
factor-α, bind to EGFR, which induce receptor dimerization and
autophosphorylation and/or transphosphorylation of tyrosine
residues in the cytoplasmic domain (10). Phosphorylation of EGFR evokes its
kinase activity, which subsequently activates several downstream
signaling molecules, including phospholipase C-γ, Ras,
phosphatidylinositol-3 kinase (PI-3K), Janus kinase (JAK), signal
transducers and activators of transcription (STAT)s, protein kinase
B (AKT/PKB) and mitogen-activated protein kinases (MAPK) involved
in regulation of many cellular processes, including cell
proliferation, survival, migration and apoptosis (11). The oncogenic mechanisms to drive
EGFR constitutively active are gene amplification and/or gene
mutations. Two most common EGFR-activating mutations are short
in-frame deletions in exon 19, E746-A750, and point mutation in
exon 21, L858R, which have been demonstrated to be useful
biomarkers and therapeutic targets to treat NSCLC (12). Gefitinib, a first-generation
EGFR-TKI, is a reversible inhibitor to competes with ATP to bind
its binding pocket, which prevents EGFR-induced activation of
downstream signaling (13). It has
been shown to be dramatically effective in NSCLC patients with
EGFR-activating mutations (14).
However, most patients eventually develop resistance (15).
Celastrol a pentacyclic triterpenoid
(10-Hydroxy-2,4a,6a,9,12b,14a-hexamethyl-11-oxo-1,2,3,4,4a,5,6,6a,11,12b,
13,14,14a,14b-tetradecahydro-picene-2-carboxylic acid), extracted
from roots of Tripterygium wilfordii Hook.f. and
Celastrus regelii, L. has been reported to have antioxidant
and anti-inflammatory activities (16–18).
The anticancer effect of celastrol has also been demonstrated by
induction of apoptosis and inhibition of cell growth, proliferation
and metastasis (19–22).
In this study, we first examined the effect of
celastrol on Axl protein level and then if Axl inhibition by
celastrol could reverse gefitinib sensitivity of EGFR mutant NSCLC
cells with the acquired resistance. We observed the effect of
celastrol, gefitinib, or both drugs on Axl protein level, cell
proliferation, and migration in gefitinib-resistant cells.
Materials and methods
Reagents and antibodies
Celastrol was obtained from Sigma-Aldrich (Merck
KGaA, Darmstadt, Germany). PC-9 cells were purchased from the
American Type Culture Collection (Manassas, VA, USA). Primers for
Axl were synthesized by the domestic company, Bioneer Corp.
(Daejeon, Korea). TRI reagent was obtained from Solgent Co., Ltd.
(Daejeon, Korea). AmpliTaq DNA polymerase and Lipofectamine 2000
were obtained from Roche Diagnostics Corp. (Indianapolis, IN, USA)
and Invitrogen (Thermo Fisher Scientific, Inc., Waltham, MA, USA),
respectively. For Western blot analysis, specific antibodies
against Axl and GAPDH, as well as secondary antibodies were
obtained from Santa Cruz Biotechnology, Inc., (Dallas, TX,
USA).
Cell culture and establishment of
gefitinib-resistant cells
The subline of PC-9 cells which are resistant to
gefitinib (PC-9/GR) were established by stepwise exposure of the
parental cells to escalating concentrations of gefitinib (ranging
from 0.5 to 10 µM). Both PC-9 and PC-9/GR cells were grown in
RPMI-1640 (Gibco BRL) containing 10% FBS, 2 mM L-glutamine, 10 U/ml
penicillin and 10 g/ml streptomycin at 37°C in 5% CO2 in a
water-saturated atmosphere.
Cell viability measurement
To measure cell viability, the number of viable
cells was counted using Trypan blue. Briefly, 3×103 cells were
seeded into 60-mm culture dish, grown overnight and then treated
with the indicated concentrations of gefitinib alone, celastrol
alone, or the two drugs in combination for 24 h. After treatment,
cells were harvested and stained with 0.4% Trypan blue solution.
Dye-excluding viable cells were counted under the microscope. Cell
viability was also expressed as a percentage of the viable cells
with respect to untreated control cells.
Colony formation assay
PC-9 or PC-9/GR cells were seeded into 24 well
plates (1×102 cells/well) and treated with the indicated
concentrations of gefitinib alone, celastrol alone, or the two
drugs in combination. Cells were then cultured for the next 7 to 10
days to form colonies. Colonies of >50 cells were stained with
crystal violet (in 60% methanol; Junsei Chemical Co., Tokyo, Japan)
and images were acquired using the RAS-3000 Image Analysis System
(FujiFilm, Tokyo, Japan).
Western blot analysis
Total cell lysates from PC-9 and PC-9/GR treated
with the indicated concentrations of celastrol alone, gefitinib
alone or the two drugs in combination were prepared using lysis
buffer [1% Triton X-100, 50 mM Tris (pH 8.0), 150 mM NaCl, 1 mM
PMSF, 1 mM Na3VO4, and protease inhibitor cocktail]. Untreated
cells were used as controls. Protein concentrations were determined
using Bio-Rad protein assays. Proteins from the cell lysates were
separated by 10% SDS-PAGE, and electrotransferred onto
nitrocellulose membranes. The membranes were blocked for 30 min at
room temperature in Tris-buffered saline with 0.05% Tween-20 (TTBS)
containing 5% non-fat dry milk, and then incubated with TTBS
containing a primary antibody for 4 h at room temperature. After
3×10 min washes in TTBS, the membranes were incubated with
peroxidase-conjugated secondary antibody for 1 h. Following 3
additional 10-min washes with TTBS, the protein bands of interest
were visualized using an enhanced chemiluminescence detection
system (Amersham™ ECL™ Prime Western Blotting Detection Reagent; GE
Healthcare, Piscataway, NJ, USA).
Reverse transcription-polymerase chain
reaction (RT-PCR)
Using TRI reagent, total RNAs from PC-9 and PC-9/GR
cells were extracted and subjected to cDNA synthesis and PCR. The
specific primers were as follows: Axl sense,
5′-AACCTTCAACTCCTGCCTTCTCG-3′ and antisense,
5′-CAGCTTCTCCTTCAGCTCTTCAC-3′; GAPDH sense,
5′-GGAGCCAAAAGGGTCATCAT-3′ and antisense,
5′-GTGATGGCATGGACTGTGGT-3′. The mRNA level of Axl was normalized to
that of GAPDH.
Flow cytometry
To determine Axl protein level present in plasma
membrane, FACS analysis was performed. Cells (1×106) were stained
with 5 ug/ml anti-Axl PE-conjugated antibody for 15 min and
subjected to flow cytometry using a Becton-Dickinson FACS Caliber
and analyzed by Cell Quest software (Becton-Dickinson, San Jose,
CA, USA).
In vitro migration assay
Cells (2-4×104 cells/well) were seeded into collagen
I-coated 96-well plates (Essen BioScience, Ann Arbor, MI, USA) and
grown overnight to form monolayer. According to the manufacturer's
instructions, scratch wound was made onto monolayer and the
detached cells were removed by washing with PBS. Cells were then
treated with the indicated concentrations of gefitinib alone,
celastrol alone, or the two drugs in combination for 48 h and the
wound width was measured every 4 h using IncuCyte software (Essen
BioScience, Ann Arbor, MI, USA).
Statistical analysis
Data were expressed as the means ± SD of triplicate
samples or at least three independent experiments. To determine
statistical significance, Student's t-test or one-way analysis of
variance followed by Dunnett's multiple comparisons test was
performed using GraphPad Prism version 8.0.0 (GraphPad Software,
San Diego, California USA) with a P-value threshold of
<0.05.
Results
Axl expression is upregulated in
gefitinib-resistant cells
Majority of patients treated with gefitinib, an
epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor
(TKI), develop resistance (15,23).
To investigate the characteristics and underlying mechanisms of the
acquired resistance of gefitinib, we first established
gefitinib-resistant cells, PC-9/GR, by treating parental PC-9 cells
with increasing concentrations of gefitinib in stepwise manner.
PC-9/GR cells were found to have no prominent
changes in size and shape compared to the parental PC-9 cells,
indicating that in morphological features, PC-9/GR cells are not
quite different from parental cells (data not shown). The
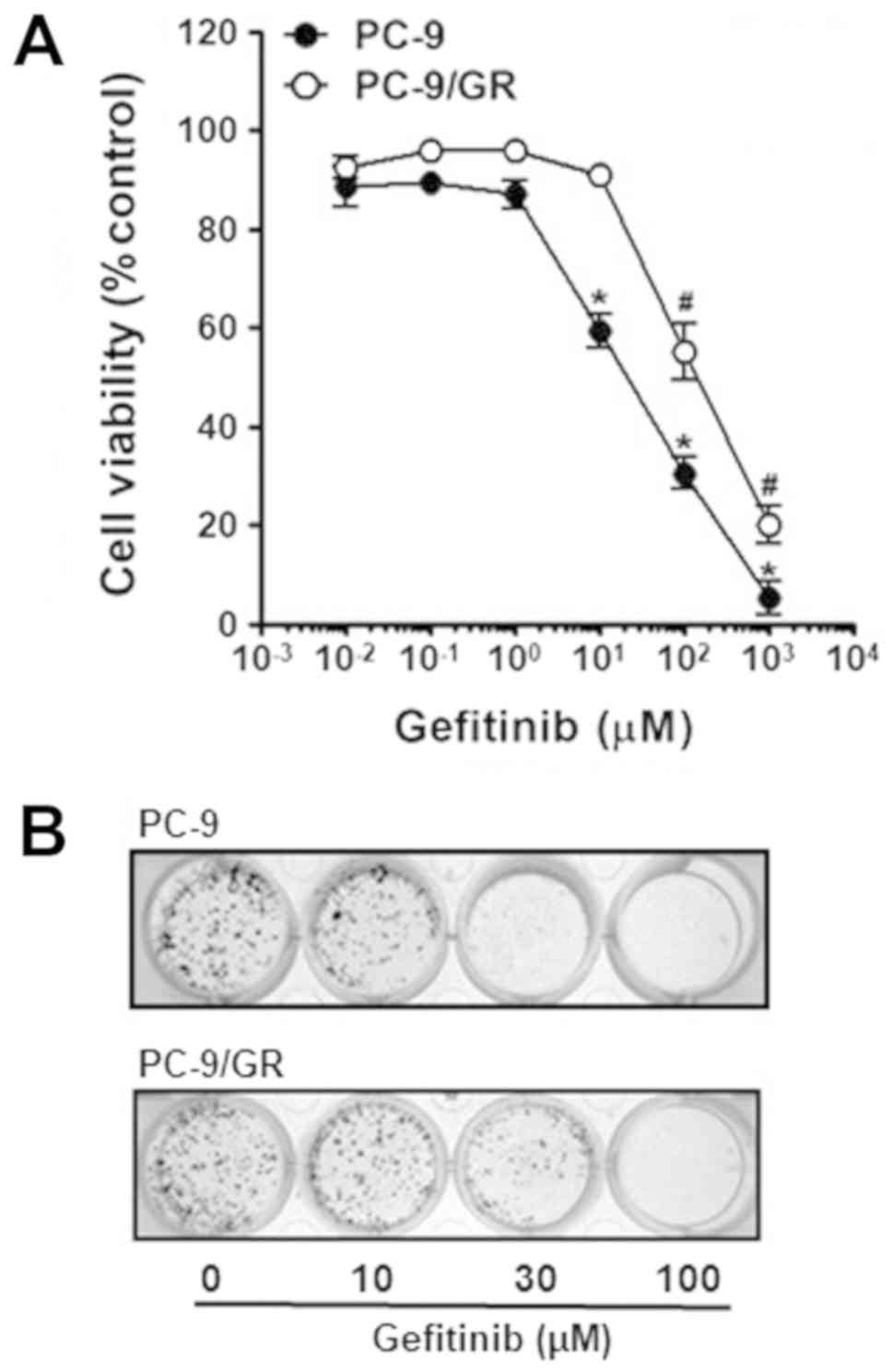
sensitivities to gefitinib of these cells were assessed by the
comparison of the viability of PC-9 and PC-9/GR cells under
gefitinib treatment ranging from 0.01 to 1,000 µM. Cell
proliferation of both PC-9 and PC-9/GR cells was decreased in
dose-dependent manner, but upon higher than 10 M gefitinib, PC-9
cells were far more sensitive to gefitinib than PC-9/GR cells,
indicating that PC-9/GR cells had acquired chemoresistance to
gefitinib (Fig. 1A).
Colony formation assay was further confirmed the
acquired gefitinib resistance of PC-9/GR cells. Both PC-9 and
PC-9/GR cells were treated with the indicated concentrations of
gefitinib and then grown for next 7 days. As shown in Fig. 1B, gefitinib treatment decreased the
clonogenicity of these cells in a dose-dependent manner. Of note,
parental PC-9 was found to form significantly less number of
colonies, in contrast to PC-9/GR cells in response to gefitinib
treatment. Taken together, these results indicate PC-9/GR cells
with gefitinib resistance was established.
Since overexpression and activation of Axl, a
receptor tyrosine kinase (RTK), have been reported to be associated
with resistance to various TKIs (24–27),
we next examined if the acquisition of gefitinib resistance has an
effect on the expression of Axl in PC-9/GR cells. Both protein and
mRNA levels of Axl in PC-9 and PC-9/GR cells were examined. As
shown in Fig. 1C, Western blot
analysis revealed that in PC-9/GR cells, Axl protein level was
significantly increased compared to parental PC-9 cells.
Up-regulation of Axl expression in PC-9/GR cell was further
confirmed by RT-PCR. Axl mRNA level in PC-9/GR cells was found to
be much higher than that in PC-9 cells, which is consistent with
Western blot analysis result.
Additionally, FACS analysis was performed to
determine Axl protein level present in plasma membrane, since Axl
is a cell surface receptor tyrosine kinase with single
transmembrane domain. We found that membrane-bound Axl protein
level in PC-9/GR cells was higher than that of parental PC-9 cells
(Fig. 1D). All these results
indicate that Axl expression is increased in gefitinib-resistant
PC-9/GR cells.
Celastrol reduces Axl expression and
proliferation in both parental and gefitinib-resistant cells
There have been several molecular targets of
celastrol to regulate various cellular responses such as cell
cycle, angiogenesis, and inflammation (19), but its effect on Axl expression was
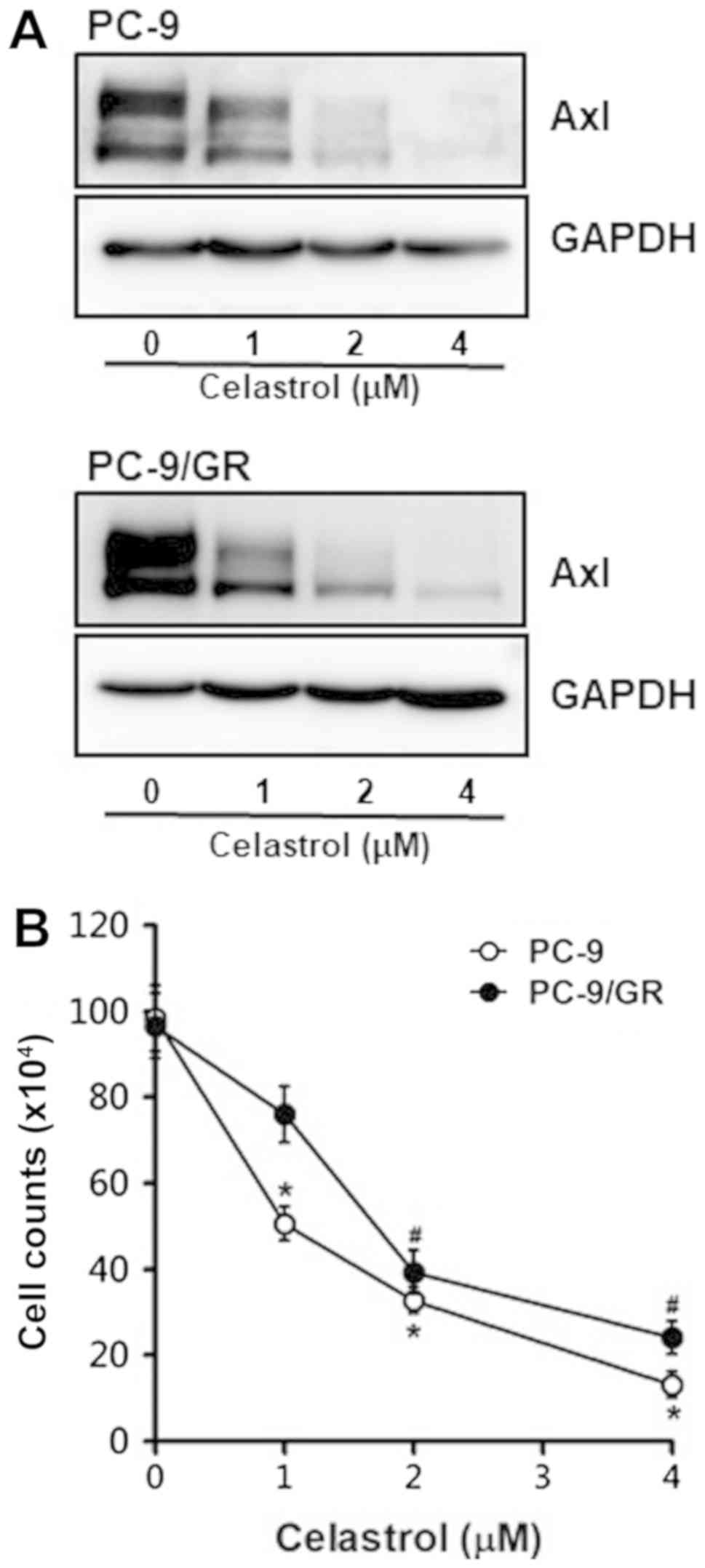
not examined yet. We first observed Axl protein level in PC-9 and
PC-9/GR cells treated with celastrol. As shown in Fig. 2A, Axl protein levels in these cells
treated with celastrol was significantly and dose-dependently
reduced, indicating that Axl is a novel target of celastrol.
In consistent with the Western blot result shown the
inhibitory effect of celastrol on Axl expression, celastrol
treatment of these cells was found to result in the dose-dependent
reduction of cell viability (Fig.
2B). Of note, survival rate of PC-9 and PC-9/GR cells treated
with 4 µM celastrol were found to be only 13 and 24%,
respectively.
The anti-proliferative effect of celastrol was
further confirmed by colony formation assay. Treatment of cells
with celastrol suppressed the clonogenicity in dose-dependent
manner (Fig. 2C). Interestingly,
PC-9/GR cells exposed to 4 µM celastrol formed more colonies
compared to parental PC-9 cells, which seemed to be resulted from
the elevated Axl level of PC-9/GR cells.
Celastrol and gefitinib cooperatively
diminish Axl protein level, cell proliferation, and cell
migration
Since Axl protein level is pretty elevated in
PC-9/GR cells, we investigated if Axl inhibition by celastrol could
reverse gefitinib sensitivity of PC-9/GR cells. To do this end, we
observed the effect of celastrol and gefitinib in combination on
Axl protein level, cell viability and cell migration.
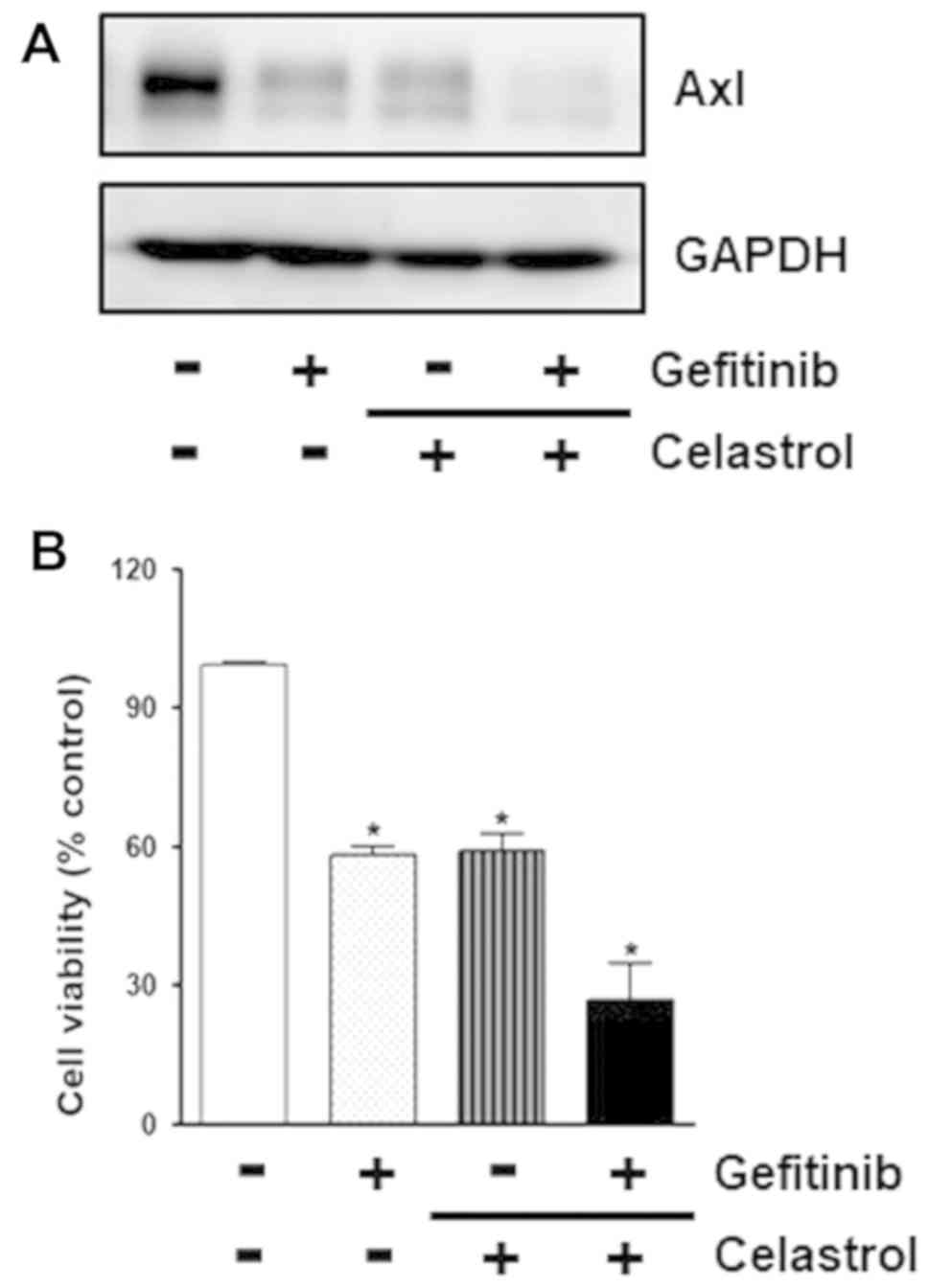
As shown in Fig.
3A, cotreatment of PC-9/GR cells with celastrol and gefitinib
synergistically decreased Axl protein level compared to treatment
of the cells with celastrol or gefitinib alone, indicating that
celastrol augments gefitinib activity to reduce Axl expression.
Cell viability was also found to be additionally attenuated by
treatment of cells with celastrol and gefitinib in combination
compared to treatments of cells with celastrol or gefitinib alone
(Fig. 3B). This combinatorial
effect of celastrol and gefitinib to inhibit cell proliferation was
further confirmed by colony formation assay. As shown in Fig. 3C, PC-9/GR cells exposed to
celastrol and gefitinib formed less colonies than cells treated
with single drug. Taken together, these results indicate that
celastrol might sensitize cytotoxicity of gefitinib to overcome
gefitinib resistance of PC-9/GR cells via Axl targeting.
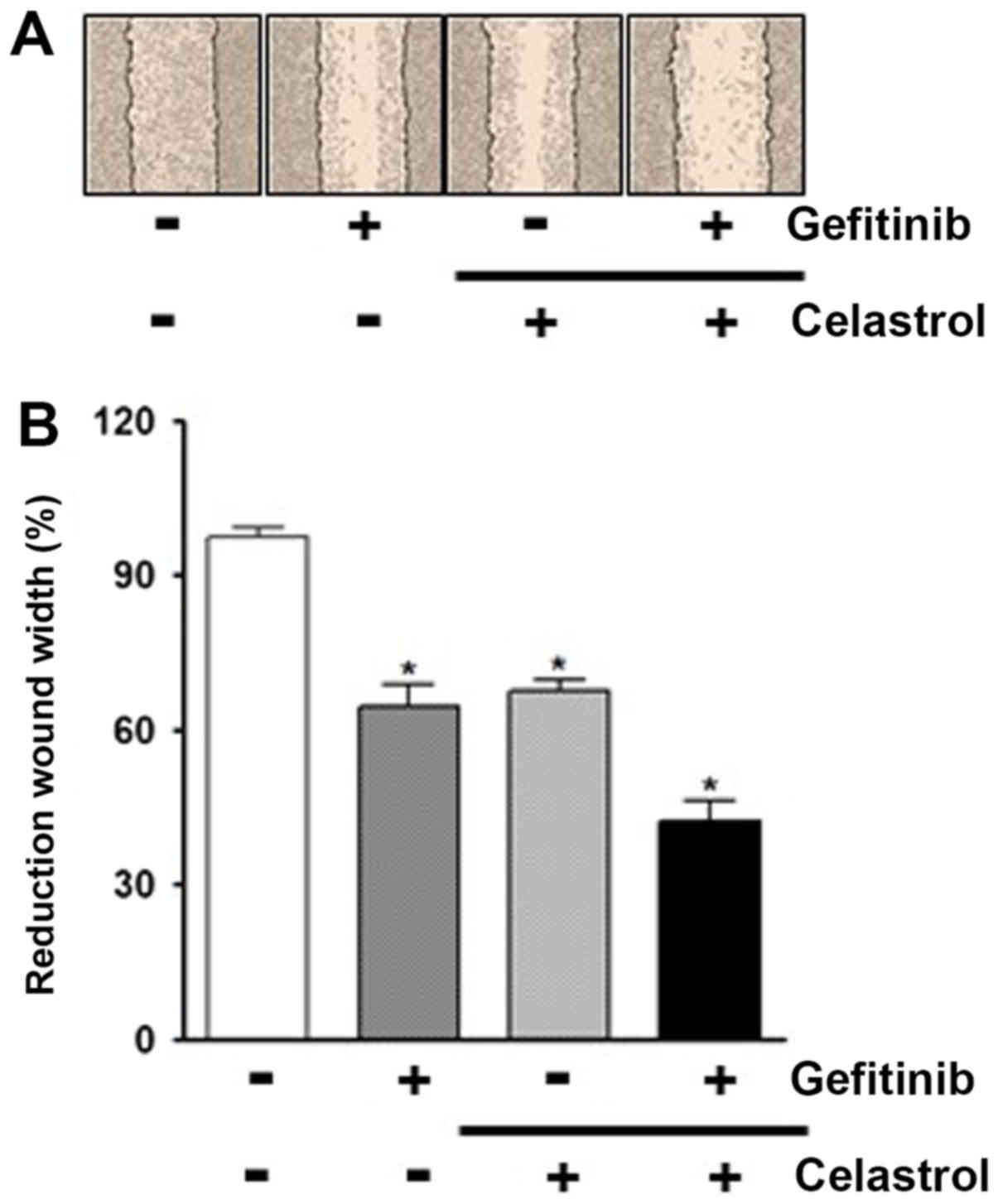
We then examined the effect of combination of
celastrol and gefitinib on cell migration via wound healing assays.
Compared to treatments of PC-9/GR cells with celastrol or gefitinib
alone, the combined treatment of cells with celastrol and gefitinib
was found to result in much less reduction in wound surface area
over time (Fig. 4A). Of note, the
percentage reduction in wound area was 67.6% (celastrol alone),
64.6% (gefitinib alone), and 42.3% (cotreatment of two drugs),
respectively (Fig. 4B). Taken
together, these results indicate that celastrol seems to increase
the susceptibility of PC-9/GR cells to gefitinib via Axl
targeting.
Discussion
Non-small cell lung cancer (NSCLC) patients with
activating mutation in the epidermal growth factor receptor (EGFR)
gene are a much more sensitive to EGFR tyrosine kinase inhibitors
(TKIs) compared to patients with wild-type EGFR (6). The first-generation EGFR-TKIs,
gefitinib and erlotinib, has been demonstrated to be highly
effective and can even be prescribed as first-line therapy of NSCLC
harboring activating EGFR mutation. However, the resistance to
these TKIs ultimately emerged in majority of patients. Thus,
identification of secondary mutations and understanding of the
molecular mechanisms associated with the acquired resistance are
urgent and essential to overcome relapsed NSCLCs.
In this study, PC-9 cells with targetable driver
mutation that is activating in-frame deletion in exon 19 of EGFR
were exposed to increasing concentrations of gefitinib to establish
gefitinib-resistant, PC-9/GR, cells. As shown in Fig. 1C and D, Axl protein and mRNA levels
in PC-9/GR cells were found to be significantly increased,
indicating that long-term exposure of PC-9 cells resulted in
outgrowth of gefitinib-resistant cells by up-regulation of Axl
expression. Axl is a member of Tyro/Axl/Mer (TAM) family of
receptor tyrosine kinases (RTKs) (28). Activation of Axl has been known to
be required for cell growth, migration, proliferation, and
inhibition of apoptosis in many cancers (26,29–31).
In consistant with our results, the induction of Axl expression has
already been demonstrated in gefitinib- or erlotinib-resistant
EGFR-mutant NSCLC cells or NSCLC tumor xenografts, respectively
(25,32,33).
In addition, a number of studies reported that the acquisition of
resistance to other TKIs, including imatinib and nilotinib is
correlated with abnormal activation and/or overexpression of TAM
RTKs in various cancers such as chronic myeloid leukemia cells
(26,34). Therefore, the inhibition of Axl or
other members of TAM RTKs which were aberrantly regulated in
TKI-resistant cells might recover susceptibility to TKIs, which
subsequently overcome the acquired resistance.
Celastrol was found to inhibit proliferation of both
parental cells, PC-9, and gefitinib-resistant cells, PC-9/GR
(Fig. 2A and B). In consistent
with our data, the anticancer effect of celastrol was observed in
many cancers, including breast, cervical, colon, gastric, prostate
cancers (35–39). Furthermore, chemosensitizing effect
of celastrol have also been revealed in cisplatin- and
gefitinib-resistant cells (19,40–42),
since PC-9/GR cells treated with celastrol and gefitinib in
combination synergistically decreased Axl protein level, cell
proliferation, and cell migration (Figs. 3 and 4). The cooperative efficacy of combined
therapy with celastrol and several anticancer agents has been
reported in doxorubicin-resistant colon cancer cells,
cisplatin/gefitinib-resistant NSCLC cells via induction of
apoptosis, inhibition of JNK/ATF2 pathway, and autophagic
degradation of EGFR, respectively (40,42).
Interestingly, Wang et al (41) reported that the combined treatment
with celastrol and EGFR-TKIs such as gefitinib and erlotinib was
found to be more effective to inhibit invasion of NSCLC cells with
an EGFR T790M mutation, while EGFR-TKIs alone had little effect on
it. Increasing evidence indicates that celastrol might be a potent
candidate to augment the sensitivity of conventional
chemotherapeutic agents in numerous cancers.
In summary, our data indicate that celastrol
downregulates Axl expression in parental and gefitinib-resistant
NSCLC cells. Thus, Axl seems to be a novel target of celastrol to
inhibit cell proliferation and to increase the susceptibility of
gefitnib-resistance cells to gefitinib, which lead to overcome
chemoresistance.
Acknowledgements
Not applicable.
Funding
The present study was supported by the 2015 Yeungnam
University Research Grant (grant no. 215A480013).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YJL, SYK and CL were equally responsible for the
establishment of the conception and design of study, and the
acquisition, analysis and interpretation of data. YJL, SYK and CL
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ettinger DS, Akerley W, Borghaei H, Chang
AC, Cheney RT, Chirieac LR, D'Amico TA, Demmy TL, Govindan R,
Grannis FW Jr, et al: Non-small cell lung cancer, version 2.2013. J
Natl Compr Canc Netw. 11:645–653; quiz 653. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hamilton G and Rath B: Pharmacogenetics of
platinum-based chemotherapy in non-small cell lung cancer:
Predictive validity of polymorphisms of ERCC1. Expert Opin Drug
Metab Toxicol. 14:17–24. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Forde PM and Ettinger DS: Targeted therapy
for non-small-cell lung cancer: Past, present and future. Expert
Rev Anticancer Ther. 13:745–758. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cabanero M, Sangha R, Sheffield BS, Sukhai
M, Pakkal M, Kamel-Reid S, Karsan A, Ionescu D, Juergens RA, Butts
C and Tsao MS: Management of EGFR-mutated non-small-cell
lung cancer: Practical implications from a clinical and pathology
perspective. Curr Oncol. 24:111–119. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
González-Larriba JL, Lázaro-Quintela M,
Cobo M, Domine M, Majem M and García-Campelo R: Clinical management
of epidermal growth factor receptor mutation-positive non-small
cell lung cancer patients after progression on previous epidermal
growth factor receptor tyrosine kinase inhibitors: The necessity of
repeated molecular analysis. Transl Lung Cancer Res. 6 (Suppl
1):S21–S34. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wada T, Qian XL and Greene MI:
Intermolecular association of the p185neu protein and EGF receptor
modulates EGF receptor function. Cell. 61:1339–1347. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Carraway KL III and Cantley LC: A neu
acquaintance for erbB3 and erbB4: A role for receptor
heterodimerization in growth signaling. Cell. 78:5–8. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yarden Y: The EGFR family and its ligands
in human cancer. Signaling mechanisms and therapeutic
opportunities. Eur J Cancer. 37 (Suppl 4):S3–S8. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang H, Berezov A, Wang Q, Zhang G,
Drebin J, Murali R and Greene MI: ErbB receptors: From oncogenes to
targeted cancer therapies. J Clin Invest. 117:2051–2058. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Seshacharyulu P, Ponnusamy MP, Haridas D,
Jain M, Ganti AK and Batra SK: Targeting the EGFR signaling pathway
in cancer therapy. Expert Opin Ther Targets. 16:15–31. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Arteaga C: Targeting HER1/EGFR: A
molecular approach to cancer therapy. Semin Oncol 30 (3 Suppl 7).
S3–S14. 2003.
|
|
13
|
Pao W, Miller V, Zakowski M, Doherty J,
Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, et al:
EGF receptor gene mutations are common in lung cancers from ‘never
smokers’ and are associated with sensitivity of tumors to gefitinib
and erlotinib. Proc Natl Acad Sci USA. 101:13306–13311. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Araki T, Yashima H, Shimizu K, Aomori T,
Hashita T, Kaira K, Nakamura T and Yamamoto K: Review of the
treatment of non-small cell lung cancer with gefitinib. Clin Med
Insights Oncol. 6:407–421. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu Y, Liu H, Chen J and Zhou Q: Acquired
resistance of lung adenocarcinoma to EGFR-tyrosine kinase
inhibitors gefitinib and erlotinib. Cancer Biol Ther. 9:572–582.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Guan Y, Cui ZJ, Sun B, Han LP, Li CJ and
Chen LM: Celastrol attenuates oxidative stress in the skeletal
muscle of diabetic rats by regulating the AMPK-PGC1α-SIRT3
signaling pathway. Int J Mol Med. 37:1229–1238. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ju SM, Youn GS, Cho YS, Choi SY and Park
J: Celastrol ameliorates cytokine toxicity and pro-inflammatory
immune responses by suppressing NF-κB activation in RINm5F beta
cells. BMB Rep. 48:172–177. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li X, Wu N, Zou L and Jia D: Protective
effect of celastrol on myocardial ischemia-reperfusion injury.
Anatol J Cardiol. 18:384–390. 2017.PubMed/NCBI
|
|
19
|
Kashyap D, Sharma A, Tuli HS, Sak K,
Mukherjee T and Bishayee A: Molecular targets of celastrol in
cancer: Recent trends and advancements. Crit Rev Oncol Hematol.
128:70–81. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mi C, Shi H, Ma J, Han LZ, Lee JJ and Jin
X: Celastrol induces the apoptosis of breast cancer cells and
inhibits their invasion via downregulation of MMP-9. Oncol Rep.
32:2527–2532. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rajendran P, Li F, Shanmugam MK, Kannaiyan
R, Goh JN, Wong KF, Wang W, Khin E, Tergaonkar V, Kumar AP, et al:
Celastrol suppresses growth and induces apoptosis of human
hepatocellular carcinoma through the modulation of STAT3/JAK2
signaling cascade in vitro and in vivo. Cancer Prev Res (Phila).
5:631–643. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Abbas S, Bhoumik A, Dahl R, Vasile S,
Krajewski S, Cosford ND and Ronai ZA: Preclinical studies of
celastrol and acetyl isogambogic acid in melanoma. Clin Cancer Res.
13:6769–6778. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yano S, Yamaguchi M and Dong RP: EGFR
tyrosine kinase inhibitor ‘gefitinib (Iressa)’ for cancer therapy.
Nihon Yakurigaku Zasshi. 122:491–497. 2003.(In Japanese).
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu F, Li J, Jang C, Wang J and Xiong J:
The role of Axl in drug resistance and epithelial-to-mesenchymal
transition of non-small cell lung carcinoma. Int J Clin Exp Pathol.
7:6653–6661. 2014.PubMed/NCBI
|
|
25
|
Bae SY, Hong JY, Lee HJ, Park HJ and Lee
SK: Targeting the degradation of AXL receptor tyrosine kinase to
overcome resistance in gefitinib-resistant non-small cell lung
cancer. Oncotarget. 6:10146–10160. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dufies M, Jacquel A, Belhacene N, Robert
G, Cluzeau T, Luciano F, Cassuto JP, Raynaud S and Auberger P:
Mechanisms of AXL overexpression and function in imatinib-resistant
chronic myeloid leukemia cells. Oncotarget. 2:874–885. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rho JK, Choi YJ, Kim SY, Kim TW, Choi EK,
Yoon SJ, Park BM, Park E, Bae JH, Choi CM and Lee JC: MET and AXL
inhibitor NPS-1034 exerts efficacy against lung cancer cells
resistant to EGFR kinase inhibitors because of MET or AXL
activation. Cancer Res. 74:253–262. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lemke G: Biology of the TAM receptors.
Cold Spring Harb Perspect Biol. 5:a0090762013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Martinelli E, Martini G, Cardone C,
Troiani T, Liguori G, Vitagliano D, Napolitano S, Morgillo F,
Rinaldi B, Melillo RM, et al: AXL is an oncotarget in human
colorectal cancer. Oncotarget. 6:23281–23296. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qu X, Liu J, Zhong X, Li X and Zhang Q:
Role of AXL expression in non-small cell lung cancer. Oncol Lett.
12:5085–5091. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rankin EB, Fuh KC, Taylor TE, Krieg AJ,
Musser M, Yuan J, Wei K, Kuo CJ, Longacre TA and Giaccia AJ: AXL is
an essential factor and therapeutic target for metastatic ovarian
cancer. Cancer Res. 70:7570–7579. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Giles KM, Kalinowski FC, Candy PA, Epis
MR, Zhang PM, Redfern AD, Stuart LM, Goodall GJ and Leedman PJ: Axl
mediates acquired resistance of head and neck cancer cells to the
epidermal growth factor receptor inhibitor erlotinib. Mol Cancer
Ther. 12:2541–2558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang Y, Xia H, Zhuang Z, Miao L, Chen X
and Cai H: Axl-altered microRNAs regulate tumorigenicity and
gefitinib resistance in lung cancer. Cell Death Dis. 5:e12272014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gioia R, Tregoat C, Dumas PY, Lagarde V,
Prouzet-Mauléon V, Desplat V, Sirvent A, Praloran V, Lippert E,
Villacreces A, et al: CBL controls a tyrosine kinase network
involving AXL, SYK and LYN in nilotinib-resistant chronic myeloid
leukaemia. J Pathol. 237:14–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tang WJ, Wang J, Tong X, Shi JB, Liu XH
and Li J: Design and synthesis of celastrol derivatives as
anticancer agents. Eur J Med Chem. 95:166–173. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kuchta K, Xiang Y, Huang S, Tang Y, Peng
X, Wang X, Zhu Y, Li J, Xu J, Lin Z and Pan T: Celastrol, an active
constituent of the TCM plant Tripterygium wilfordii Hook.f.,
inhibits prostate cancer bone metastasis. Prostate Cancer Prostatic
Dis. 20:156–164. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bufu T, Di X, Yilin Z, Gege L, Xi C and
Ling W: Celastrol inhibits colorectal cancer cell proliferation and
migration through suppression of MMP3 and MMP7 by the PI3K/AKT
signaling pathway. Anticancer Drugs. 29:530–538. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee HW, Jang KS, Choi HJ, Jo A, Cheong JH
and Chun KH: Celastrol inhibits gastric cancer growth by induction
of apoptosis and autophagy. BMB Rep. 47:697–702. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhou Y, Li W, Wang M, Zhang H, Tong X and
Xiao Y: Competitive profiling of celastrol targets in human
cervical cancer HeLa cells via quantitative chemical proteomics.
Mol Biosyst. 13:83–91. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xu SW, Law BY, Mok SW, Leung EL, Fan XX,
Coghi PS, Zeng W, Leung CH, Ma DL, Liu L and Wong VK: Autophagic
degradation of epidermal growth factor receptor in
gefitinib-resistant lung cancer by celastrol. Int J Oncol.
49:1576–1588. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang Y, Liu Q, Chen H, You J, Peng B, Cao
F, Zhang X, Chen Q, Uzan G, Xu L and Zhang D: Celastrol improves
the therapeutic efficacy of EGFR-TKIs for non-small-cell lung
cancer by overcoming EGFR T790M drug resistance. Anticancer Drugs.
29:748–755. 2018.PubMed/NCBI
|
|
42
|
Lo Iacono M, Monica V, Vavalà T, Gisabella
M, Saviozzi S, Bracco E, Novello S, Papotti M and Scagliotti GV:
ATF2 contributes to cisplatin resistance in non-small cell lung
cancer and celastrol induces cisplatin resensitization through
inhibition of JNK/ATF2 pathway. Int J Cancer. 136:2598–2609. 2015.
View Article : Google Scholar : PubMed/NCBI
|