Introduction
Diabetes mellitus is a metabolic disorder
characterized by hyperglycemia as a result of β-cell dysfunction
and insulin resistance (1,2). The pathogenesis of diabetes is not
fully understood, but includes insulin resistance and a deficit of
β-cell mass. Increasing attention has been paid to the significant
role of mitochondrial energy metabolism in the development of
diabetes mellitus (3,4). Mitochondria convert energy-storing
molecules, such as glucose, free fatty acids and amino acids, into
ATP via oxidative phosphorylation, and are involved in β-cell
proliferation, and the synthesis and release of insulin (5).
Mitochondria are widely distributed in eukaryotic
cells, providing energy for cellular activities, participating in
important metabolic pathways, maintaining the cell ion balance and
participating in signal transduction, all of which are important
processes to maintain normal cellular activity (6).
Mitochondrial regulation controls various aspects of
cellular homeostasis in the organism (7–9).
Furthermore, the regulation of mitochondria contributes to the
pathogenesis of metabolic disorders, including obesity, metabolic
syndrome, type 2 diabetes mellitus (T2DM) and cancer (10,11).
Previous studies have indicated that dysfunction in mitochondrial
energy metabolism may be the fundamental cause of impaired insulin
secretion by β-cells (11,12).
The primary role of β-cells is to release insulin in
response to postprandial blood glucose levels (2). Glucose is transported into β-cells by
diffusion and is subsequently metabolized during glycolysis and the
tricarboxylic acid (TCA) cycle (5,7).
Glycolysis leads to the production of pyruvate, which enters the
mitochondria to be oxidized (8).
Pyruvate is metabolized by the pyruvate dehydrogenase (PDH) complex
(PDHc), a controller of glucose oxidation, which is present in the
mitochondrial matrix, and is further converted to acetyl-coenzyme A
(CoA) to obtain citrate that participates in the TCA cycle
(12–14). This increases the ATP/ADP ratio and
causes the plasma ATP-dependent K+ channel to close. The
voltage-dependent Ca2+ channel opens and triggers the
exocytosis of insulin (12,15).
The metabolic coupling factor amplifies insulin secretion, meaning
that glucose and products of glucose metabolism potentiate the
exocytosis of insulin molecules without any further increase in
cytosolic Ca2+ concentrations (11).
The most important enzyme to determine which path to
take is PDHc, which catalyzes the initiating reaction in the TCA
cycle and directly regulates insulin secretion. Impairment of the
PDHc directly affects the growth and function of β-cells (7,9).
The PDHc is a composite of the mitochondrial enzyme
system and a member of the 2-oxygen (generation) acid dehydrogenase
complex family. PDHc is composed of thiamine-dependent tetrameric
(α2β2) PDH (PDHA1; encoded by the PDHA1 gene),
dihydrolipoamide acetyltransferase and flavin adenine
dinucleotide-containing dihydrolipoamide dehydrogenase (E3), which
is attached to the complex by the E3-binding protein (16,17).
PDH catalyzes the irreversible oxidative
decarboxylation of pyruvate into acetyl-CoA and reduces
NAD+ to NADH, which links the aerobic oxidation of
glucose with the cyclic capacity of TCA, playing an important role
in the energy metabolism of the mitochondrial respiratory chain and
distinguishing between aerobic and anaerobic oxidation (18,19).
When the levels of PDH are reduced, the proportion of energy
supplied by glucose decreases, while the contribution of other
energy-producing molecules, such as lipids and amino acids,
increases (20). The activity of
PDH is determined by the inhibitory effect of pyruvate
dehydrogenase kinase (PDK) on the PDHc (17,21–23).
PDK phosphorylates PDH-E1α, inactivating PDH. Loss of PDH activity
leads to glucose metabolic disorders and tissue damage, which
influence the growth, differentiation and functional expression of
β-cells (4,18).
Previous studies have focused on the function of PDH
(17,24,25).
Mice with PDHA1 knocked out in the heart exhibited
ventricular dysfunction, predominantly diastolic (26,27),
while treatment with dichloroacetic acid has been reported to
reverse ventricular dysfunction (28). The hyperinsulinemia-positive
glucose clamping test in obese Wistar rats revealed higher plasma
lactate levels (17,29). In the liver, under insulin
resistance or obese conditions, PDH activity is abnormally reduced
(30), glucose utilization is
reduced and hepatic glycogen production is increased, leading to
high levels of blood glucose (23,25,31).
Therefore, mitochondrial metabolism plays a
significant role in the onset and development of diabetes. Previous
studies have shown that the expression of PDHc is reduced in rodent
models of T2DM and the human body (22,32),
indicating the central role played by PDHc in the development of
diabetes. However, the effect of PDHA1 on pancreatic β-cells has
not been extensively explored. The present study aimed to clarify
the association between PDHA1 and diabetes, assess the effect of
PDHA1 on β-cell morphology and function, and elucidate the possible
mechanism guiding PDHA1 action. The present study may provide a new
theoretical framework to explain diabetes development and proposes
a potential molecular target for the treatment of this
disorder.
Materials and methods
Animals
B6.Cg-Tg (Ins1-cre/ERT) 1 lphi/J mice (cat. no.
024709; hereafter referred to as Ins-cre+/− mice) and
B6.129P2-Pdha1tm1Ptl/J mice (cat. no. 017443; hereafter referred to
as PDHA1flox/floxmice) were obtained from the Jackson Laboratory
(n=4/group, 2 males and 2 females, ~20 g/each). Then, 5 db/db mice
and 5 C57BL/6 mice were provided by the animal laboratory of the
Southern Medical University (Guangzhou, China). All mice were given
food and water ad libitum, housed in a clean laminar animal
room at a controlled indoor temperature of 20–25°C and a humidity
of 40–70% with a 12-h light/dark cycle.
Cre-loxP system
Based on the principle of the Cre-loxP system,
PDHA1flox/flox mice were mated with
Ins-cre+/− mice to obtain the F2
PDHA1flox/−Cre+/− mouse generation. These
heterozygotes were further crossed with PDHA1flox/−
Ins-cre+/− mice to produce PDHA1flox/flox
Ins-cre+/− mice. Ins-cre recombinase effectively
eliminates the sequence between two loxP sites. Moreover, the
expression of Ins-cre recombinase can be triggered by the
simultaneous action of insulin and exogenous tamoxifen
(intraperitoneally injected tamoxifen for 7 consecutive days, 50
mg/kg) in mouse islets. Therefore, by controlling the time of
exogenous tamoxifen injection, the PDHA1 gene could be
specifically knocked out in the mouse islets (33,34).
Mice with Ins-cre+/− genotypes were selected as the
negative control (NC) group, while mice of the same age and gender
with genotype PDHA1flox/flox Ins-cre+/− were
the knockout experimental (βKO) group.
Genotypic identification
Then, ~3–4 mm length of tail was removed from the
mouse with ophthalmic scissors and 100 µl each of rat tail lysate A
(0.5% SDS, 0.1 M NaCl, 0.05 M EDTA, 0.01 M Tris-HCl pH 8.0 and
protease K 100 g/ml) and B (NaOH 1 mmol/ml) added and then heated
for 1 h. The supernatant contained mouse DNA (34). DNA was used for genotyping using
PCR with mouse-specific primers (Table
I) for the PDHA1 and Ins-cre genes. The reaction (20 µl total)
contained 10 µl PCR mix (2X Taq Plus Master Mix, Invitrogen; Thermo
Fisher Scientific, Inc.), 2 µl of each primer (primer concentration
10 µM) and 2 µl template DNA. For Ins-cre amplification, the PCR
reaction conditions were as follows: 94°C for 2 min; 10 cycles of
94°C for 20 sec, 65°C for 15 sec and 68°Cf or 10 sec; 28 cycles of
94°C for 15 sec, 50°C for 15 sec and 72°C for 10 sec; and 72°C for
2 min. For PDHA1 amplification the PCR conditions were as follows:
94°C for 2 min; 10 cycles of 94°C for 20 sec, 65°C for 15 sec, 68°C
for 10 sec; 28 cycles of 94°C for 15 sec, 60°C for 15 sec and 72°C
for 10 sec; and 72°C for 2 min. The PCR products were separated
using agarose gel electrophoresis (2%; 6 µl/well). Image analysis
was performed using an Automatic Digital Gel Imaging System (Tanon
1600; Tanon Science & Technology, Co.). Each experiment was
performed in triplicate.
 | Table I.Primers used for genotyping. |
Table I.
Primers used for genotyping.
| Primer name | Forward
(5′-3′) | Reverse
(3′-5′) |
|---|
| PDHA1 |
AGCAGCCAGCACGGACTACT |
GCAGCCAAACAGATTACACC |
| INS1-CRE |
AGCAGCCAGCACGGACTACT |
TGCGAACCTCATCACTCGT |
| INS1-CRE internal
positive control |
CTAGGCCACAGAATTGAAAGATCT |
GTAGGTGGAAATTCTAGCATCATCC |
Hematoxylin and eosin (H&E)
staining
Mice were intraperitoneally injected with
pentobarbital sodium solution at 150 mg/kg, with death determined
after cessation of breathing for 2–3 min. When mice suffered pain
and discomfort that affected their quality of life, they were
euthanized with pentobarbital solution; this was set as the humane
end point. The pancreas was removed from the mice, weighed, fixed
for 24 h in 4% paraformaldehyde at 4°C and embedded in paraffin.
The pancreas was sliced into 4-µm thick sections and stained with
hematoxylin for 5 min at room temperature, alcohol hydrochloric
acid for 1 sec and eosin for 1 sec. The tissue was dehydrated using
graded ethanol, cleared in dimethylbenzene, sealed and observed
under an inverted fluorescence microscope (IX53; Olympus
Corporation, magnification, ×200; 3 fields analyzed per sample).
Image-Pro Plus 5 (Media Cybernetics, Inc.) was used to count cells
and measure cell size.
Western blot analysis
Pancreas tissue was collected, weighed and ground
into a powder under liquid nitrogen. The samples were mixed with
RIPA lysis buffer [50 mM Tris (pH 7.4), 150 mM NaCl, 1% NP-40, 0.5%
sodium deoxycholate]. The protein concentration was determined
using the bicinchoninic acid method. Samples (20 µl) were separated
using 10% SDS-PAGE, transferred to nitrocellulose membranes, then
blocked with 5% skimmed milk at room temperature, incubated with
the following primary antibodies for 14 h at 4°C: PDHA1 (1:1,000;
cat. no. ab110334; Abcam) and GAPDH (1:1,000; cat. no. ab9484;
Abcam). Membranes were washed three times with TBS with Tween 20
for 5 min each time and incubated with an horseradish peroxidase
(HRP)-conjugated secondary antibody (1:500; cat. no. ab97023;
Abcam) for 2 h at room temperature. Chemiluminescence was performed
using Pierce™ Fast Western Blot ECL substrate (cat. no. 35055;
Thermo Fisher Scientific, Inc.). Protein bands were visualized
using the Odyssey Infrared Imaging System (LI-COR Biosciences). The
results were quantitatively analyzed using Image-Pro Plus 6.0
software (Media Cybernetics, Inc.). The experiments were repeated
three times for each sample.
Immunohistochemistry
The tissue samples were cut into 3–4-µm thick
sections, dewaxing with xylene and hydration with alcohol (xylene1
10 min, xylene2 10 min, 95, 90, 80, 70 and 50% alcohol, each 5
min). Following permeabilization with 0.1% Triton X-100 and 3%
H2O2, blocking with normal goat serum (cat.
no. MP20008; Shanghai Yuanye Science & Technology) at room
temperature for 30, 30 and 60 min, respectively. The slides were
incubated with a primary antibody (PDHA1; 1:500; cat. no. ab110334;
Abcam) at 4°C in a wet box for 14–16 h. Following this, the slides
were washed with PBS for 3 min three times. A secondary
HRP-conjugated antibody (1:300, cat. no. ab97023; Abcam) was added
dropwise to each section, followed by incubation in a wet box at
37°C for 2 h. After washing for 3 min three times with PBS, the DAB
color solution (OriGene Technologies, Inc.) was added, and tap
water was used to stop development. The slides were counterstained
with hematoxylin at room temperature for 3 min and washed with tap
water until a blue color developed. The slides were observed and
images were captured using a light microscope (Zeiss AG). As a
negative control the primary antibody was substituted with PBS in
parallel experiments.
Immunofluorescence
Tissue samples were cut into sections and processed
as aforementioned. Slides were incubated with mouse anti-PDHA1
(1:200; cat. no. ab110334; Abcam) and rabbit anti-insulin (1:200;
cat. no. ab63820; Abcam) overnight at 4°C in a wet box. The slides
were washed for 3 min three times and incubated in the dark with
the corresponding diluted secondary antibodies [1:500; cat. no.
ab150077 (Alexa Fluor® 488-conjugated goat anti-rabbit
IgG) and ab150116 (Alexa Fluor 594-conjugated goat anti-mouse IgG;
Abcam) at 37°C for 2 h. The secondary antibody solution was
discarded and the slides were washed three times. Slides were
incubated with DAPI (1drop: 1 ml, ~1 min at room temperature.
Images were captured under a laser confocal microscope and 5 visual
fields containing pancreatic islet cells were selected under a the
microscope (magnification, ×400); the number of PDHA1 positive
cells and total number of cells in each visual field were counted
by Image-Pro Plus 6.0 software (Media Cybernetics, Inc.). The rate
of PDHA1 positive cells in islet cells was calculated.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA from the islets, liver and adipose tissues
were extracted using TRIzol (Vazyme). Absorbance at 260 and 280 nm
(A260 and A280, respectively) was determined using an ultraviolet
spectrophotometer, and the ratio of the optical density (OD) at
these wavelengths (ODA260/A280) was 1.8–2.0.
Complementary DNA was reverse transcribed using Hiscript II Q RT
SuperMix for qPCR (Nanjing KeyGen Biotech Co., Ltd.), according to
the manufacturer's instructions in a final volume of 20 µl. qPCR
was performed using real-time PCR Master Mix (SYBR Green; KGA1339-1
200T, Nanjing KeyGen Biotech Co, Ltd.). The primers for PDHA1 were:
Forward, 5′-GAGCTGAGCAGCTGTGTAAC-3′ and reverse,
5′-TGCCAATCGTTACAGGTATTACAG-3′. GAPDH forward,
5′-ACCACAGTCCATGCCATCAC-3′ and reverse, 5′-TCCACCACCCTGTTGCTGTA-3′.
The thermocycling conditions were: 95°C for 30 sec, 95°C 10 sec;
followed by 45 cycles of 60°C for 30 sec, 72°C for 30 sec; 60°C for
60 sec and 95°C for 15 sec. The gene expression was quantified with
the 2−∆∆Cq method (35). Data were normalized to GAPDH mRNA
levels. All reactions were performed in triplicate.
Intraperitoneal glucose tolerance test
(IPGTT) and intraperitoneal insulin tolerance test (IPITT)
βKO and NC mice were selected (5/group) of the same
age (8 weeks) and weight. Their body weight and fasting blood
glucose levels were measured every 2 weeks. The food intake and
nutritional status of the mice were also monitored. The mice were
weighed and glucose was administered intraperitoneally (1.5 g/kg
body weight) after 14 h fasting. Following this, blood samples (1–2
µl) were collected through the tail vein at 0, 15, 30, 60 and 120
min. The glucose levels were measured using a glucometer (Accu-Chek
Active; Roche Diagnostics). At 1 week, the mice were fasted for 4
h, weighed and injected intraperitoneally with insulin (0.6 U/kg
body weight). Blood samples (1–2 µl) were subsequently withdrawn
from the tail vein at 0, 15, 30 and 60 min for blood glucose
measurements.
Insulin concentration measurement
Glucose solution (5%; 1.5 g/kg) was injected into
mice that had been fasting for 14 h. Blood samples (0.8–1 ml) were
collected from the hearts of mice under anesthesia. Blood was
collected from the heart at the moment of the injection or 30 min
later, then euthanasia. The serum was blood was allowed to settle
and separate for 30 min prior to centrifugation at 1,680 × g for 10
min, both at 4°C. An insulin ELISA kit (cat. no. EZRMI-13K; EMD
Millipore) was used to measure the content of insulin in the mouse
serum. Each sample was analyzed in triplicate. The samples and
reaction solutions were added and mixed in the dark for 30 min. The
OD value was determined using a UV spectrophotometer. A standard
curve was constructed and the corresponding conversion of sample
concentration was carried out.
Primary islet extraction
All experiments were repeated 3 times. The mouse
epidermis was disinfected with ethanol, and the abdominal cavity
was opened to allow isolation of the pancreatic tissue. The
pancreases were rinsed with pre-cooled PBS solution, visible blood
and nerves were removed and the pancreas was placed in a pre-cooled
petri dish. Collagenase P (0.3 mg) was injected in each pancreas
tissue. The tissue was then digested at 37°C for 15 min. The
digestion was terminated using pre-cooled Hank's buffer (Gino
Biomedical Technology Co., Ltd.), and after centrifugation (430 ×
g; 30 sec, 4°C), the digested tissue was filtered with a 40-mesh
screen. Pancreatic islet cells (brown, cell mass) were selected
with a 10 µl pipette under stereomicroscope. The selected islet
cells used to extract RNA.
Statistical analysis
All experiments were repeated 3 times. Statistical
analysis was performed using GraphPad 5 (GraphPad Software, Inc.),
Image-Pro Plus 6.0 (Media Cybernetics, Inc.) and SPSS 22 (IBM
Corp). Paired t-tests and ANOVA followed by a Bonferroni post-hoc
analysis were used to determine differences between the βKO and NC
mice. Data are presented as the mean ± SD. P<0.05 was considered
to indicate a statistically significant difference.
Results
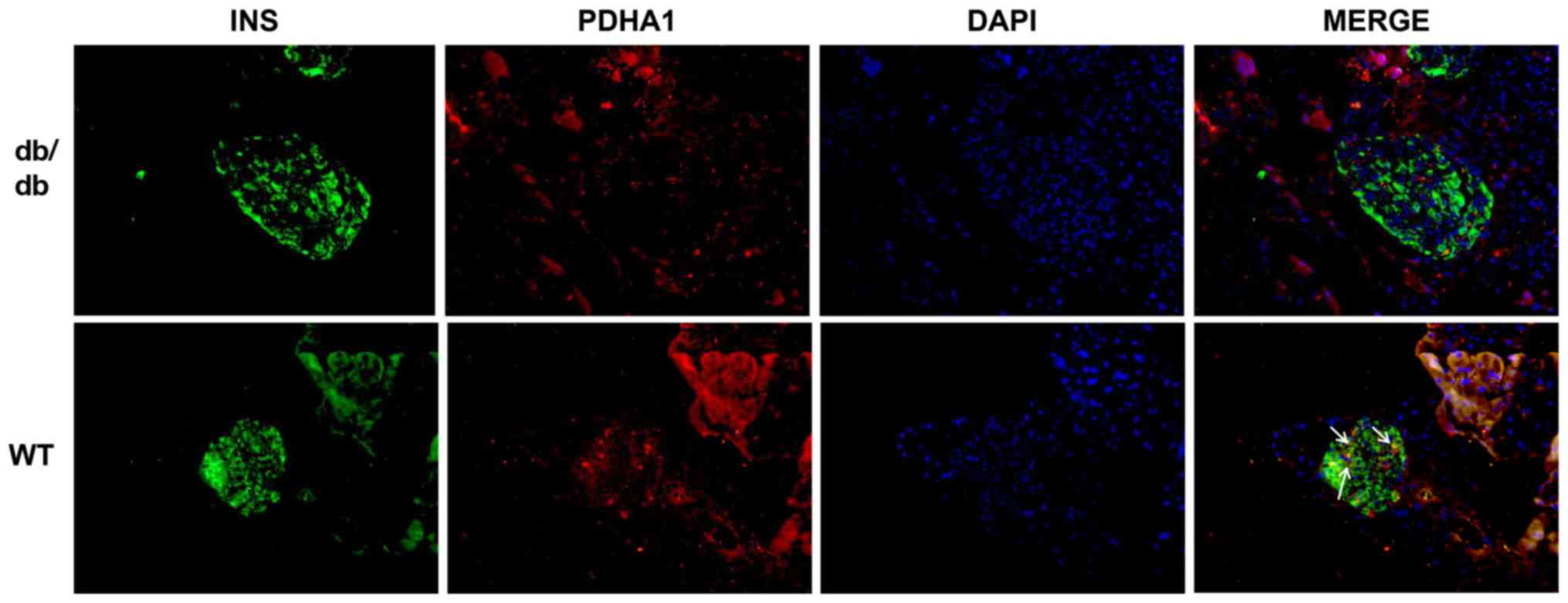
PDHA1 levels are lower in db/db
mice
db/db mice are an animal model of T2DM that exhibit
elevated blood glucose and insulin resistance in their adulthood
(36). To investigate the
expression of PDHA1 in diabetic mice, pancreas sections of db/db
mice and C57BL/6 mice were analyzed using immunofluorescence. The
cells that secreted insulin had green fluorescence, while cells
expressing PDHA1 exhibited red fluorescence. The expression of
PDHA1 in the islets of db/db mice was markedly downregulated
compared with in C57BL/6 mice (Fig.
1), which was consistent with histology results showing that
PDHA1 expression was decreased in T2DM mammals (36,37).
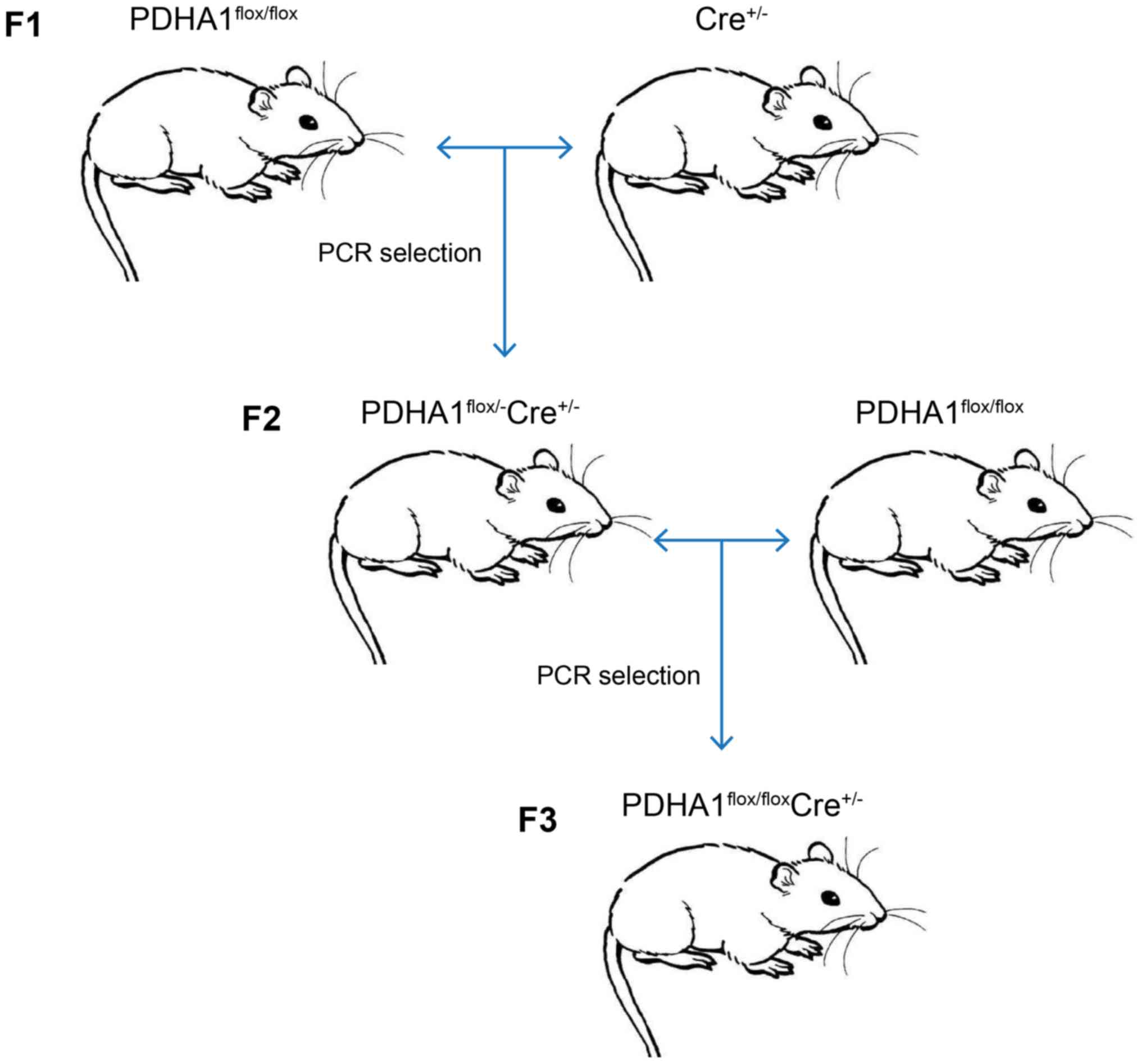
Generation and validation of the βKO
mouse model
To further investigate the role of PDHA1 in the
pancreas, a βKO mouse model was established. As the homozygous loss
of PDHA1 may be embryonically lethal (37,38),
the Cre-loxP method was used to generate mice with a pancreatic
β-cell-specific knockout of PDHA1. Following the steps shown in
Fig. 2, PDHA1flox/flox
Ins-cre+/− mice were obtained by crossing
PDHA1flox/− Ins-cre+/− mice and
PDHA1flox/flox mice.
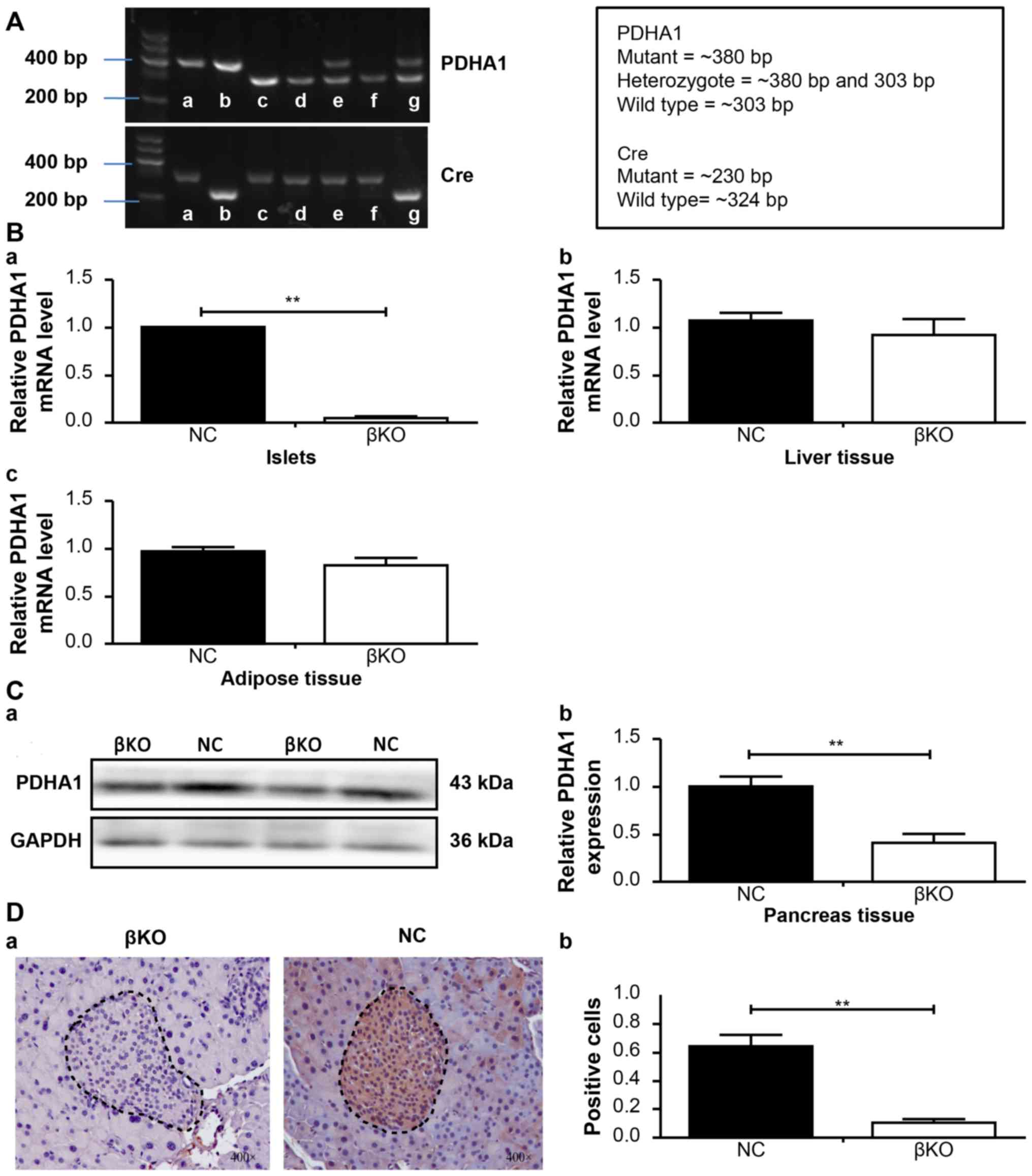
PCR analysis of DNA prepared from tail snips of the
male knockout mice (4 weeks old) confirmed the recessiveness of the
PDHA1 gene (303 bp fragment) and the dominance of PDHA1 (380
bp fragment), as well as that of the Ins-cre transgene (230 and 324
bp fragment; Fig. 3A).
Subsequently, tamoxifen (5 mg/ml) was injected to induce the
removal of the DNA between the loxP sites. PDHA1+/+
Ins-cre+/− mice were designated as βKO mice, while the
PDHA1+/+ Ins-cre− mice from the same litter
were referred to as NC mice. After 10 months of breeding, a total
of 17 βKO mice were obtained, which were bright black and showed
excellent growth and nutritional status.
RT-qPCR and western blotting was used to verify the
effect of PDHA1 knockout on the islets. RT-qPCR revealed that the
relative PDHA1 mRNA levels in the pancreas islets were
significantly reduced in the βKO mice (P<0.001) compared with
the NC mice (Fig. 3B). Similar
results were obtained in the western blot analysis; the expression
of the PDHA1 protein was significantly lower in the βKO mice than
in the NC mice (P<0.001; Fig.
3C), indicating that the expression of PDHA1 was almost
completely abolished in the pancreatic islets of βKO mice.
To exclude the possible effect of loxP sites on
target gene expression in other tissues or organs (14), the mRNA levels of PDHA1 in the
liver and adipose tissue of βKO and NC mice were determined
(Fig. 3B). The results showed no
significant difference in PDHA1 expression in the liver and adipose
tissues between the groups. To observe the distribution of PDHA1 in
the pancreas of βKO and NC mice, immunohistochemistry was performed
(Fig. 3D). It was found that the
PDHA1 protein was predominantly distributed in the gland cells and
the islets of the pancreas in the NC group; however, in the βKO
group there was almost no brown-red PDHA1 staining in the islets
(P<0.001), indicating that the expression of PDHA1 in the islets
was significantly reduced. These results indicated that the mouse
model had been successfully established.
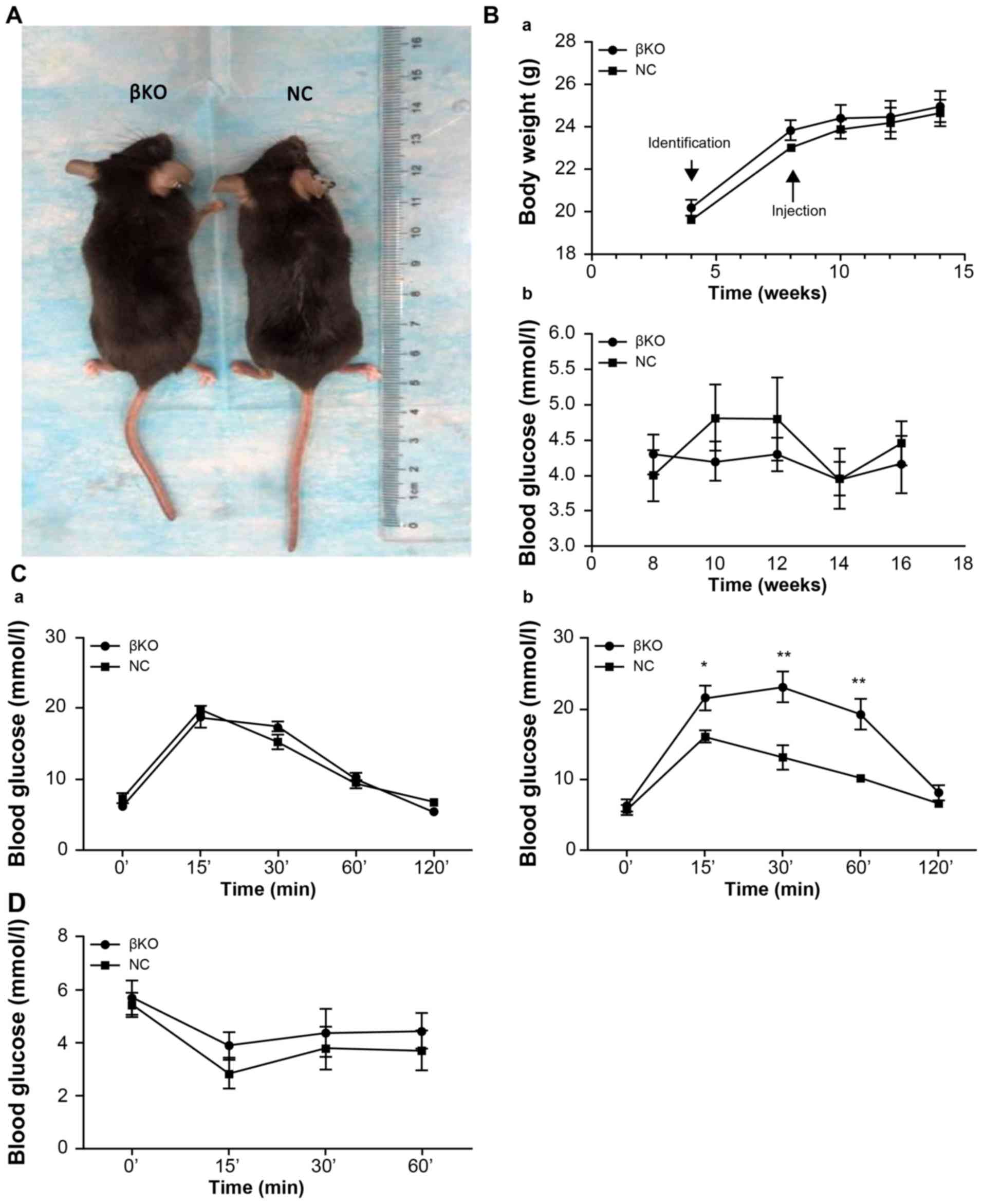
βKO mice exhibit impaired glucose, but
not insulin, tolerance
Age- and weight-matched βKO and NC mice were
compared. There were no differences in hair, morphology and
nutritional status between the two groups (Fig. 4A). The weight of the mice increased
with time; however, there were no significant differences between
the two groups. Moreover, there were no significant alterations in
fasting blood glucose levels (Fig.
4B). An IPGTT was performed in 8-week-old mice (Fig. 4Ca) and 12-week-old mice (Fig. 4Cb). βKO mice were less tolerant to
glucose than the NC group (Fig.
4Cb). However, the IPITT values subsequently obtained showed no
difference between the groups (Fig.
4D), indicating that the peripheral tissues of the βKO mice had
similar levels of insulin sensitivity and showed no signs of
insulin resistance compared with the NC mice. This indicated that
the cause of hyperglycemia in the βKO mice was not related to
insulin resistance in peripheral tissues, but instead was related
to cellular dysfunction.
Impaired glucose-stimulated insulin
secretion in βKO mice
To investigate the variation in the function of the
β-cells in knockout mice, serum insulin levels were measured at
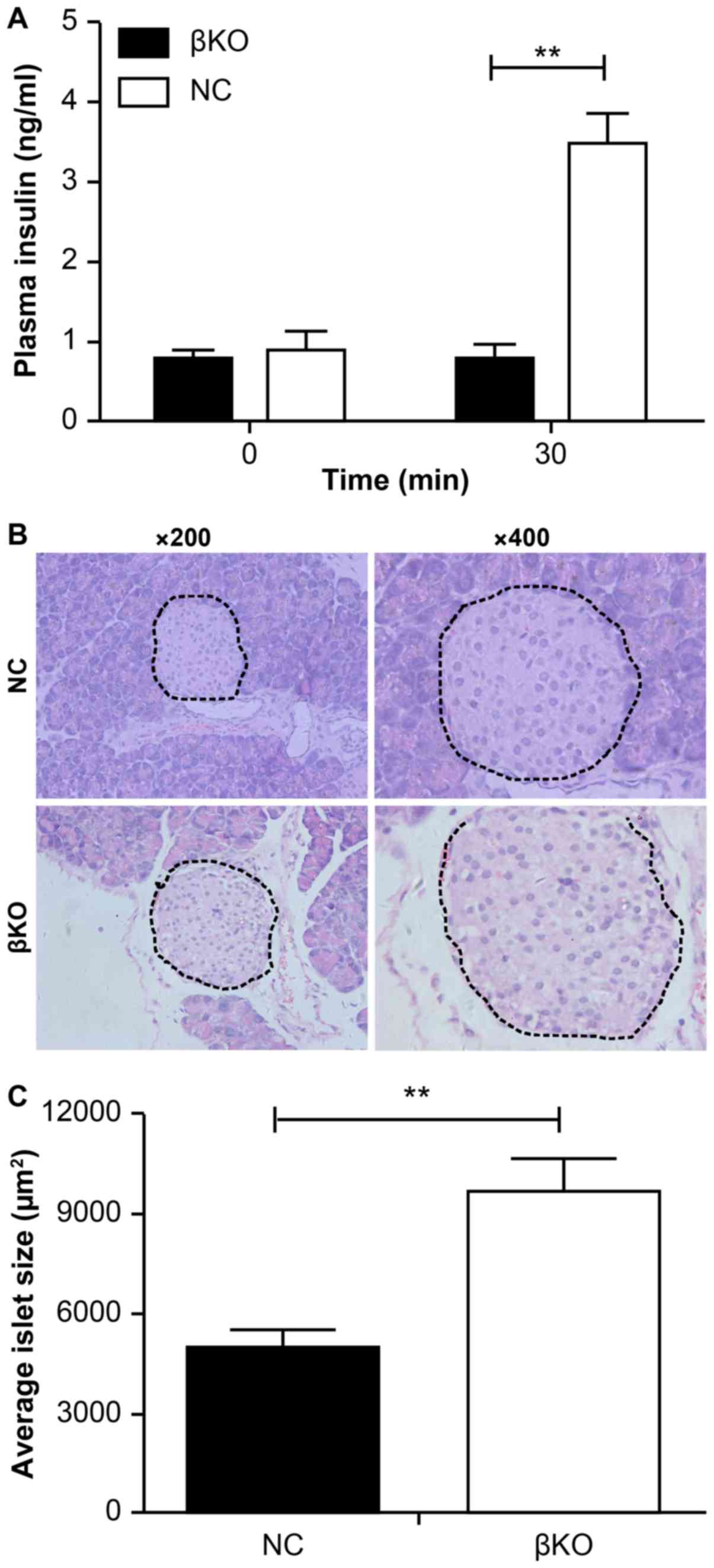
different times after glucose stimulation. It was found that serum
insulin levels were significantly increased at 30 min (3.5±0.52
ng/ml) after glucose injection in NC mice. However, there was
little change (0.78±0.26 ng/ml) in the levels of insulin in the βKO
mice after glucose challenge (Fig.
5A).
This suggested that the impaired glucose tolerance
in βKO mice may be related to impaired insulin secretion by
β-cells. Considering that an abnormality in islet cells may impair
secretion from islets, islet paraffin sections from βKO mice were
analyzed. H&E staining (Fig.
5B) showed that the islets of the βKO mice were abnormally
large compared with those of the NC mice. Most islets in the
control group were tightly clustered and numerous, whereas those of
the βKO mice were distributed loosely with an increased volume. The
mean size of islets was significantly increased in βKO mice at 16
weeks compared with the NC mice (Fig.
5C). These results suggested that PDHA1 serves a significant
role in the development of pancreas islets and that without PDHA1,
the islet cells undergo significant changes in either morphology or
function.
Discussion
Diabetes is a condition that affects individuals
worldwide and can be caused by multiple factors, including
hereditary, environmental and lifestyle factors, and can severely
and negatively affect quality of life for patients (39). Interactions between various genes
and environmental factors result in mutual causality, which can
often aggravate symptoms (40,41).
A number of previous medical studies have attempted to elucidate
the effects of various factors on the growth and development of
β-cells and insulin secretion (42) in order to explore the possible
pathogenesis of diabetes.
The PDHA1 gene, which encodes PDHA1, is
located on the short arm of the X chromosome (Xp22.12) and is ~17.5
kb in length (43,44), and contains a conservative thiamine
diphosphate-binding region (2,44).
PDH is a heterogeneous tetramer, whose components are encoded by
the PDHA1 and PDHB genes (26). The expression level of PDHA1
is the primary determinant of PDH activity (45,46).
When the activity of PDHA1 is impaired, pyruvate can no longer be
metabolized into acetyl-CoA. This causes an increased flux in the
production of acetyl-CoA from fats, amino acids and proteins,
subsequently affecting metabolic states, such as fats and proteins,
resulting in impaired cell growth and function (45). Another pathway by which
mitochondria can be supplied with pyruvate is through pyruvate
carboxylase (PC) (47), which
catalyzes the conversion of pyruvate to oxaloacetic acid, which
then either enters the TCA cycle or is used in gluconeogenesis
(48). The Cre/loxP recombinase
system is a conditional, inducible and spatiotemporal-specific gene
targeting technology that is widely used in basic research into
gene function (49). In the
present study, this technique was used to construct a mouse model
with an islet cell-specific knockout of the PDHA1 gene using adult
male mice by regulating the injection time of tamoxifen. The
expression levels of the PDHA1 protein were determined in pancreas
sections of T2DM (db/db) and wild-type mice. The results of the
present study suggested an association between the PDHA1
gene and T2DM, which is consistent with previous studies (5,22,24).
To verify the causal relationship between changes in
PDHA1 protein levels and the development of T2DM, the Cre/loxP
recombinase system was used (50,51)
to construct transgenic mice with a specific knockout of the PDHA1
gene in β-cells. This model was then used to further investigate
the role of PDHA1 in the development of T2DM by comparing the
phenotypes of mice in the βKO and NC groups.
Homozygous Ins-cre mice exhibit embryonic lethality,
and cannot survive and reproduce (34). Therefore, PDHA1flox/flox
mice were bred with Cre+/− mice and tail tissue samples
from the offspring were used to verify successful crossing.
PDHA1flox/− Ins-cre+/− mice were then bred
with PDHA1flox/flox mice; PDHA1flox/flox
Ins-cre+/− mice were selected as the experimental group.
Subsequently, tissues and cells were evaluated via RT-qPCR and
western blot analysis. The experimental results indicated that
β-cell-specific PDHA1 gene knockout mice had been
successfully established.
Knocking out the PDHA1 gene in the β-cells of
adult male mice did not significant alter the body weight, fasting
blood glucose, body shape or nutritional status between the βKO and
NC groups, which was consistent with the results of a study by
Srinivasan et al (52).
However, the authors of this previous study directly knocked out
the PDHA1 gene in specific islet cells of embryos without
tamoxifen induction, primarily studied the gene in immature mice,
and did not discuss the morphology or phenotype of the βKO mice
cells.
In the present study, the relationship between the
PDHA1 gene and T2DM in adult mice (8 weeks old) was
assessed. As the knockout affects only the β-cells of the islets,
other tissues and organs may have compensated for the reduced
expression of the PDHA1 gene. However, no differences in
body weight and fasting blood glucose were observed between the two
groups. It was found that the glucose tolerance of βKO mice was
impaired compared with the control group, as determined by the
higher blood glucose levels after injection of a glucose solution.
The results of the present study suggested that the deletion of
PDHA1 affected the homeostasis of glucose metabolism and the
TCA cycle. Subsequent experiments should determine the levels of
acetyl-CoA and the fatty acid content, to verify this
conjecture.
To investigate whether the impaired homeostasis of
glucose metabolism is due to the effects of insulin resistance in
peripheral tissues (including the liver, fat and skeletal muscles)
or the β-cells themselves, IPITT was performed. The results of the
IPITT indicated that the elevated blood glucose level was
associated with impaired islet function, not with peripheral
resistance. The hypothesis that β-cell insulin secretion was
functionally impaired was confirmed by ELISAs, contrary to the
results of Nicholls et al (53). This discrepancy may be as a result
of the models used; the study by Nicholls et al (53) was conducted at the cellular level
and did not test insulin levels in animals. Furthermore, the
previous study indirectly influenced the activity of PDH through
PDK and did not directly interfere with the expression of PDH.
Other studies have shown that PDH regulates insulin secretion in
vitro (20,45,54).
The islets of the βKO group were irregularly
enlarged with clear internal vacuoles, but no islet atrophy or
necrosis was observed. These results, together with those from the
glucose tolerance experiment, suggested that the morphological
changes seen in the cells may be due to a compensatory increase in
early diabetic islet cells, and an overall decrease in insulin
secretion. The present study suggested that the PDHA1 gene
plays an important role in the maintenance of β-cell status and
insulin secretion in the islets, laying a foundation for further
studies into mitochondrial energy metabolism.
The present study used knockout mice to verify the
function of PDH in β-cells in adult mice, conducting for the first
time, to the best of our knowledge, a comprehensive islet function
test in knockout mice. Primary islet β-cells were extracted to
allow further functional and morphological analyses. In conclusion,
the present study successfully established the βKO model and
explored the association between PDH and the morphology, and
phenotype, of β-cells. The restoration of PDH expression in the
pancreas of diabetic animals and patients may have clinically
beneficial effects on β-cell function. The mechanisms responsible
for the dysfunction of insulin secretion should be further explored
and could aid in the development of a novel clinical therapy,
providing new avenues for the diagnosis and treatment of
diabetes.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant awarded
by the National Natural Science Foundation of China (grant. no.
81770878); the Science and Technology Program of Guangzhou (grant
no. 201604020007).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XW and JS conceived and designed the study. XW, SL,
YY and YH performed the experiments. XB provided the mutants. XW
drafted the manuscript. SL, DP, XB and JS analyzed the data. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The animal experiments were revised and approved by
the Ethics Committee of the Southern Medical University (grant no.
L2015069).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
IPGTT
|
intraperitoneal glucose tolerance
test
|
|
IPITT
|
intraperitoneal insulin tolerance
test
|
|
PC
|
pyruvate carboxylase
|
|
PDH
|
pyruvate dehydrogenase
|
|
PDHA1
|
pyruvate dehydrogenase E1-alpha
subunit
|
|
PDHc
|
PDH complex
|
|
PDK
|
pyruvate dehydrogenase kinase
|
|
T2DM
|
type 2 diabetes mellitus
|
|
TCA
|
tricarboxylic acid cycle
|
|
βKO
|
mouse PDHA1 knockout in β-cells
|
References
|
1
|
Aamodt KI and Powers AC: Signals in the
pancreatic isletmicroenvironment influence β-cell proliferation.
Diabetes Obes Metab. 19 (Suppl 1):S124–S136. 2017. View Article : Google Scholar
|
|
2
|
Seino S, Sugawara K, Yokoi N and Takahashi
H: β-Cell signalling and insulin secretagogues: A path for improved
diabetes therapy. Diabetes Obes Metab. 19 (Suppl 1):S22–S29. 2017.
View Article : Google Scholar
|
|
3
|
Perry RJ, Zhang D, Zhang XM, Boyer JL and
Shulman GI: Controlled-release mitochondrial protonophore reverses
diabetes and steatohepatitis in rats. Science. 347:1253–1256. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rovira-Llopis S, Bañuls C, Diaz-Morales N,
Hernandez-Mijares A, Rocha M and Victor VM: Mitochondrial dynamics
in type 2 diabetes: Pathophysiological implications. Redox Biol.
11:637–645. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kaufman BA, Li C and Soleimanpour SA:
Mitochondrial regulation of β-cell function: Maintaining the
momentum for insulin release. Mol Aspects Med. 42:91–104. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Morrow RM, Picard M, Derbeneva O, Leipzig
J, McManus MJ, Gouspillou G, Barbat-Artigas S, Dos Santos C, Hepple
RT, Murdock DG and Wallace DC: Mitochondrial energy deficiency
leads to hyperproliferation of skeletal muscle mitochondria and
enhanced insulin sensitivity. Proc Natl Acad Sci USA.
114:2705–2710. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang H, Lu J, Dolezal J, Kulkarni S, Zhang
W, Chen A, Gorka J, Mandel JA and Prochownik EV: Inhibition of
hepatocellular carcinoma by metabolic normalization. PLoS One.
14:e02181862019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Aleem S, Iqbal R, Shar T, Noreen S, Rafiq
N, Javed I, Kosar S, Majeed HN, Sattar NA and Abid MK:
Complications of Diabetes: An insight into genetic polymorphism and
role of insulin. Recent Pat Inflamm Allergy Drug Discov. 12:78–86.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bulthuis EP, Adjobo-Hermans MJW, Willems
PHGM and Koopman WJH: Mitochondrial morphofunction in mammalian
cells. Antioxid Redox Signal. 30:2066–2109. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu YQ, Jetton TL and Leahy JL: Beta-cell
adaptation to insulin resistance. Increased pyruvate carboxylase
and malate-pyruvate shuttle activity in islets of nondiabetic
Zucker fatty rats. J Biol Chem. 277:39163–39168. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Srinivasan S, Guha M, Kashina A and
Avadhani NG: Mitochondrial dysfunction and mitochondrial
dynamics-The cancer connection. Biochim Biophys Acta Bioenerg.
1858:602–614. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mulder H: Transcribing β-cell mitochondria
in health and disease. Mol Metab. 6:1040–1051. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marchetti P, Bugliani M, De Tata V,
Suleiman M and Marselli L: Pancreatic Beta cell identity in humans
and the role of Type 2 diabetes. Front Cell Dev Biol. 5:552017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ciccarone F, Vegliante R, Di Leo L and
Ciriolo MR: The TCA cycle as a bridge between oncometabolism and
DNA transactions in cancer. Semin Cancer Biol. 47:50–56. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Williams M and Caino MC: Mitochondrial
dynamics in type 2 diabetes and cancer. Front Endocrinol
(Lausanne). 9:2112018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Adeva M, González-Lucán M, Seco M and
Donapetry C: Enzymes involved in l-lactate metabolism in humans.
Mitochondrion. 13:615–629. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wijenayake S, Tessier SN and Storey KB:
Regulation of pyruvate dehydrogenase (PDH) in the hibernating
ground squirrel, (Ictidomys tridecemlineatus). J Therm Biol.
69:199–205. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Arumugam R, Horowitz E, Noland RC, Lu DH,
Fleenor D and Freemark M: Regulation of islet β-cell pyruvate
metabolism: interactions of prolactin, glucose, and dexamethasone.
Endocrinology. 151:3074–3083. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu J, Han J, Epstein PN and Liu YQ:
Regulation of PDK mRNA by high fatty acid and glucose in pancreatic
islets. Biochem Biophys Res Commun. 344:827–833. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kono M, Yoshida N, Maeda K, Skinner NE,
Pan W, Kyttaris VC, Tsokos MG and Tsokos GC: Pyruvate dehydrogenase
phosphatase catalytic subunit 2 limits Th17 differentiation. Proc
Natl Acad Sci USA. 115:9288–9293. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jeoung NH: Pyruvate dehydrogenase kinases:
Therapeutic targets for diabetes and cancers. Diabetes Metab J.
39:188–197. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang S, Hulver MW, McMillan RP, Cline MA
and Gilbert ER: The pivotal role of pyruvate dehydrogenase kinases
in metabolic flexibility. Nutr Metab (Lond). 11:102014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Krus U, Kotova O, Spégel P, Hallgard E,
Sharoyko VV, Vedin A, Moritz T, Sugden MC, Koeck T and Mulder H:
Pyruvate dehydrogenase kinase 1 controls mitochondrial metabolism
and insulin secretion in INS-1 832/13 clonal beta-cells. Biochem J.
429:205–213. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tso SC, Lou M, Wu CY, Gui WJ, Chuang JL,
Morlock LK, Williams NS, Wynn RM, Qi X and Chuang DT: Development
of dihydroxyphenyl sulfonylisoindoline derivatives as
liver-targeting pyruvate dehydrogenase kinase inhibitors. J Med
Chem. 60:1142–1150. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hanson BJ, Capaldi RA, Marusich MF and
Sherwood SW: An immunocytochemical approach to detection of
mitochondrial disorders. J Histochem Cytochem. 50:1281–1288. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gopal K, Almutairi M, Al Batran R, Eaton
F, Gandhi M and Ussher JR: Cardiac-specific deletion of pyruvate
dehydrogenase impairs glucose oxidation rates and induces diastolic
dysfunction. Front Cardiovasc Med. 5:172018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sidhu S, Gangasani A, Korotchkina LG,
Suzuki G, Fallavollita JA, Canty JM Jr and Patel MS:
Tissue-specific pyruvate dehydrogenase complex deficiency causes
cardiac hypertrophy and sudden death of weaned male mice. Am J
Physiol Heart Circ Physiol. 295:H946–H952. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu CY, Tso SC, Chuang JL, Gui WJ, Lou M,
Sharma G, Khemtong C, Qi X, Wynn RM and Chuang DT: Targeting
hepatic pyruvate dehydrogenase kinases restores insulin signaling
and mitigates ChREBP-mediated lipogenesis in diet-induced obese
mice. Mol Metab. 12:12–24. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Small L, Brandon AE, Quek LE, Krycer JR,
James DE, Turner N and Cooney GJ: Acute activation of pyruvate
dehydrogenase increases glucose oxidation in muscle without
changing glucose uptake. Am J Physiol Endocrinol Metab.
315:E258–E266. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Perry RJ, Samuel VT, Petersen KF and
Shulman GI: The role of hepatic lipids in hepatic insulin
resistance and type 2 diabetes. Nature. 510:84–91. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gudiksen A, Bertholdt L, Stankiewicz T,
Tybirk J, Plomgaard P, Bangsbo J and Pilegaard H: Effects of
training status on PDH regulation in human skeletal muscle during
exercise. Pflugers Arch. 469:1615–1630. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou YP, Priestman DA, Randle PJ and Grill
VE: Fasting and decreased B cell sensitivity: Important role for
fatty acid-induced inhibition of PDH activity. Am J Physiol.
270:E988–E994. 1996.PubMed/NCBI
|
|
33
|
Andersson KB, Winer LH, Mørk HK, Molkentin
JD and Jaisser F: Tamoxifen administration routes and dosage for
inducible Cre-mediated gene disruption in mouse hearts. Transgenic
Res. 19:715–725. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Valny M, Honsa P, Kirdajova D, Kamenik Z
and Anderova M: Tamoxifen in the mouse brain: Implications for
Fate-Mapping studies using the Tamoxifen-Inducible Cre-loxP system.
Front Cell Neurosci. 10:2432016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Grosbellet E, Dumont S, Schuster-Klein C,
Guardiola-Lemaitre B, Pevet P, Criscuolo F and Challet E: Circadian
phenotyping of obese and diabetic db/db mice. Biochimie.
124:198–206. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dugan LL, You YH, Ali SS, Diamond-Stanic
M, Miyamoto S, DeCleves AE, Andreyev A, Quach T, Ly S, Shekhtman G,
et al: AMPK dysregulation promotes diabetes-related reduction of
superoxide and mitochondrial function. J Clin Invest.
123:4888–4899. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Miki Y, Tanji K, Mori F, Kakita A,
Takahashi H and Wakabayashi K: Alteration of mitochondrial protein
PDHA1 in Lewy body disease and PARK14. Biochem Biophys Res Commun.
489:439–444. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
American Diabetes Association: (2)
Classification and diagnosis of diabetes. Diabetes Care. 38
(Suppl):S8–S16. 2015. View Article : Google Scholar :
|
|
40
|
Karaa A and Goldstein A: The spectrum of
clinical presentation, diagnosis, and management of mitochondrial
forms of diabetes. Pediatr Diabetes. 16:1–9. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Pastors JG, Warshaw H, Daly A, Franz M and
Kulkarni K: The evidence for the effectiveness of medical nutrition
therapy in diabetes management. Diabetes Care. 25:608–613. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bugger H and Abel ED: Molecular mechanisms
of diabetic cardiomyopathy. Diabetologia. 57:660–671. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mayers RM, Leighton B and Kilgour E: PDH
kinase inhibitors: A novel therapy for Type II diabetes? Biochem
Soc Trans. 33:367–370. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu YQ, Moibi JA and Leahy JL: Chronic
high glucose lowers pyruvate dehydrogenase activity in islets
through enhanced production of long chain acyl-CoA: Prevention of
impaired glucose oxidation by enhanced pyruvate recycling through
the malate-pyruvate shuttle. J Biol Chem. 279:7470–7475. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Park S, Jeon JH, Min BK, Ha CM, Thoudam T,
Park BY and Lee IK: Role of the pyruvate dehydrogenase complex in
metabolic remodeling: Differential pyruvate dehydrogenase complex
functions in metabolism. Diabetes Metab J. 42:270–281. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu Z, Yu M, Fei B, Fang X, Ma T and Wang
D: miR-21-5p targets PDHA1 to regulate glycolysis and cancer
progression in gastric cancer. Oncol Rep. 40:2955–2963.
2018.PubMed/NCBI
|
|
47
|
Gray LR, Tompkins SC and Taylor EB:
Regulation of pyruvate metabolism and human disease. Cell Mol Life
Sci. 71:2577–2604. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jitrapakdee S, St Maurice M, Rayment I,
Cleland WW, Wallace JC and Attwood PV: Structure, mechanism and
regulation of pyruvate carboxylase. Biochem J. 413:369–387. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Branda CS and Dymecki SM: Talking about
arevolution: The impact of site-specific recombinases on genetic
analyses in mice. Dev Cell. 6:7–28. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yang F, Liu C, Chen D, Tu M, Xie H, Sun H,
Ge X, Tang L, Li J, Zheng J, et al: CRISPR/Cas9-loxP-mediated gene
editing as a novel site-specific genetic manipulation tool. Mol
Ther Nucleic Acids. 7:378–386. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Aguirre AJ, Meyers RM, Weir BA, Vazquez F,
Zhang CZ, Ben-David U, Cook A, Ha G, Harrington WF, Doshi MB, et
al: Genomic copy number dictates a gene-independent cell response
to CRISPR/Cas9 targeting. Cancer Discov. 6:914–929. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Srinivasan M, Choi CS, Ghoshal P, Pliss L,
Pandya JD, Hill D, Cline G and Patel MS: ß-cell-specific pyruvate
dehydrogenase deficiency impairs glucose-stimulated insulin
secretion. Am J Physiol Endocrinol Metab. 299:E910–E917. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Nicholls LI, Ainscow EK and Rutter GA:
Glucose-stimulated insulin secretion does not require activation of
pyruvate dehydrogenase: Impact of adenovirus-mediated
overexpression of PDH kinase and PDH phosphate phosphatase in
pancreatic islets. Biochem Biophys Res Commun. 291:1081–1088. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Xu J, Han J, Long YS, Epstein PN and Liu
YQ: The role of pyruvate carboxylase in insulin secretion and
proliferation in rat pancreatic beta cells. Diabetologia.
51:2022–2030. 2008. View Article : Google Scholar : PubMed/NCBI
|