Introduction
Cerebral stroke remains the most common cause of
mortality and a major cause of neurological disability, resulting
in notable societal burden worldwide (1). Ischemia restricts the blood supply to
certain regions of the brain, resulting in acute glucose and oxygen
deprivation (2). Accumulating
evidence demonstrated that ischemia/reperfusion (I/R) stress causes
oxidative damage and cell apoptosis (3,4).
Increasing efforts have been made to determine the mechanism
underlying I/R-associated cell stress and injury (5,6);
however, whether I/R injury dysregulates the metabolic profile
remains unknown.
In traditional Chinese medicine, safflower
(Carthamus tinctorius) has been widely used for the
treatment of cerebrovascular and cardiovascular diseases (7). The pigments of the Carthamus
tinctorius extract contain hydroxysafflor yellow A (HSYA),
safflor yellow B, safflomin A, safflomin C and other components
(8). As the most important and
representative component of safflower, HSYA has a notable
therapeutic and protective effects against cerebral and myocardial
I/R injury (9). HSYA can inhibit
cell apoptosis and reduce the levels of reactive oxygen species
(ROS), which helps to rescue damaged neurons and promote cell
survival (10). Thus, HSYA is
widely used for the neuroprotection for cerebral stroke and I/R in
clinic (11).
The ‘Warburg effect’ has been widely accepted as a
common feature of cancer cells that occurs via metabolic
reprogramming (12). Of note,
metabolic reprogramming is not only limited to cancer cells but has
been reported in disorders of the brain, gut and lungs associated
with I/R stress (13–15). In addition, cerebral infarction
also caused metabolite alterations and reduced the levels of
branched-chain amino acid in ischemic stroke (16). It has been demonstrated that HSYA
can activate cell survival signaling via the PI3K/AKT pathway, as
well as other pathways (17);
however, whether HSYA protects against I/R injury via metabolic
alterations requires further investigation.
In the present study, we first identified the
metabolic amino acid profile in I/R mouse model. I/R injury notably
disturbed the metabolic flux and induced a significant increase in
levels of plasma phenylalanine. Notably, treatment with HSYA could
recover the alterations in phenylalanine levels induced by I/R
stress, and regulate the expression of key enzymes, including
phenylalanine hydroxylase (PAH), tyrosine aminotransferase (TAT)
and aspartate aminotransferase (GOT1), which are responsible for
phenylalanine transformation (18). Notably, HSYA was observed to rescue
mitochondrial function and glucose uptake ability. Additionally,
HSYA could promote mitochondria biogenesis by increasing the
expression of fission-associated dynamin-1-like protein (DRP1),
which was downregulated via oxygen and glucose
deprivation/reoxygenation (OGD/R) and phenylalanine treatment.
The findings of the present study demonstrated the
novel mechanism of I/R injury via upregulating the levels of
phenylalanine; HSYA was reported to inhibit increases in
phenylalanine levels for neuroprotection via enhancing
mitochondrial function and biogenesis. The present study proposed a
novel metabolite as a biomarker for cerebral I/R injury and the
evaluated the efficacy of HSYA treatment.
Materials and methods
Mouse model of ischemia and
reperfusion
Male C57BL/6 mice (age, 6–8 weeks; weight, 18–20 g)
were obtained from the animal center of The Fourth Military Medical
University, and housed with a 12-h light/dark cycle at 18–23°C,
with a humidity of 40–60% and ad libitum access to food and
water. A total of 24 mice were used to generate a model of cerebral
I/R. The mice were divided into four groups: Sham operation and
I/R, and I/R model treatment groups, which were treated with HSYA
(5 or 20 mg/kg). Transient focal cerebral ischemia was induced by
right middle cerebral artery occlusion (MCAO) as described
(19). After 2 h, the monofilament
was withdrawn to establish the I/R model. Then, two I/R groups of
mice were treated with 5 or 20 mg/kg/day HSYA (intraperitoneal
injection) for 3 days. Following treatment and analysis of
neurological behavior, 50 µl plasma samples were extracted from
mouse tail veins, and the mice were subsequently sacrificed. All
experiments were approved by The Animal Care Committees of The
Fourth Military Medical University (Xi'an, China).
Neurological behavior deficit
evaluation
Modified scoring systems for neurological deficits
were used to evaluate the neuroprotection effect of HSYA for 72 h
with I/R injury. With slight modifications, a 0–4 point scale was
used to evaluate the neurological deficits observed in all groups
(20). The scoring system used was
the following: 0, no deficit; 1, forelimb flexion; 2, forelimb
flexion and decreased resistance to lateral push; 3, unidirectional
circling; 4, longitudinal spinning, seizure or no movement.
Measurement of infarct zone in the
brain
The brain tissues were collected, resected and cut
into 2 mm thick slices using a brain matrix, and treated with 2, 3,
5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) at 37°C for 30 min and then fixed in 10%
phosphate-buffered formalin for 45 min at room temperature. The
tissues were imaged with a light microscope (Olympus CX31; Olympus
Corporation, Tokyo, Japan; magnification, 4×). The infarction zone
was calculated as the percentage of infarction area compared with
the ipsilateral hemisphere. The infarct zone was analyzed in five
fields of view using MPLAS-500 multimedia color pathological
graphic analysis system (Wuhan Qingping Imaging Technology Co.,
Ltd., Wuhan, China) (21).
Metabolic profiling and
quantification
Samples were analyzed using liquid
chromatography-tandem mass spectroscopy (MS/MS). A liquid
chromatography system (Shimadzu Corporation, Kyoto, Japan) was used
to flow the sample into the MS/MS system. A mixture of acetonitrile
and water (70:30, v/v) was used as the carrier solution at a flow
rate of 0.14 ml/min. In total, 18 µl of the sample was added to the
carrier solution; subsequently, the liquid was delivered to the ion
source of the MS/MS system without column separation. The MS/MS
analysis was conducted with an API 4000+ tandem mass spectrometer
(AB Sciex LLC, Framingham, MA, USA) in positive ionization mode and
electrospray ionization was performed with nitrogen gas at 400°C
and the nebulizer pressure was 30 psi. In total, two alternating
scan modes were defined in the MS setup. Neutral loss scan of m/z
102 was used for neutral amino acids (25V, 13–17 eV). All data
acquisition and processing were performed with Analyst software
(version 1.5.2; AB Sciex LLC).
Primary culture of mouse cortex
neurons
Pregnant female C57BL/6 mice (age, 8–10 weeks;
weight, 18–20 g) were obtained from the animal center of The Fourth
Military Medical University (Xi'an, China), housed with a 12-h
light/dark cycle at 18–23°C, with a humidity of 40–60% and ad
libitum access to food and water. The mice were sacrificed via
50–70% CO2 inhalation with a fill rate of CO2
displacement ~20% of the chamber volume/min. Cerebral cortical
neurons from the cerebrum of mice at embryonic age 18.5 (E18.5)
were prepared. The cerebral cortex of the brains was dissected out
in cold PBS and the tissues were cut into small pieces (1
mm3) and digested with 0.25% trypsin + 0.04% EDTA at
37°C for 15 min. The cell suspension was centrifuged at 500 × g on
4°C for 5 min, and the cell pellet was resuspended in Neurobasal +
B27 (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Following harvest, cells were incubated for 7 days and were
subsequently exposed to OGD for 120 min and treated with or without
HSYA at a concentration of 1 or 10 µM for 20 h at 37°C.
OGD/R in primary mouse neurons
OGD was induced as previously described (22). The cells were cultured with
glucose-free Dulbecco's modification of Eagle medium (DMEM; Thermo
Fisher Scientific, Inc.) under 5% CO2 and 95%
N2 at 37°C for 120 min. Then, the culture medium was
replaced with DMEM, and the sample was incubated in regular
conditions, with 5% CO2 at 37°C for 20 h. In all
experiments, the pH of the medium was maintained at 7.2 under OGD
conditions.
OGD/R in PC12 cells
Similar to the primary mouse neuronal cells, PC12
cells (American Type Culture Collection, Manassas, VA, USA) were
cultured with glucose-free DMEM and infused with 5% CO2
and 95% N2 at 37°C for 12 h. Then, the culture medium
was replaced with DMEM, and the sample was returned to the regular
conditions of 5% CO2 and 95% air at 37°C for 24 h. In
all experiments, the pH of the medium was maintained at 7.2 under
OGD conditions.
Treatment with phenylalanine in PC12
cells
PC12 cells were treated with various concentrations
of phenylalanine (0, 0.5, 1, 2, 4, 8, 16 and 32 mM; Sigma-Aldrich;
Merck KGaA) for 48 or 96 h at 37°C prior to MTT assay. The cells
were treated with 8 mM phenylalanine for 48 h in JC-1 prior to
western blotting analysis.
Western blot analysis
Radioimmunoprecipitation assay protein extraction
buffer was purchased from The Beyotime Institute of Biotechnology
(Haimen, China). The total protein was extracted from mouse brain
tissues or cultured cells and was subsequently quantified using a
bicinchoninic acid assay kit or Bradford's assay kit (Thermo Fisher
Scientific, Inc.), respectively. A total of 40 µg protein was
loaded per lane. Proteins were separated by 12% SDS-PAGE for 60–80
min. The gel was subsequently transferred on nitrocellulose
membranes for 90 min. The membrane was blocked with 5% fat-free
milk for 1 h at room temperature. Antibodies against phosphorylated
protein kinase B (p-Akt; 1:1,000; cat. no. 4058; Cell Signaling
Technology, Inc., Danvers, MA, USA), anti-Akt (1:1,000; cat. no.
9272; Cell Signaling Technology, Inc.), anti-cleaved caspase-3 (of
c-Casp3; 1:1,000; cat. no. 9664; Cell Signaling Technology, Inc.),
anti-β-catenin (1:1,000; cat. no. 8480; Cell Signaling Technology,
Inc.), anti-B-cell lymphoma 2 (BCL2; 1:1,000; cat. no. 2870; Cell
Signaling Technology, anti-mitochondrial dynamin like GTPase (OPA1;
1:1,000; cat. no. 67589; Cell Signaling Technology, Inc.),
anti-voltage-dependent anion channel (VDAC; 1:1,000; cat. no. 4661;
Cell Signaling Technology, Inc.), anti-mitofusin 2 (MFN2; 1:1,000;
cat. no. 11925; Cell Signaling Technology, Inc.), anti-fission,
mitochondrial 1 (Fis1; 1:1,000, ab71498; Abcam, Cambridge, UK) and
anti-neuronal nuclei (NeuN; 1:500; cat. no. 26975-1-AP; ProteinTech
Group, Inc., Chicago, IL, USA) were applied overnight at 4°C.
β-actin (1:2,000; cat. no. AB8227; Abcam) served as the loading
control. Secondary antibodies were incubated for 1 h at room
temperature (1:2,000; cat. nos. 7074 and 7076; Cell Signaling
Technology, Inc.). Membranes were incubated with chemiluminescent
reagents (Pierce; Thermo Fisher Scientific, Inc.) for detecting
horseradish peroxidase-labeled antibodies at room temperature prior
to x-ray exposure. Western blot analysis was repeated three times,
and similar results were obtained. Densitometry was performed using
the Image-Pro Plus software (version 6.0; Media Cybernetics, Inc.,
Rockville, MD, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RNA was extracted using TRIzol® RNA
Isolation Reagents (Invitrogen; Thermo Fisher Scientific, Inc.) and
cDNA was generated with SuperScript™ II reverse transcriptase
according to the manufacturer's protocol (Invitrogen; Thermo Fisher
Scientific, Inc.). A total of 2–5 µg RNA was mixed with reverse
transcriptase and incubated at 42°C for 50 min to synthesize cDNA.
The mRNA levels of mouse brain tissues, primary mouse neuronal
cells or PC12 cells exposed to OGD/R were evaluated using a
SYBR-Green RT-qPCR kit (Takara Biotechnology Co., Ltd., Dalian,
China). qPCR was conducted using a CFX96 Touch™ RT PCR Detection
System (Bio-Rad Laboratories, Inc., Hercules, CA, USA) as follows:
Denaturation at 95°C for 10 sec, primer annealing at 60°C for 20
sec and elongation at 72°C for 30 sec, for 40 cycles. The mouse and
rat-specific primers designed are presented in Table I. The relative quantification of
each target gene was normalized to GAPDH using the
2−∆∆Cq quantification method (23), and the fold change between sham and
control group was calculated with three replicates in each
group.
 | Table I.Primers for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primers for reverse
transcription-quantitative polymerase chain reaction.
| Gene name | Forward
sequence | Reverse
sequence |
|---|
| Mouse
Pah |
5′-TTGTCCTGGAGAACGGAGTC-3′ |
5′-CTGGATTCAATGTGTGTCAGGTT-3′ |
| Mouse
Got1 |
5′-GCGCCTCCATCAGTCTTTG-3′ |
5′-ATTCATCTGTGCGGTACGCTC-3′ |
| Mouse
Tat |
5′-TGCTGGATGTTCGCGTCAATA-3′ |
5′-CGGCTTCACCTTCATGTTGTC-3′ |
| Mouse
GAPDH |
5′-CTTCACCACCATGGAGGAGGC-3′ |
5′-GGCATGGACTGTGGTCATGAG-3′ |
| Rat Pah |
5′-ACCCTCTAGGGGTAAATCTTTCA-3′ |
5′-GAAGCCCCAATGACACAAGC-3′ |
| Rat
Got1 |
5′-TCTGCACGTTGCTTGAGTCT-3′ |
5′-GCCGAGAGACAGAGACGATG-3′ |
| Rat Tat |
5′-AATGCGGACCTCTGCTATGG-3′ |
5′-GACAAGCCATCTCCTGTGCT-3′ |
| Rat
GAPDH |
5′-GTGAAGCTCATTTCCTGGTATG-3′ |
5′-AACTGACGGCCTCTCTCTTG-3′ |
Flow cytometry and apoptosis
analysis
Cell apoptosis was analyzed using an Annexin
V-fluorescein isothiocyanate (FITC) Apoptosis Detection kit
(eBioscience; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. PC12 or primary mouse neuronal cells at
80% confluency were harvested using 0.25% trypsin for 5 min at 37°C
and washed twice with PBS. Following centrifugation at 500 × g for
5 min at 4°C, cells were resuspended in solution containing Annexin
V-FITC and propidium iodide for 15 min at room temperature.
Subsequently, the cells were analyzed with a flow cytometer and the
FACSCanto™ Plus Software (version 3.0; Becton, Dickinson and
Company, Franklin Lakes, NJ, USA).
Dichloro-dihydro-fluorescein diacetate
(DCFH-DA) cellular ROS detection
PC12 cells at 80% confluency were seeded in 6-well
plates and stained with 2.5 µM DCFH-DA in 1X dilution buffer
provided in the kit (eBioscience; Thermo Fisher Scientific, Inc.)
for 20 min at 37°C; the cells were washed with 1X Buffer three
times. Subsequently, cells were harvested with 0.25% trypsin for 5
min at 37°C and washed with PBS twice, followed by analysis with a
flow cytometer for the detection of FITC, using the FACSCanto™ Plus
Software (version 3.0; Becton, Dickinson and Company).
JC-1 mitochondrial membrane potential
and MitoTracker red assay
PC12 cells were seeded in 6 well plates and labeled
according to the manufacturer's protocols (cat. no. ab113850,
Abcam). Cells were then cultured for 20 min at 37°C and washed with
1X dilution buffer provided in the kit (eBioscience; Thermo Fisher
Scientific, Inc.) three times. Subsequently, cells were harvested
and analyzed with a flow cytometer using the FACSCanto™ Plus
Software (version 3.0; Becton, Dickinson and Company). Fluorescence
in FL-1 channel and lacking fluorescence in FL-2 channel is
considered to indicate mitochondria with depolarized Δψ, suggesting
apoptosis or dysfunction (24). As
for the MitoTracker labelling assay, PC12 cells were incubated with
200 nM MitoTracker Red (Cell Signaling Technology, Inc.; cat. no.
9082) for 20 min at 37°C. Following incubation, the cells were
rinsed with PBS three times and incubated with regular DMEM culture
medium. Subsequently, MitoTracker Red was detected with confocal
immunofluorescence analysis using an Olympus FV1000 confocal
microscope (Olympus Corporation). In total, five fields per view
were imaged (magnification, ×400).
2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxyglucose
(2-NBDG) uptake assay
PC12 cells were seeded at 1×105/well in
6-well plate and cultured for 24 h. Then, the cell culture medium
was removed and starved for 2 h without glucose and serum. The
culture medium was replaced with serum with or without 25 µM 2-NBDG
(Sigma-Aldrich; Merck KGaA), and incubated for 40 min at 37°C.
Subsequently, the cells were washed with ice-cold PBS three times
and collected for analysis with a flow cytometer with settings for
applied for FITC detection.
MTT assay
A total of 3,000 cells/well were cultured with 100
µl DMEM medium in 96-well plate at 37°C and incubated overnight.
Various concentrations of phenylalanine (0, 0.5, 1, 2, 4, 8, 16 and
32 mM) were applied to the cells for 2 or 4 days at 37°C. Then, 20
µl of 5 mg/ml MTT was added to each well and incubated for 4 h at
37°C. The medium was removed and 150 µl dimethyl sulfoxide was
added; the absorbance at 590 nm was detected with a microplate
reader (BioTek Instruments, Inc., Winooski, VT, USA).
Statistical analysis
Data are expressed as the mean ± standard error.
Statistical analysis was performed using a Student's t-test,
one-way analysis of variance followed by a Dunnett's post hoc test.
P<0.05 was considered to indicate a statistically significant
difference. Analyses were performed using Prism (version 7.0;
GraphPad Software, Inc., La Jolla, CA, USA). The experiments were
performed three times and at least three replicates were present in
each group.
Results
Protective effects of HSYA on
neurologic behavior and infarction size with TTC staining
As presented in Fig.
1A, an experimental mouse model of cerebral I/R was generated
and the protective effect of HSYA was investigated. After 3 days
following 2 h of I/R treatment, significant increases in
neurological behavior deficits in the I/R group compared with the
sham group were observed (P<0.001). Of note, administration of 5
mg/kg HSYA notably alleviated the neurological deficits in the I/R
+ HSYA group compared with the I/R group; however, a significant
effect was observed in response to 20 mg/kg HSYA (P<0.01;
Fig. 1B). TTC staining revealed
that the infarct area of the I/R group was ~37% of that of the sham
group; however, 5 and 20 mg/kg HSYA significantly decreased the
size to 7.2 and 22.4%, respectively (P<0.05; Fig. 1C and D). The results indicated that
HSYA had an notable protective effect against cerebral I/R injury.
To further confirm the neuroprotective effect of HSYA, western
blotting was performed to detect the expression of apoptosis and
proliferation-associated proteins. The expression of the
proliferation markers p-Akt, β-catenin and BCL2 were notably
decreased and that of c-Casp3 increased following I/R treatment;
however, HSYA rescued the expression of p-Akt, β-catenin and BCL2,
and decreased that of c-Casp3 compared with the control (Fig. 1E). In particular, treatment with 20
mg/kg HSYA significantly increased the expression of neuronal cell
marker NeuN (Fig. 1E and F), which
indicated the neuroprotective effect of HSYA against I/R.
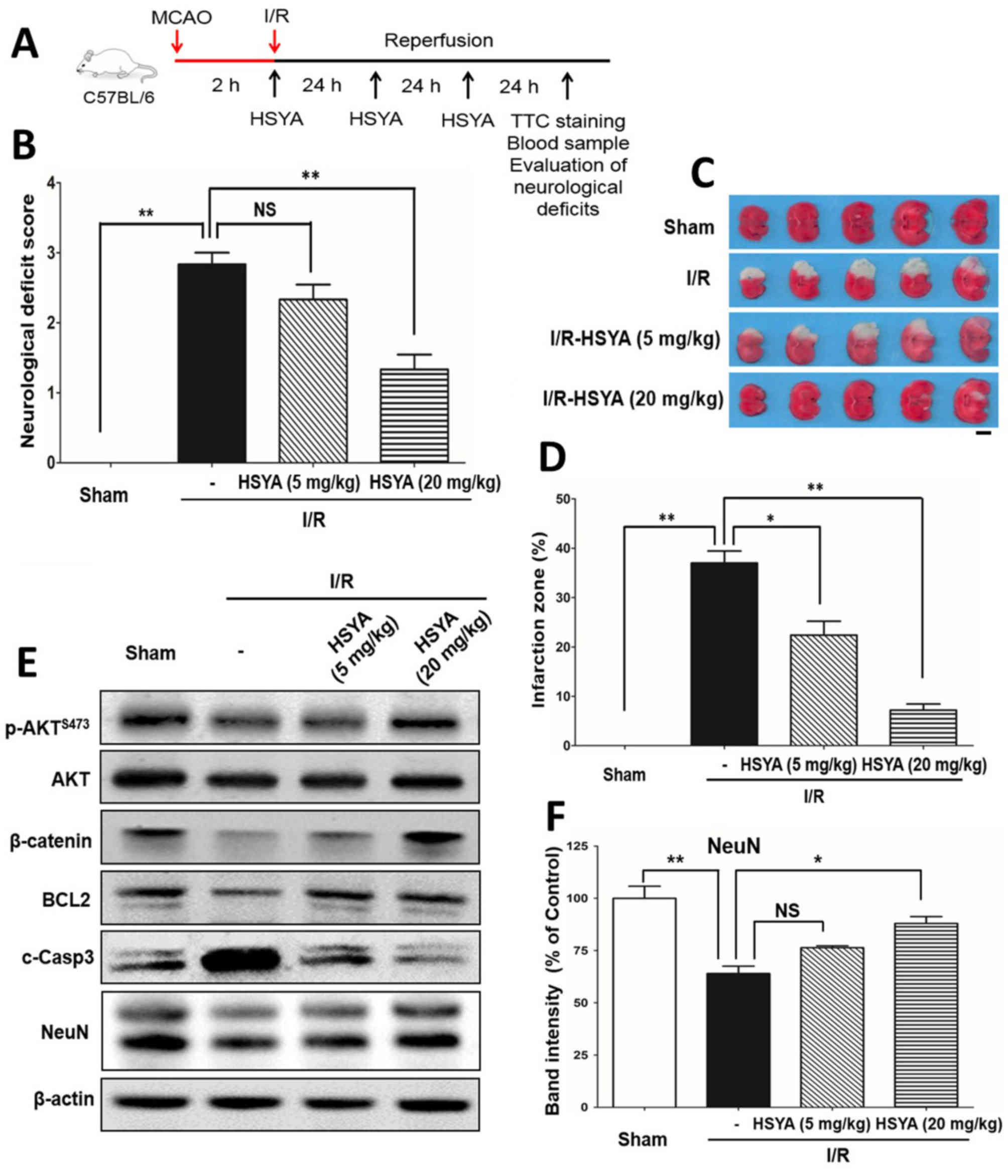 | Figure 1.HSYA alleviates neurological deficits
and reduces the infarct size induced by cerebral I/R. (A)
Experimental scheme for the experimental mice cerebral I/R.
Briefly, mice received ischemia via middle cerebral artery
occlusion for 2 h and reperfusion for 3 days, with or without HSYA
(5 or 20 mg/kg) treatment. (B) Neurological behavior deficit
evaluation. A scale of 0–4 points was used to evaluate the
neurological deficits. The values are expressed as the mean ±
standard error of the mean of 6 mice in each group. I/R
significantly increased deficit score, but was reduced following
treatment with HSYA. (C) HSYA reduced the infarct zone. TTC
staining was used to monitor the infarct area (white). I/R resulted
in an infarct zone; HSYA decreased the size of the area. Scale
bar=5 mm. (D) Results of statistical analysis of the TTC staining
zone with three mice of each group (n=3). (E) Western blotting was
conducted to analyze the expression of proliferation and
apoptosis-associated molecules with antibodies against p-Akt,
β-catenin, BCL2, cleaved caspase-3 and neuron marker NeuN. β-actin
was the internal control. (F) Band intensity quantification of
NeuN. *P<0.05, **P<0.01. Akt, protein kinase B; BCL2, B-cell
lymphoma 2; c-Casp3, cleaved caspase 3; HYSA, hydroxysafflor yellow
A; I/R, ischemia/reperfusion; NeuN, neuronal nuclei; p,
phosphorylated; TTC, 2, 3, 5-triphenyltetrazolium chloride. |
HSYA regulates the expression of
enzymes of phenylalanine metabolism to reprogram the metabolic
amino acid profile associated with cerebral I/R
To investigate whether I/R and HSYA alter the
metabolic profile, plasma samples from the sham and I/R groups
treated with or without HSYA were extracted from mouse tail veins
and centrifuged at 10,500 × g at room temperature for 5 min for
metabolic screening. The results revealed a significant increase in
the levels of plasma phenylalanine (from 56.69±1.097 to 95.55±13.01
µmol/l) with I/R stress compared with the sham group Treatment with
HSYA significantly reduced the levels of phenylalanine (P<0.05;
Fig. 2A). The data suggested that
HSYA could abrogate increases in phenylalanine induced by cerebral
I/R. Additionally, whether HSYA affects the expression of key
enzymes for the metabolism of phenylalanine was investigated. As
presented in Fig. 2B,
phenylalanine can be metabolized into tyrosine via PAH;
phenylalanine can also be transformed into phenylpyruvate or vice
versa via TAT and GOT1. Then, the mRNA expression of these three
enzymes were analyzed; significantly decreased Pah and
Got1 expression, and increased Tat expression were
observed in the I/R group compared with the sham group (Fig. 2C-E). Most importantly, HSYA could
reverse the altered expression of these three enzymes caused by I/R
(Fig. 2C-E). These findings
suggested that HSYA may reprogram the metabolism of phenylalanine
by regulating the expression of key enzymes.
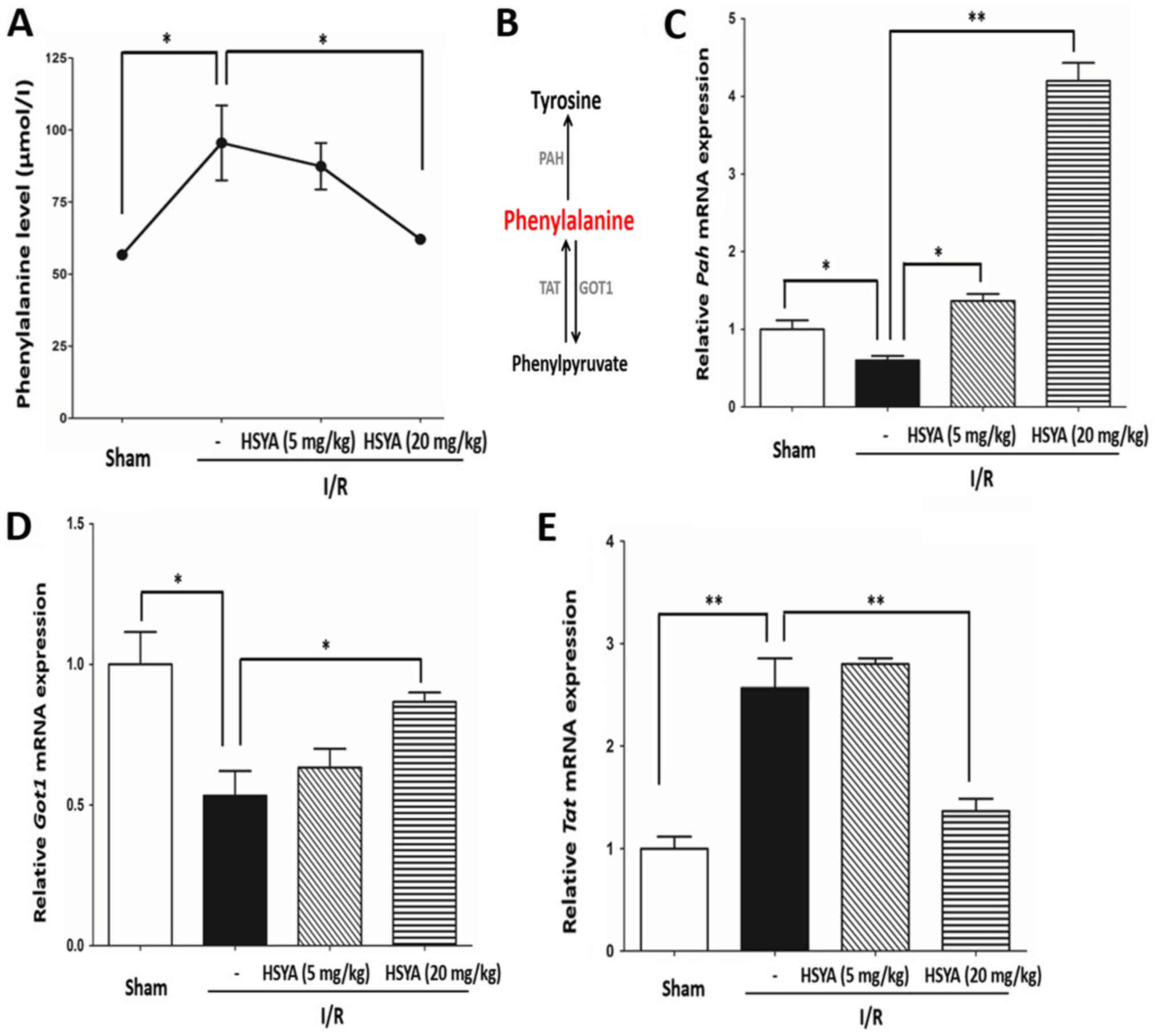 | Figure 2.HSYA regulates the levels of
phenylalanine and the metabolic enzymes expression caused by
cerebral I/R in vivo. (A) I/R stress increased the plasma
levels of phenylalanine, which was reversed by HSYA. (B)
Transformation flux of phenylalanine. Phenylalanine can be
metabolized to phenylpyruvate and tyrosine via GOT1 and TAT, and
PAH, respectively. HSYA reduced the levels of phenylalanine by
regulating the expression of key metabolic enzymes. mRNA expression
and relative quantification of (C) Pah, (D) Got1 and
(E) Tat. The data are expressed as the mean ± standard error
of the mean. *P<0.05, **P<0.01. GOT1, aspartate
aminotransferase; HYSA, hydroxysafflor yellow A; I/R,
ischemia/reperfusion; PAH, phenylalanine hydroxylase; TAT, tyrosine
aminotransferase. |
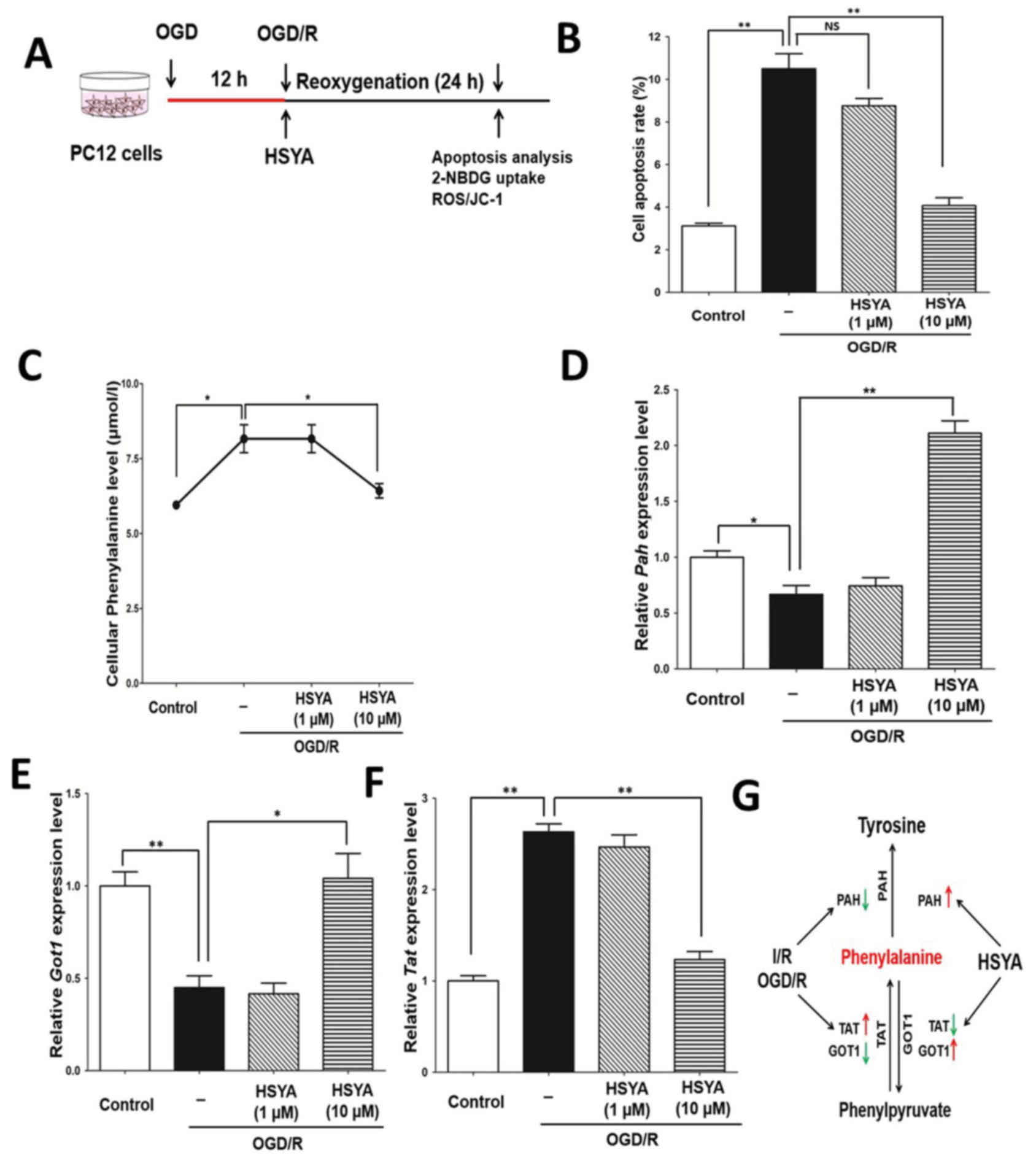
HSYA inhibits metabolic increases in
phenylalanine caused by OGD/R stress in primary mouse neuronal
cells
Furthermore, primary mouse neuronal cells were
exposed to OGD/R stress to evaluate the protective effects of HSYA.
OGD/R treatment was performed as presented in Fig. 3A. Western blotting demonstrated
that HSYA significantly inhibited the expression of c-casp3; the
expression of p-Akt, BCL2 and NeuN had recovered in response to
HSYA following I/R (Fig. 3B).
Additionally, as expected, HSYA could significantly inhibit
neuronal cell apoptosis induced by OGD/R treatment; the effect was
notably greater in response to 10 µM HSYA than 1 µM (Fig. 3C and D). Therefore, HSYA may
protect mouse neuronal cells from OGD/R stress.
 | Figure 3.HSYA regulates the levels of
phenylalanine in primary mouse neuronal cells with OGD/R stress.
(A) Experimental scheme for primary mouse neurons with OGD/R
treatment. (B) Analysis of p-Akt, BCL2, cleaved caspase-3 and NeuN
levels by means of western blotting in primary mouse neuron with
OGD/R treatment. (C) Analysis of apoptosis via flow cytometry. (D)
Quantification of apoptosis. (E) Quantification of phenylalanine
levels in the control and OGD/R groups treated with or without
HSYA. HSYA regulates the expression of key metabolic enzymes in
primary mouse neuronal cells. mRNA expression and relative
quantification of (F) Pah, (G) Got1 and (H)
Tat. The data are expressed as the mean ± standard error of
the mean. *P<0.05, **P<0.01. Akt, protein kinase B; BCL2,
B-cell lymphoma 2; c-Casp3, cleaved caspase 3; GOT1, aspartate
aminotransferase; HYSA, hydroxysafflor yellow A; PAH, phenylalanine
hydroxylase; NeuN, neuronal nuclei; OGD/R, oxygen and glucose
deprivation/reoxygenation; p, phosphorylated; TAT, tyrosine
aminotransferase. |
To evaluate whether HSYA also alters the metabolic
profile associated with OGD/R in primary neurons, metabolic
screening was performed. The results revealed alterations in
phenylalanine levels, which was similar to the in vivo model
(I/R injury). HSYA also restricted the increase of phenylalanine in
primary neurons with OGD/R. Thus, alterations in the levels of
phenylalanine were markedly coincident in vivo and in
vitro (Figs. 2A and 3E). Furthermore, the mRNA expression
levels of Pah, Got1 and Tat in the mouse neuron model
was similar to the results of the in vivo model (Fig. 3F-H). Therefore, HSYA may recover
the levels of phenylalanine by regulating the expression of key
metabolic enzymes in a mouse model of I/R and in primary mouse
neuronal cells exposed to OGD/R stress. Thus, the neuroprotective
properties of HSYA may occur by recovering the levels of
phenylalanine dysregulated by I/R injury.
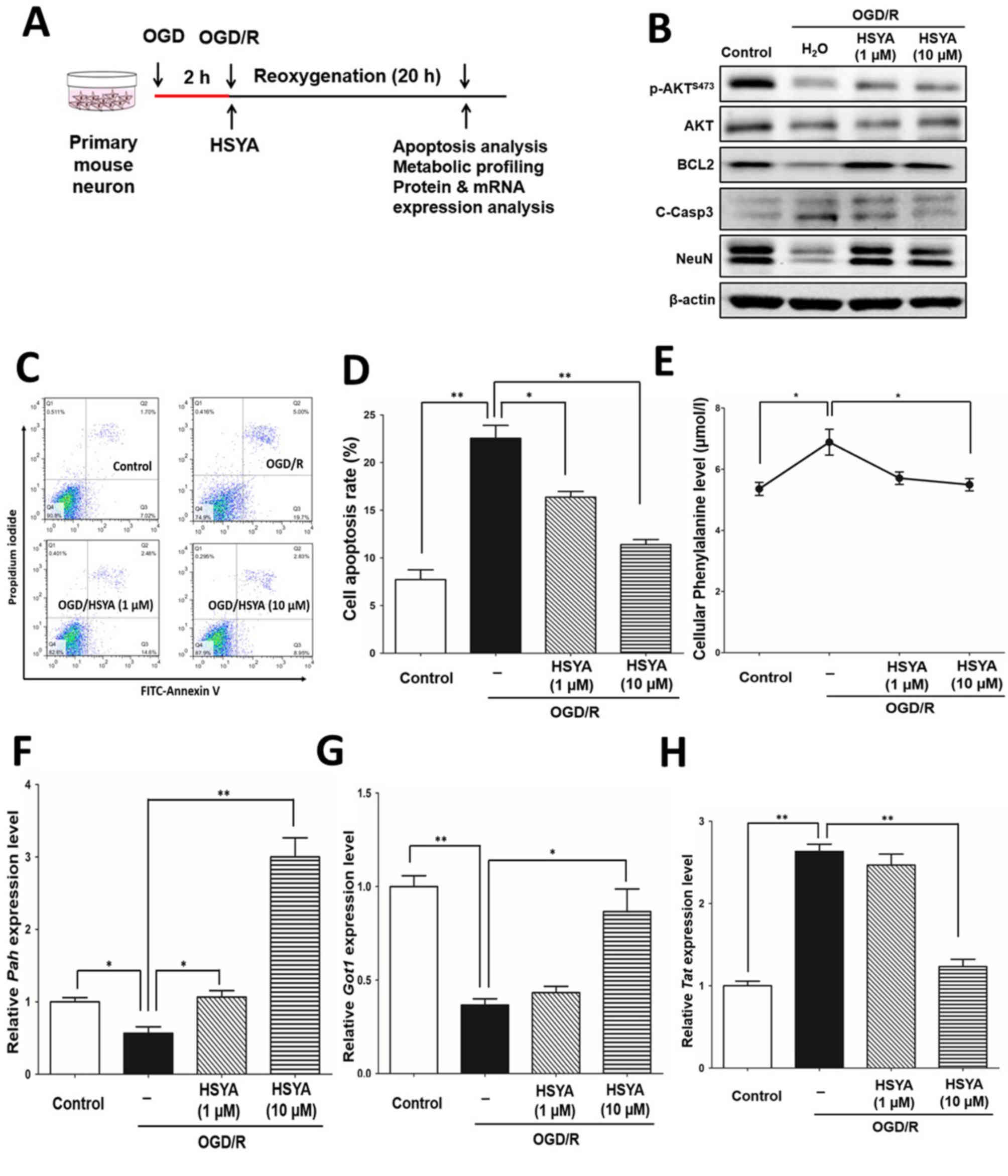
HSYA protects PC12 cells exposed to
OGD/R stress via the regulation of phenylalanine metabolism
In addition, to further confirm the neuroprotective
effects of HSYA, as previously reported (25), an OGD/R model in PC12 cells was
generated (Fig. 4A). Similar to
the primary mouse neuronal cells, OGD/R significantly induced the
apoptosis of PC12 cells compared with the control; HSYA
significantly protected against apoptosis caused by OGD/R stress
(P<0.01; Fig. 4B). Of note, the
levels of phenylalanine in PC12 cells were significantly increased
in response to OGD/R compared with the control, whereas the levels
were significantly reduced following treatment with HSYA
(P<0.05; Fig. 4C).
Furthermore, alterations in the mRNA expression
levels of Pah, Got1 and Tat in PC12 cells were
similar within the in vivo model and primary mouse neuronal
cells (Fig. 4D-F). Therefore,
in vivo, mouse I/R injury and neuronal cells in vitro
exposed to OGD/R stress exhibited increased levels of phenylalanine
by suppressing the expression of key metabolic enzymes, pah
and Got1, and upregulating Tat expression (Fig. 4G). HSYA may reduce the levels of
phenylalanine by regulating the expression of these three key
enzymes (Fig. 4G). Thus, the
neuroprotective properties of HSYA may occur by recovering the
levels of phenylalanine induced by I/R injury and OGD/R stress
(Fig. 4G).
HSYA promotes mitochondrial function
and biogenesis associated with its neuroprotective effect against
OGD/R stress in PC12 cells
To clarify the neuroprotective mechanism of HSYA,
whether HSYA affected the mitochondrial function in PC12 cells was
investigated. As presented in Fig.
5A, OGD/R stress significantly increased the ROS levels of PC12
cells compared with the control, as detected by DCFH-DA analysis.
As expected, HSYA significantly reduced the ROS levels induced by
OGD/R compared with the OGD/R group (P<0.01; Fig. 5A). JC-1 staining revealed that HSYA
(10 µM) significantly promoted mitochondrial function by increasing
membrane potential suppressed by OGD/R stress (P<0.01; Fig. 5B). Additionally, HSYA significantly
enhanced the glucose uptake ability of PC12 cell as determined via
2-NBDG analysis (P<0.01; Fig.
5C).
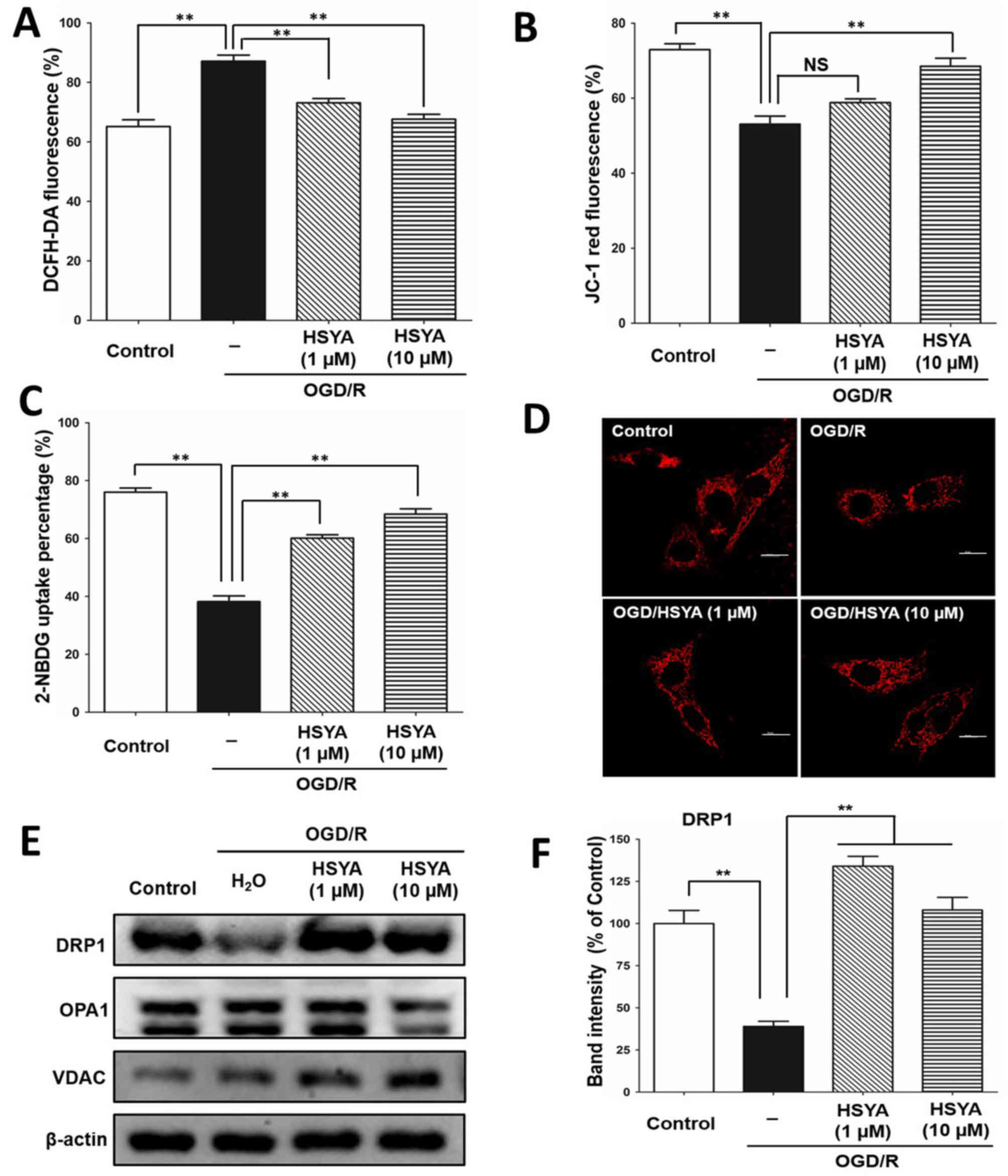 | Figure 5.HSYA promotes mitochondrial function
and biogenesis in PC12 cells exposed to OGD/R stress. (A) DCFH-DA
analysis in PC12 cells with OGD/R stress and HSYA treatment as
indicated. (B) JC-1 red fluorescence assay in PC12 cells with OGD/R
stress and HSYA treatment. (C) Glucose uptake ability in PC12 cells
exposed to OGD/R stress and HSYA as detected by 2-NBDG labelling.
(D) MitoTracker Red fluorescence in PC12 cells was detected with a
confocal immunofluorescence microscope. Mitochondrial shape was
compared in each group. Scale bar=50 µm. (E) Western blotting
analysis of mitochondrial fission protein DRP1, fusion protein OPA1
and mitochondrial marker VDAC. β-actin was the internal control.
(F) Band intensity quantification of DRP1 in (E). All data are
expressed as the mean ± standard error of the mean. **P<0.01.
2-NBDG,
2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxyglucose;
DCFH-DA, dichloro-dihydro-fluorescein diacetate; DRP1,
dynamin-1-like protein; HYSA, hydroxysafflor yellow A; OPA1,
dynamin-like GTPase; NS, not significant; VDAC, voltage-dependent
anion-selective channel 1; OGD/R, oxygen and glucose
deprivation/reoxygenation. |
Furthermore, via MitoTracker red assay, it was
demonstrated that mitochondria were small and rod-like following
OGD/R (Fig. 5D); however, HSYA
promoted the morphological alteration of mitochondria to a narrow
and elongated shape, indicating mitochondria fission and biogenesis
potential (26). Most importantly,
HSYA treatment significantly increased the expression of
mitochondria fission protein DRP1 and notably inhibited that of the
mitochondria fusion protein OPA1 compared with the OGD/R group
(Fig. 5E and F). Additionally,
treatment with HSYA increased the protein expression level of VDAC,
a mitochondrial marker, suggesting an increase in mitochondrial
number (Fig. 5E). These data
demonstrated HSYA could promote mitochondria function and
biogenesis for its neuroprotective effects in PC12 cells exposed to
OGD/R stress.
HSYA recovers the function of
mitochondria suppressed by phenylalanine in PC12 cells
As an endogenous metabolite, it is difficult to
inhibit the production of phenylalanine. To evaluate whether
phenylalanine can directly impair mitochondrial function in
neuronal cells, the proliferation of PC12 cells treated with
phenylalanine was analyzed. As presented in Fig. 6A, higher concentrations of
phenylalanine inhibited the proliferation of PC12 cells (16 and 32
mM). Analysis of JC-1 red fluorescence demonstrated that
phenylalanine significantly inhibited the mitochondrial function in
PC12 cells, and HSYA (10 µM) was able to increased mitochondrial
function impaired upon treatment with phenylalanine (P<0.05;
Fig. 6B). In addition,
phenylalanine significantly reduced the expression of the
mitochondrial fission protein DRP1 expression compared with the
control, which was rescued by HSYA treatment (Fig. 6C and D). Treatment with
phenylalanine did not alter the protein expression level of Fis1.
However, the protein expression level of MFN2 was slightly
increased upon treatment with phenylalanine, although the mechanism
underlying MFN2 upregulation remains to be investigated. The
findings suggested that HSYA may promote mitochondrial function and
biogenesis suppressed by phenylalanine in PC12 cells.
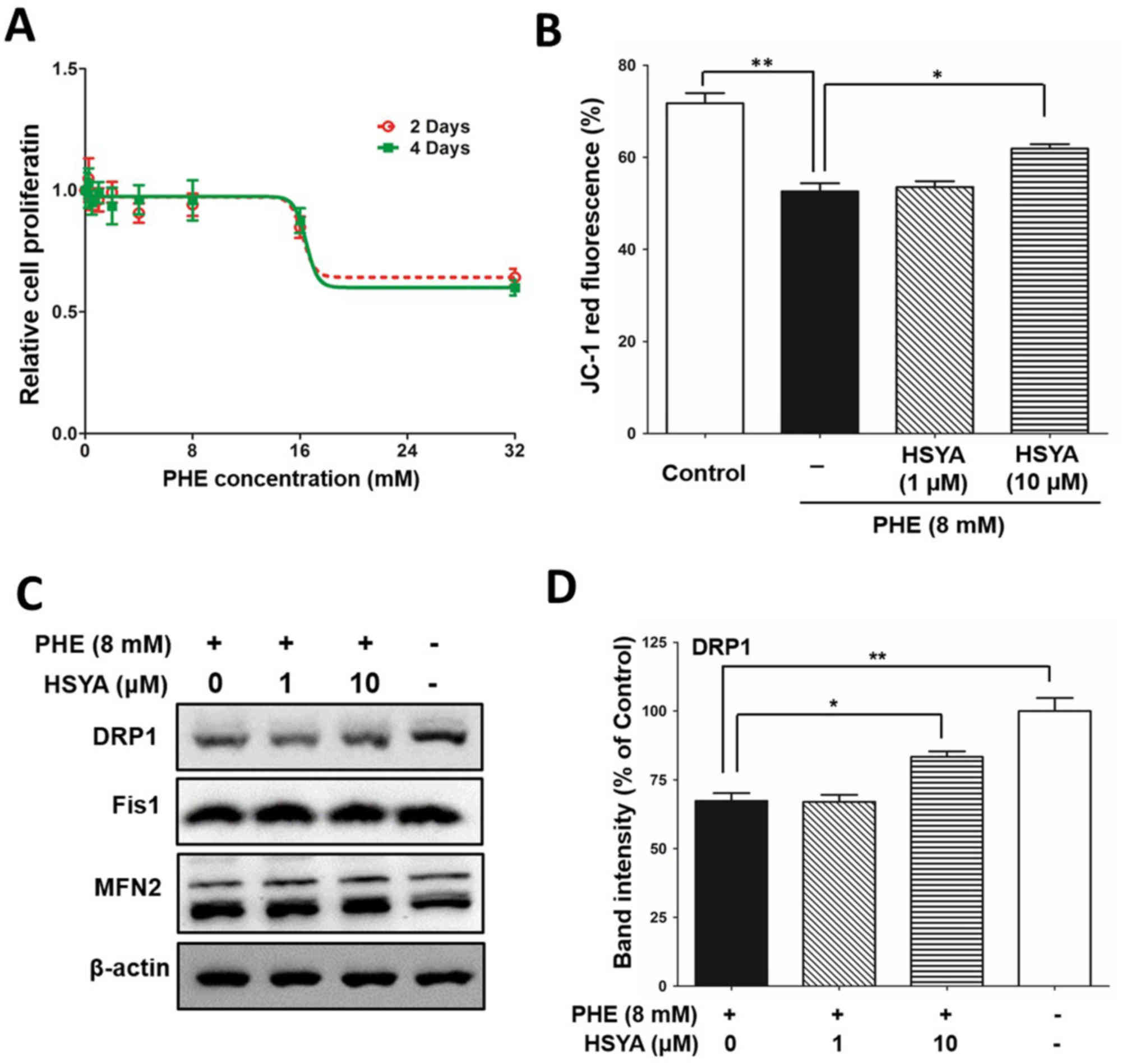 | Figure 6.HSYA protects mitochondrial function
suppressed by PHE in PC12 cells. (A) MTT assay in PC12 cells with
PHE treatment for 2 and 4 days with increased concentration (0,
0.5, 1, 2, 4, 8, 16 and 32 mM). (B) JC-1 red fluorescence assay in
PC12 cells with 8 mM PHE and HSYA (1 and 10 µM) treatment as
indicated. (C) Western blotting analysis of mitochondrial fission
protein DRP1 and Fis1, and fusion protein MFN2. β-actin was the
internal control. (D) Band intensity quantification of DRP1. All
data are expressed as the mean ± standard error of the mean.
*P<0.05, **P<0.01. DRP1, dynamin-1-like protein; Fis1,
mitochondrial fission 1 protein; HYSA, hydroxysafflor yellow A;
MFN2, mitofusin-2; OGD/R, oxygen and glucose
deprivation/reoxygenation; PHE, phenylalanine. |
Discussion
Stroke is the second most common cause of mortality
worldwide, causing ~6.2 million cases of mortality annually
(27). There are two major types
of stroke: Ischemic and hemorrhagic. Ischemic stroke, which occurs
in ~87% of patients, is mainly caused by an interruption of the
blood supply to certain regions of the brain (28). Despite major advances in stroke
imaging and treatment, stroke continues to threaten patients and
causes familial and societal burden. The initial goal of therapy in
ischemic stroke is to restore blood flow. The phenomenon that
successful alleviation of regional tissue hypoxia exacerbates
reperfusion injury in the form of cell death requires further
investigation. Numerous studies using cerebral I/R animal models
have identified the mechanism of I/R injury and provided a wide
array of neuroprotective strategies to reduce the deleterious
effects of reperfusion injury (3);
however, the approaches for predicting the efficacy of these
strategies are limited.
Several studies reported that the levels of numerous
metabolites were altered with cerebral ischemia (16). In particular, branched chain amino
acids are notably reduced in ischemic stroke, and the extent
correlates with poor neurological outcome (29). In the present study, alterations in
the metabolic amino acid profile due to cerebral I/R injury were
observed. I/R stress increased the levels of phenylalanine and
altered metabolic flux. The findings of the present study
demonstrated that I/R and OGD/R stress caused a significant
increase in phenylalanine. Furthermore, the significant alterations
in the mRNA expression levels of the key metabolic enzymes Pah,
Tat and Got1 were reported, which are responsible for
the metabolism of phenylalanine (18). The underlying mechanism as to how
I/R stress regulates the expression of metabolic enzymes remains
unknown; however, the findings of the present study indicates that
the increased ROS levels induced by I/R injury may be involved.
HSYA has been widely used for the treatment of
cerebrovascular diseases in clinical practice. Our previous study
revealed that HSYA could reduce ROS levels and activate cellular
survival signaling in a myocardial I/R rat model (30). Additionally, the neuroprotective
effect of HSYA in a cerebral I/R mouse model was demonstrated in
the present study. Furthermore, HSYA reduced the levels of
malondialdehyde, and increased those of glutathione and SOD to
suppress ROS; thus, apoptosis was inhibited (data not shown). The
present study also proposed that HSYA could activate Akt and
β-catenin signaling to promote neuronal cell survival.
Metabolic screening was conducted to identify
alterations in the metabolic amino acid profile in the present
study. The results revealed that increases in the levels of
phenylalanine were induced by cerebral I/R injury. Interestingly,
the in vivo and in vitro models demonstrated that
HSYA could reduce alterations in phenylalanine levels caused by I/R
and OGD/R stress. Furthermore, we explored the potential mechanism
and reported that HSYA could regulate the expression of the key
enzymes Pah, Tat and Got1, which are responsible for
phenylalanine transformation. Finally, whether phenylalanine can
directly impair the function of mitochondria in neuronal cells was
determined. Thus, the levels of phenylalanine may be an ideal
biomarker for evaluating HSYA as a therapeutic agent.
As a metabolite, phenylalanine possesses a
physiological function associated with amino acid metabolism. A
previous study demonstrated that phenylalanine may inhibit neurite
outgrowth by interfering with L1-mediated cell adhesion (31). The results of the present study
revealed that higher concentrations of phenylalanine inhibited the
function of mitochondria and cell proliferation; however,
phenylalanine alone could not induce apoptosis. Additional
conditions, including an increased concentration of phenylalanine
or a combination of multiple metabolites, may lead to an increase
in apoptosis.
Mitochondria are critical for cellular metabolism
and the regulation of apoptosis. The biogenesis of mitochondria is
mostly dependent on the process of mitochondrial fission and fusion
(32). In the present study, I/R
and OGD/R stress increased the levels of phenylalanine, damaged
mitochondrial function and induced ROS production (Fig. 7). Whereas, HSYA could reduce
phenylalanine levels and promote mitochondrial function via the
upregulation of mitochondrial fission protein DRP1. Thus, HSYA may
promote mitochondria function and biogenesis in association with
its neuroprotective effects. The present study proposed a novel
metabolite as a biomarker for cerebral I/R injury and provided a
novel mechanism for the development of therapeutic strategies based
on HSYA to treat ischemic strokes.
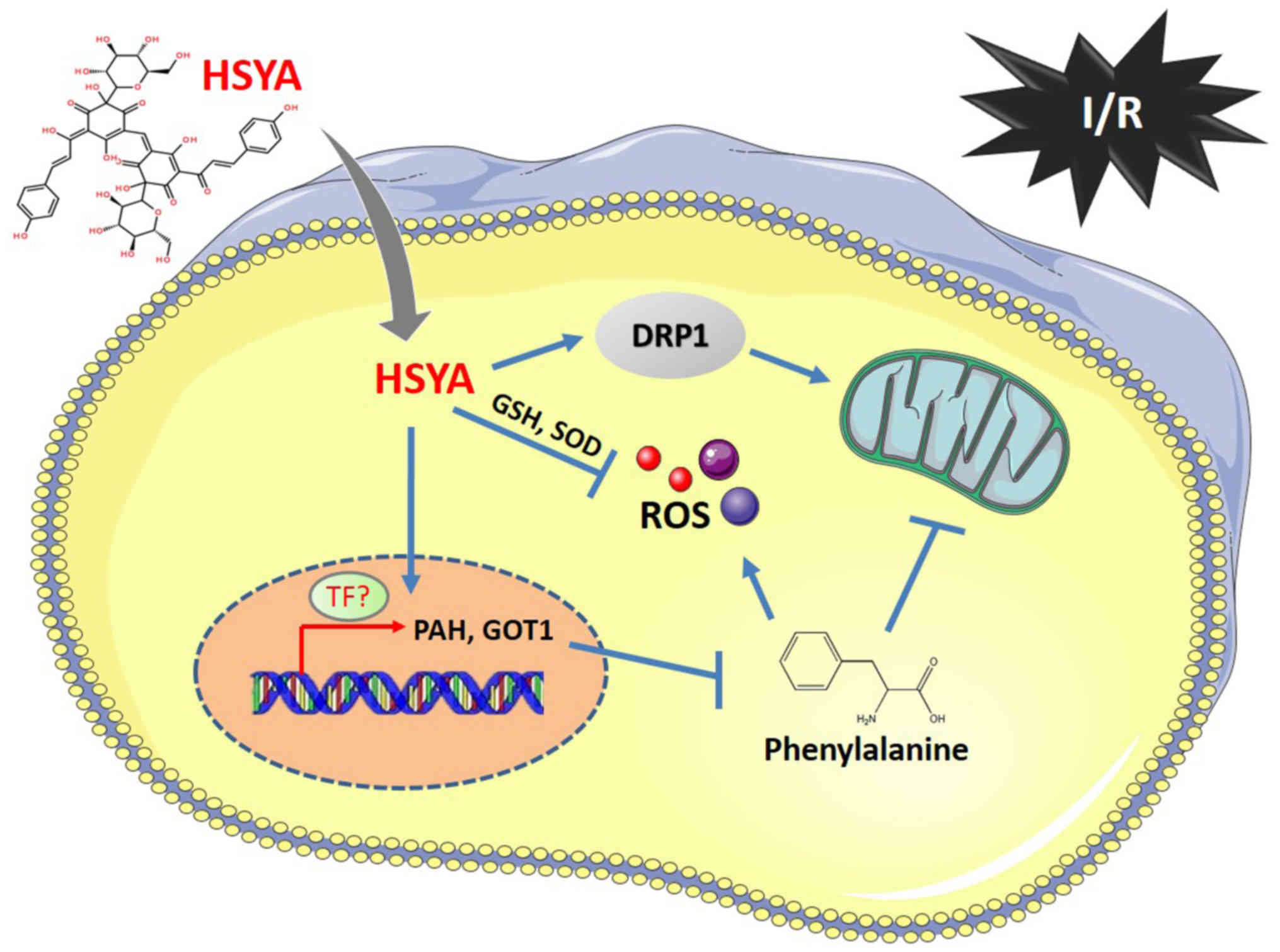 | Figure 7.Diagram of HSYA regulating the levels
of phenylalanine, mitochondrial function and biogenesis for
neuroprotection. I/R injury caused increased ROS and phenylalanine
levels to induce neuronal cell apoptosis. HSYA could reduce the
production of ROS and phenylalanine by regulating the expression of
key metabolic enzymes, including PAH, TAT and GOT1. In addition,
HSYA could promote mitochondrial function and biogenesis by
upregulating mitochondria fission protein DRP1. GOT1, aspartate
aminotransferase; GSH, glutathione; HYSA, hydroxysafflor yellow A;
PAH, phenylalanine hydroxylase; ROS, reactive oxygen species; SOD,
superoxide dismutase. |
Acknowledgements
The authors would like to thank Professor Xiaoqiang
Li (Department of Pharmacology, School of Pharmacy, Fourth Military
Medical University, Xi'an, China) for providing advice on the
experimental design and critical comments on the present
manuscript.
Funding
The present study was supported by The National
Natural Science Foundation of China (grant nos. 81573549 and
81503280).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AW and XL designed and supervised the experiments.
SC and MS conducted the mouse I/R injury, OGD/R model, metabolomics
analysis, qPCR, western blotting and the cellular experiments. XZ
performed the experiments on primary neuron cells. ZY, WL and JC
performed the cellular reactive oxygen species detection analysis
and the cell proliferation assay. YQ performed the statistical
analysis. ZY, WL, JC and YQ gave critical comments and revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Animal Care
Committees of the Fourth Military Medical University (Xi'an,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chu SF, Zhang Z, Zhang W, Zhang MJ, Gao Y,
Han N, Zuo W, Huang HY and Chen NH: Upregulating the expression of
Survivin-HBXIP complex contributes to the protective role of
IMM-H004 in transient global cerebral Ischemia/reperfusion. Mol
Neurobiol. 54:524–540. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sarami Foroshani M, Sobhani ZS, Mohammadi
MT and Aryafar M: Fullerenol nanoparticles decrease blood-brain
barrier interruption and brain edema during cerebral
ischemia-reperfusion injury probably by reduction of interleukin-6
and matrix metalloproteinase-9 transcription. J Stroke Cerebrovasc
Dis. 27:3053–3065. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wiklund L, Patnaik R, Sharma A, Miclescu A
and Sharma HS: Cerebral tissue oxidative ischemia-reperfusion
injury in connection with experimental cardiac arrest and
cardiopulmonary resuscitation: Effect of mild hypothermia and
methylene blue. Mol Neurobiol. 55:115–121. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gong L, Tang Y, An R, Lin M, Chen L and Du
J: RTN1-C mediates cerebral ischemia/reperfusion injury via ER
stress and mitochondria-associated apoptosis pathways. Cell Death
Dis. 8:e30802017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li Y, Wang M and Wang S: Effect of
inhibiting mitochondrial fission on energy metabolism in rat
hippocampal neurons during ischemia/reperfusion injury. Neurol Res.
1–8. 2016.(Epub ahead of print).
|
|
6
|
Guo J, Yong Y, Aa J, Cao B, Sun R, Yu X,
Huang J, Yang N, Yan L, Li X, et al: Compound danshen dripping
pills modulate the perturbed energy metabolism in a rat model of
acute myocardial ischemia. Sci Rep. 6:379192016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jiang S, Shi Z, Li C, Ma C, Bai X and Wang
C: Hydroxysafflor yellow A attenuates ischemia/reperfusion-induced
liver injury by suppressing macrophage activation. Int J Clin Exp
Pathol. 7:2595–2608. 2014.PubMed/NCBI
|
|
8
|
Bai Y, Lu P, Han C, Yu C, Chen M, He F, Yi
D and Wu L: Hydroxysafflor yellow A (HSYA) from flowers of
Carthamus tinctorius L. and its vasodilatation effects on
pulmonary artery. Molecules. 17:14918–14927. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun L, Yang L, Xu YW, Liang H, Han J, Zhao
RJ and Cheng Y: Neuroprotection of hydroxysafflor yellow A in the
transient focal ischemia: Inhibition of protein
oxidation/nitration, 12/15-lipoxygenase and blood-brain barrier
disruption. Brain Res. 1473:227–235. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang X, Ma Z, Fu Z, Gao S, Yang L, Jin Y,
Sun H, Wang C, Fan W, Chen L, et al: Hydroxysafflor Yellow A
protects neurons from excitotoxic death through Inhibition of
NMDARs. ASN Neuro. 8(pii)2016.
|
|
11
|
Li CY, Yin JG, Zhang J, Wang XX, Xu MJ,
Liu F, Zou JD and Ju WZ: Pharmacokinetic profiles of hydroxysafflor
yellow A following intravenous administration of its pure
preparations in healthy Chinese volunteers. J Ethnopharmacol.
162:225–230. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Svensson RU and Shaw RJ: Cancer
metabolism: Tumour friend or foe. Nature. 485:590–591. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao Q, Wu J, Hua Q, Lin Z, Ye L, Zhang W,
Wu G, Du J, Xia J, Chu M and Hu X: Resolvin D1 mitigates energy
metabolism disorder after ischemia-reperfusion of the rat lung. J
Transl Med. 14:812016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hoehn RS, Seitz AP, Jernigan PL, Gulbins E
and Edwards MJ: Ischemia/Reperfusion injury alters sphingolipid
metabolism in the gut. Cell Physiol Biochem. 39:1262–1270. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gellert L, Knapp L, Nemeth K, Herédi J,
Varga D, Oláh G, Kocsis K, Menyhárt A, Kis Z, Farkas T, et al:
Post-ischemic treatment with L-kynurenine sulfate exacerbates
neuronal damage after transient middle cerebral artery occlusion.
Neuroscience. 247:95–101. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kimberly WT, Wang Y, Pham L, Furie KL and
Gerszten RE: Metabolite profiling identifies a branched chain amino
acid signature in acute cardioembolic stroke. Stroke. 44:1389–1395.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Song Y, Long L, Zhang N and Liu Y:
Inhibitory effects of hydroxysafflor yellow A on PDGF-BB-induced
proliferation and migration of vascular smooth muscle cells via
mediating Akt signaling. Mol Med Rep. 10:1555–1560. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rodriguez B, Torres N, Rincon AR, Bourges
H, Panduro A and Tovar AR: Hepatic phenylalanine-hydroxylase and
tyrosine-aminotransferase mRNA levels in rats adapted to diets with
different concentrations of protein. Rev Invest Clin. 48:413–419.
1996.PubMed/NCBI
|
|
19
|
Zhang J, Li F, Liu X, Shen L, Liu J, Su J,
Zhang W, Deng Y, Wang L, Liu N, et al: The repression of human
differentiation-related gene NDRG2 expression by Myc via
Miz-1-dependent interaction with the NDRG2 core promoter. J Biol
Chem. 281:39159–39168. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ganan-Gomez I, Wei Y, Yang H,
Boyano-Adanez MC and Garcia-Manero G: Oncogenic functions of the
transcription factor Nrf2. Free Radic Biol Med. 65:750–764. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu Y, Wang L, Dai C, Ma G, Zhang Y, Zhang
X and Wu Z: Neuroprotection by platelet-activating factor
acetylhydrolase in a mouse model of transient cerebral ischemia.
Neurosci Lett. 558:26–30. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hu Z, Yang B, Mo X and Zhou F: HspB8
mediates neuroprotection against OGD/R in N2A cells through the
phosphoinositide 3-kinase/Akt pathway. Brain Res. 1644:15–21. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chazotte B: Labeling mitochondria with
JC-1. Cold Spring Harbor Protoc. 2011(pii): pdb.prot065490.
2011.
|
|
25
|
Liu L, Huang W, Wang J, Song H, Cen J and
Ji B: Anthraquinone derivative exerted hormetic effect on the
apoptosis in oxygen-glucose deprivation-induced PC12 cells via ERK
and Akt activated Nrf2/HO-1 signaling pathway. Chem Biol Interact.
262:1–11. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang JL, Mukda S and Chen SD: Diverse
roles of mitochondria in ischemic stroke. Redox Biol. 16:263–275.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Winterholler M, Hollander C, Kerling F,
Weber I, Dittrich S, Türk M and Schröder R: Stroke in duchenne
muscular dystrophy: A retrospective longitudinal study in 54
patients. Stroke. 47:2123–2126. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Morris NA, Merkler AE, Gialdini G and
Kamel H: Timing of incident stroke risk after cervical artery
dissection presenting without ischemia. Stroke. 48:551–555. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu Y, Luo F, Xu Y, Wang B, Zhao Y, Xu W,
Shi L, Lu X and Liu Q: Epithelial-mesenchymal transition and cancer
stem cells, mediated by a long non-coding RNA, HOTAIR, are involved
in cell malignant transformation induced by cigarette smoke
extract. Toxicol Appl Pharmacol. 282:9–19. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wei G, Yin Y, Duan J, Guo C, Zhu Y, Wang
Y, Xi M and Wen A: Hydroxysafflor yellow A promotes
neovascularization and cardiac function recovery through
HO-1/VEGF-A/SDF-1α cascade. Biomed Pharmacother. 88:409–420. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hartwig C, Gal A, Santer R, Ullrich K,
Finckh U and Kreienkamp HJ: Elevated phenylalanine levels interfere
with neurite outgrowth stimulated by the neuronal cell adhesion
molecule L1 in vitro. FEBS Lett. 580:3489–3492. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bockler S, Chelius X, Hock N, Klecker T,
Wolter M, Weiss M, Braun RJ and Westermann B: Fusion, fission, and
transport control asymmetric inheritance of mitochondria and
protein aggregates. J Cell Biol. 216:2481–2498. 2017. View Article : Google Scholar : PubMed/NCBI
|