Introduction
Peripheral arterial disease (PAD) remains a major
clinical problem that can lead to disability, limb loss and is
potentially fatal in the elderly (1,2).
Growth factors and extracellular matrix proteins are important
mediators in the pathogenesis of PAD and the development of
restenosis secondary to intimal hyperplasia (IH) after balloon
angioplasty (3,4). IH is a complex process that begins by
platelet activation (5,6). Platelets then bind to the area of
vascular injury releasing many agents including thrombospondin-1
(TSP-1) and platelet derived growth factor (PDGF) (5), which in turn cause vascular smooth
muscle (VSMC) migration into the area of injury where they begin to
proliferate and produce extracellular matrix (5). All of these processes clearly
contribute to IH by regulating the arterial response to injury.
Recently heat shock protein 90 (HSP90) has also been implicated in
pathologic vascular remodeling (7).
HSP90 is a molecular chaperone that binds many
signaling proteins regulating their final maturation (8). HSP90 is ubiquitously expressed and is
important for normal cell function (8). However, aberrant activation of HSP90
can result in increased cell migration and proliferation (8). Inhibition of HSP90 has been examined
in states of aberrant cell growth such as cancer (8). Several different HSPs have been
detected in atherosclerosis, and HSP90, in particular, is strongly
expressed in atherosclerotic plaque (9). However, the role HSP90 plays in IH
after balloon angioplasty is unknown. At least one previous study
has shown that blocking HSP90 inhibits VSMC proliferation and
migration (9). As the development
of IH depends on VSMC migration and proliferation, inhibition of
HSP90 may be an efficacious approach to this important clinical
problem. The quintessential HSP90 inhibitor is the natural product
geldanamycin; however, geldanamycin exhibits a relatively high
toxicity (10,11). Several derivatives of geldanamycin
have been created that have significantly less toxicity and are in
clinical trials for cancer therapy, two of these are
17-N-allylamino-17-demethoxygeldanamycin (17-AAG) and
17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG)
(10,12).
In the present study, we sought to understand the
contribution of HSP90 in VSMC physiologic processes and IH
formation after arterial balloon injury. We hypothesized that HSP90
inhibition would reduce agonist-induced VSMC proliferation and
migration, and that localized HSP90 inhibition would inhibit
post-angioplasty IH formation.
Materials and methods
Materials
Smooth muscle cell growth and basal medium were
purchased from Cell Applications, Inc. (San Diego, CA). Trypsin and
trypsin neutralizing solution were purchased from Cell Applications
Inc. (San Diego, CA). 17-AAG and 17-DMAG were purchased from Tocris
Bioscience (Bristol, UK). Fibronectin and PDGF were purchased from
R&D Systems (Minneapolis, MN).
Cell culture
Human aortic VSMCs (Cell Applications, Inc., San
Diego, CA, USA) were used in early passage (P3-P5). Cells were made
quiescent by incubation in serum free media (SFM) for 48 h at 37°C
in a tissue culture incubator (5% CO2).
Cell viability
Cell viability was determined with the Trypan Blue
exclusion assay (Thermo Fisher Scientific, Waltham, MA) using the
Countess Cell counter (Thermo Fisher Scientific, Waltham, MA)
according to the manufacturer's instructions. All experiments were
performed in triplicate studying 17-AAG or 17-DMAG at three
different concentrations (100 nM, 10 or 30 µM). The doses chosen
have been previously shown to be effective (13).
Migration assay
Chemotaxis was assessed in quiescent human VSMCs
incubated in a 96-well FluoroBlok migration plate (One Riverfront
Plaza, Corning, NY) according to the manufacturer's instructions
with 50,000 calcein AM labeled cells per well. Experiments were
performed in triplicate. Cells were exposed to the chemoattractant
agents: SFM (negative control), PDGF 20 ng/ml (positive control) or
Fibronectin 20 µg/ml (positive control), which were placed in the
bottom chamber. VSMCs treated with 17-AAG (100 nM, 10 or 30 µM) or
17-DMAG (100 nM, 10 µM or 30 µM) for 2 h at 37°C in a tissue
culture incubator (5% CO2) in addition to SFM (negative
control) and PDGF or fibronectin (positive control) were placed in
the top chamber. Migrated cells were measured on a fluorescent
plate reader with excitation of 480 nm and emission of 520 nm. Data
is expressed as relative fluorescence units (RFU).
Proliferation assay
Cell proliferation was assessed in growth-arrested
VSMCs incubated in a 96 well plate with 5,000 cells/well.
Experiments were performed in triplicate. Quiescent cells were
exposed to serum free media (SFM), PDGF (20 ng/ml) or Fibronectin
(20 µg/ml), 17-AAG (100 nM, 10 or 30 µM) or 17-DMAG (100 nM, 10 or
30 µM). VSMCs were incubated for 72 h at 37°C in a tissue culture
incubator (5% CO2). Proliferation was measured using a
MTS tetrazolium absorbance assay (Cell Titer 96 Aqueous Cell
Proliferation Assay, Promega, Madison, WI) according to
manufacturer's instructions.
Carotid artery balloon injury
model
This study was approved by the Syracuse VA IACUC and
the animal care complied with the Guide for the Care and Use of
Laboratory Animals. Sprague-Dawley rats at 10–12 weeks of age
(Harlan Laboratories, Indianapolis, IN) were randomized into four
groups (8–10 animals per group)-control, saline control,
intraluminal 17-DMAG or topical 17-DMAG. 17-DMAG was selected as
the HSP90 inhibitor for the in vivo work because of ease of
use (water soluble) and equivalent lack of toxicity to cells and
inhibition of VSMC proliferation in vitro. Rats were
anesthetized using inhaled isoflurane at 5% and then maintained at
2% for the surgery. The left common carotid artery (CCA) and its
bifurcation were exposed via carotid artery cutdown. A 2F Fogarty
catheter (Edwards Lifesciences, Irvine, CA) was placed into the CCA
via the external carotid artery and the balloon was inflated (5
atmospheres for 5 min). Then a polyethylene (PE) catheter attached
to a pump was placed into the vessel and the vessel gently
distended with either saline (0.9%, PH=8.5) with flow rate 10
ml/min in saline control and topical treatment groups or 17-DMAG
(500 nM) suspended in saline for 20 min (intraluminal treatment
group). The catheter was withdrawn, and the external carotid artery
ligated. For the topical group, 17-DMAG (500 nM) in 20% pluronic
gel (F-127, Anaspec, Fermont, CA) was placed on top of the vessel
prior to closing the incision. We chose 17-DMAG for the in
vivo part of the study over 17-AAG as it is more water soluble
and has better bioavailability than 17-AAG (12) Thereby, delivery formulations of
17-DMAG do not require organic solubilizing agents which have their
own limitations. After 14 days, the animals were euthanized and
bilateral CCAs were perfusion fixed, harvested, sectioned, stained
with hematoxylin and eosin and morphometric analysis was performed
to assess severity of IH. Two groups have been assigned as control
groups for this particular study, isolated angioplasty injury
(control) and angioplasty plus intraluminal saline infusion (saline
control). The reason behind using dual control groups is to exclude
the assumption of concomitant saline injury to the vessel wall.
Morphometric analysis
The left CCA was fixed in paraffin, sectioned,
stained with hematoxylin and eosin for cross- sectional
morphometric analysis. The medial thickness (M) was quantified
between the internal (IEL) and external elastic (EEL) laminae,
i.e., M=EEL-IEL. The intimal thickness (I) was measured between the
endothelial cell layer and the internal elastic lamina, i.e.,
I=IEL-Luminal area. The ratio of intimal area to medial area was
calculated as the intimal thickness divided by the combined intimal
and medial thickness (14).
Statistical analysis
Data are presented as the mean ± standard error of
the mean. Data was tested via ANOVA with post-hoc testing with
Fisher's PLSD in StatView (SAS Institute, Cary, NC) or a Student's
t-test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Cell viability
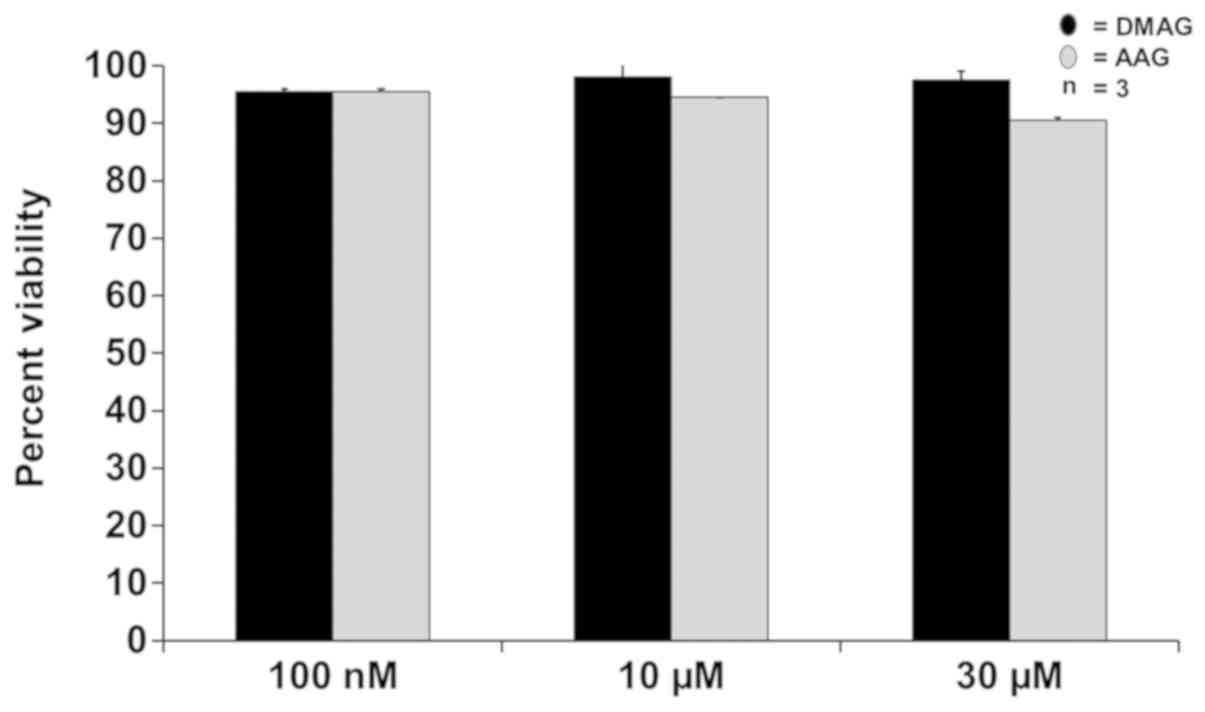
17-AAG and 17-DMAG had no effect on VSMC viability.
After 24-hour incubation, VSMC viable cell count did not
significantly change in all tested concentrations (100 nM, 10 µM or
30 µM) of either drug. At 100 nM, the cells were 95.5% viable with
both 17-AAG and 17-DMAG treatment. At 10 µM the viability was 94.5%
for 17-AAG and 98% for 17-DMAG. At the highest tested dose-30
µM-cell viability was determined to be 90.5% for 17-AAG and 97.5%
for 17-DMAG (Fig. 1).
Migration
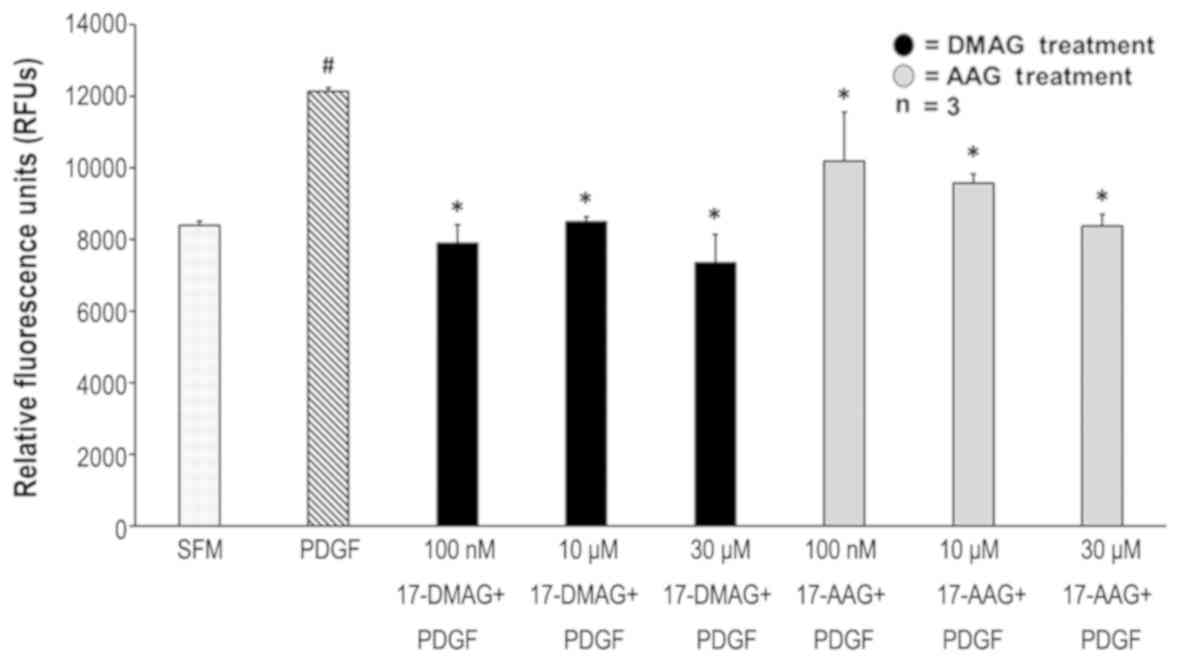
17-AAG and 17-DMAG decreased VSMC migration in
response to PDGF. PDGF at a concentration of 20 ng/ml increased
smooth muscle migration by 30% compared to SFM (P=0.0001). At the
concentration of 100 nM, 17-AAG decreased migration by 16%
(P=0.0048) while 17-DMAG decreased migration by 34.9% (P=0.0003).
Ten µM 17-AAG and 17-DMAG decreased migration by 21.1% (P=0.003)
and 29.9% (P=0.0002) respectively. At 30 µM, 17-AAG and 17-DMAG
were found to decrease migration by 30.8% (P=0.0003) and 39.4%
(P=0.0003) respectively. There was no difference in inhibition of
VSMC migration between 17-AAG and 17-DMAG (Fig. 2).
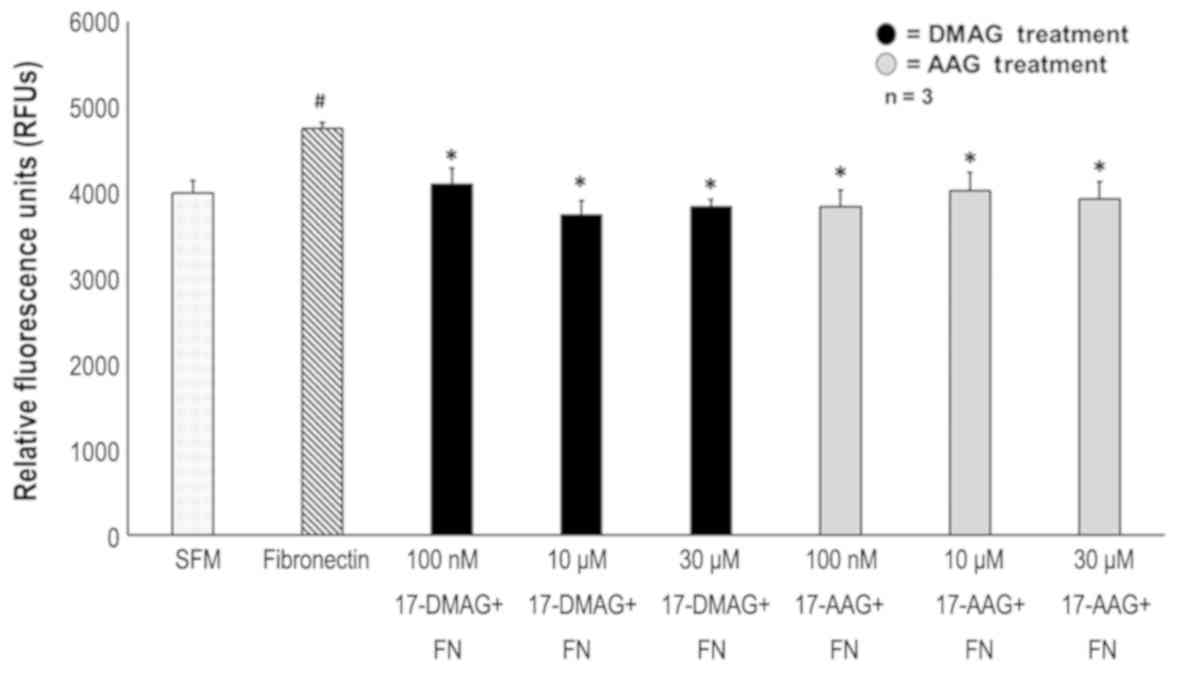
17-AAG and 17-DMAG decreased VSMC migration in
response to Fibronectin. Fibronectin at a concentration of 20 µg/ml
increased smooth muscle cell migration by 16% compared to SFM
(P=0.01). At a concentration of 100 nM, 17-AAG decreased migration
by 19.21% (P=0.015) while 17-DMAG decreased migration by 13.78%
(P=0.03). Ten µM 17-AAG and 17-DMAG decreased smooth muscle
migration by 15.35 (P=0.01) and 21.3% (P=0.01) respectively. At 30
µM, 17-AAG and 17-DMAG were found to decrease migration by 17.3
(P=0.004) and 19.2% (P=0.01) respectively. There was no difference
in inhibition of VSMC migration between 17-AAG and 17-DMAG
(Fig. 3).
Proliferation
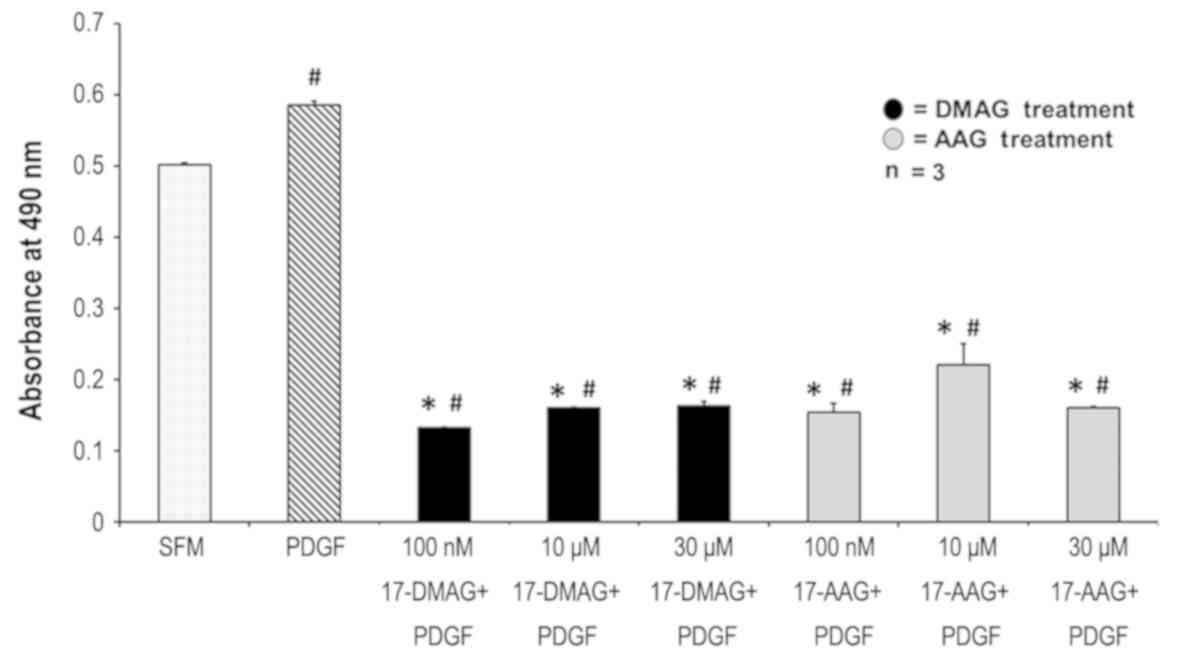
17-AAG and 17-DMAG decreased VSMC proliferation in
response to PDGF. PDGF at a concentration of 20 ng/ml increased
smooth muscle cell proliferation by 15.42% compared to SFM
(P=0.006). At a concentration of 100 nM, 17-AAG decreased
proliferation by 73.9% (P=0.001) while 17-DMAG decreased
proliferation by 77.5% (P=0.0002). Ten µM 17-AAG and 17-DMAG
decreased proliferation by 62.6% (P=0.01) and 72.8% (P=0.0002)
respectively. At 30 µM, 17-AAG and 17-DMAG were found to decrease
proliferation by 72.8% (P=0.0002) and 72.4% (P=0.0006)
respectively. There was no difference in VSMC proliferation between
17-AAG and 17-DMAG by PDGF (Fig.
4).
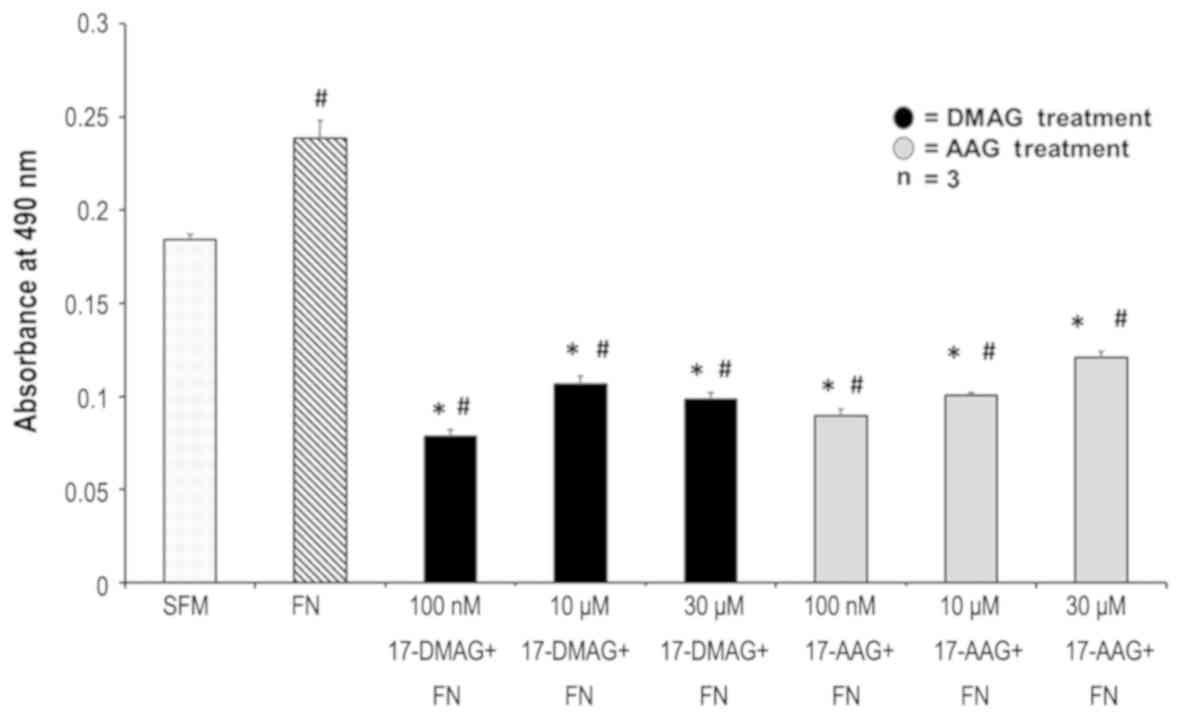
17-AAG and 17-DMAG decreased VSMC proliferation in
response to Fibronectin. Fibronectin at a concentration of 20 µg/ml
increased smooth muscle proliferation by 25% absorbance at 490 nm
from 0.18 to 0.24 (P<0.0001). At a concentration of 100 nM,
17-AAG decreased proliferation by 62.9% (decreased absorbance to
0.089, P=0.0001) while 17-DMAG decreased proliferation by 67.5%
(decreased absorbance to 0.078, P=0.0001). At 10 µM 17-AAG and
17-DMAG decreased proliferation by 58.1% (P=0.0001) and 55.6%
(P=0.0001) respectively. At 30 µM, 17-AAG and 17-DMAG were found to
decrease proliferation by 49.6% (P=0.0001) and 59% (P=0.0001)
respectively. There was no difference in the degree of inhibition
of VSMC proliferation between 17-AAG and 17-DMAG (Fig. 5).
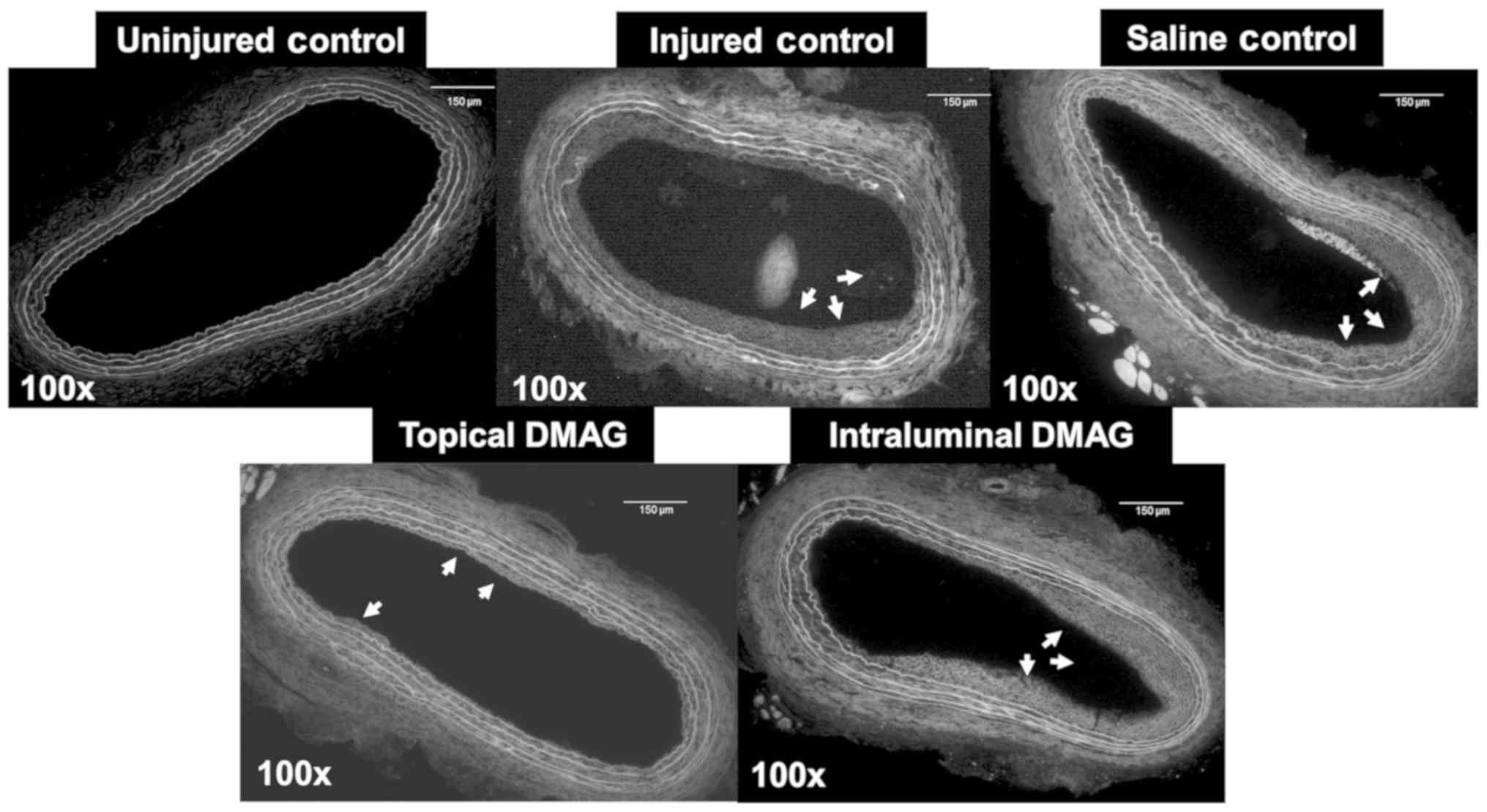
Morphometric analysis
Topical 17-DMAG reduces IH after
arterial injury in Sprague-Dawley rats
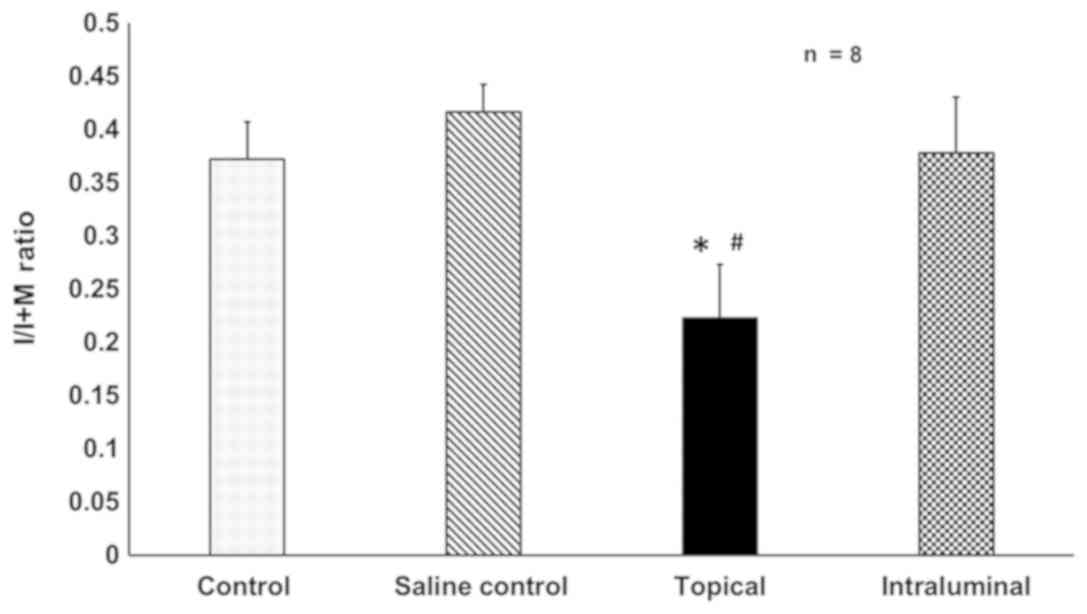
Rats that were treated with adventitial 17-DMAG
dissolved in 20% pluronic gel had 46.5% less IH when compared to
the saline control group (0.22±0.02 vs. 0.42±0.05, P=0.001).
Intra-luminal delivery of 17-DMAG had no effect on IH, with the
control group I/M ratio being 0.41±0.22 and the intra-luminal group
being 0.38±0.05. No difference was determined between control and
saline control IH (Figs. 6 and
7).
Discussion
IH is a major cause of long-term failure after
either open or endovascular revascularization (15). Despite all the technological
advances in endovascular revascularization modalities, restenosis
remains a significant risk, particularly within the first 12 months
after angioplasty (16). A fuller
understanding of the cellular processes behind VSMC migration and
proliferation (key processes in IH development), is critical to
developing new therapies to attenuate restenosis. The present study
examined the role of HSP90 in IH after balloon arterial injury and
the potential therapeutic role of HSP90 inhibitors, 17-AAG and
17-DMAG, in attenuating or even preventing IH. Our results
demonstrate that 17-DMAG dissolved in 20% pluronic gel, applied
topically on the adventitia of the common carotid artery of
Sprague-Dawley rats reduced the amount of IH after injury. We
further found that intra-luminal delivery of 17-DMAG dissolved in
saline to the injured artery didn't attenuate the IH in the
affected animals.
HSPs are a family of conserved intracellular
proteins that are involved in proper protein folding which helps
both in protein function and intracellular targeting (17). HSP90 is a common member of the HSP
family that was found to be overexpressed in atherosclerotic plaque
in patients and is associated with plaque instability (18). The essential function of HSP90 is
believed to be the interaction with proteins known as clients
(19). Many of the clients are
protein kinases and transcription factors that regulate cell
differentiation, proliferation and apoptosis (20,21).
Proliferation of VSMCs is crucial in the
pathogenesis of IH and restenosis (9,22,23).
After arterial injury, VSMCs, originating primarily from the
adventitia, migrate into the area of vascular injury and
proliferate, thus initiating the process of IH. HSP90 has been
shown to be involved in the proliferation of many cell types
including VSMCs (21,24–26).
The current study demonstrates that HSP90 does play a broad role in
VSMC proliferation, as 17-AAG and 17-DMAG were able to inhibit VSMC
proliferation in response to two functionally different agonists,
PDGF and fibronectin.
Geldanamycin, was the first HSP90 inhibitor to be
developed; however, this inhibitor has poor solubility and is
highly toxic to various human cell types (27). Two main geldanamycin
derivatives-17-AAG and 17-DMAG-have less toxicity and have been
tested in phase II/III clinical trials as novel antineoplastic
agents (9). In the present study,
we needed to determine if either geldanamycin derivative has the
same cellular toxic effects as geldanamycin (11,27,28).
Our data shows that VSMC viability was determined to be above 90%
at any tested concentration of 17-AAG or 17-DMAG used. Both 17-AAG
and 17-DMAG work by inhibiting the binding of ATP to HSP90, which
is necessary for HSP90 function (18).
The current study further investigated whether HSP90
contributes to the development of IH after arterial injury in an
animal model. At least one other study has demonstrated the HSP90
inhibition can attenuate IH (7).
However in that study the authors used a peptide inhibitor which
can be immunogenic in humans, our study used FDA approved HSP90
inhibitors that have been shown to be safe in humans (7,12,29).
Given that our in vitro data demonstrated no differences in
efficacy between 17-AAG and 17-DMAG, we chose to use 17-DMAG in
vivo as this drug is more water soluble and therefore easier to
use. We found topical (periadventitial) delivery of 17-DMAG
significantly reduced the formation of IH. The specific downstream
effects of HSP90 that account for its role in IH development are
unknown. Recent studies showed that HSP90 inhibition downregulates
cyclinD3, PCNA, and pRb, leading to cell cycle arrest which could
contribute to the antiproliferative effect of either 17-DMAG or
17-AAG (27). These effects and
their role in IH will need further study. Our findings indicate
that HSP90 is a vital factor in the process of post-arterial
injury, including VSMC migration, proliferation and IH.
The present study does have limitations that will
need further study. Since HSP90 inhibition affects several
intracellular proteins, kinases and intracellular signaling
pathways at the same time, other pathophysiological processes such
as apoptosis, angiogenesis or oxidative stress could also be
affected by the drug used in the study and warrants further
investigation to accurately clarify the role of HSP90 in human
arterial disease. Another limitation of our study is how to
optimize the delivery route of the HSP90 inhibitor agent, as in our
model only the periadventitial topical route was found to
significantly decrease IH. This method may have been effective
because of the longer residence time of the inhibitor with the
vessel. Periadventitial delivery is relevant for open
revascularization, but may not be practical for endovascular
procedures, mitigating a need for further technological
advancements to optimize the intraluminal 17-DMAG delivery method.
The method used for intraluminal delivery in the current study may
have been inadequate for absorbance by the vessel wall.
Nanotechnology for the improved delivery of 17-DMAG could be one
option. Endovascular adventitial delivery with an infusion device
through the arterial wall (e.g., Bullfrog Micro-Infusion Device,
Mercator MedSystems) would be an alternative method to endovascular
delivery.
In conclusion, the present study demonstrates the
significance of HSP90 in the development of IH. In addition, these
studies showed no toxic effects of either 17-AAG or 17-DMAG on VSMC
viability, indicating possible safety of these agents if used as
anti-intimal hyperplastic agents in revascularization procedures.
Interestingly, both agents were able to markedly reduce
PDGF-induced and fibronectin-induced VSMC migration and
proliferation. Topical periadventitial delivery of the HSP90
inhibitor was found to attenuate post-balloon injury IH. The
current study provides a foundation for future explorations into
potential therapeutic targets for reducing the damaging effects of
IH.
Acknowledgements
The abstract was presented at the 32nd Eastern
Vascular Society annual meeting Sep. 6-Sep 8 2018 in Washington,
D.C. and published as abstract no. 3734 in J Vasc Surg 68 e20:
2018.
Funding
The present study was supported by a grant obtained
from the United States Department of Defense (grant no. PR
152338).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MMK performed the experiments, analyzed the data and
prepared the manuscript for submission in this work. FM and MD
performed the experiments and analyzed data. DB performed all
surgical procedures. VG assisted in experimental design, data
interpretation and manuscript editing. KGM was the principal
investigator of the grant, who designed the present study,
interpreted the data, and wrote and edited the manuscript.
Ethics approval and consent to
participate
All animal studies in this manuscript were approved
by The Syracuse Department of Veterans Affairs Institutional Animal
Care and Use Committee in accordance with AAALAC guidelines.
Patient consent for publication
Not applicable
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Valentine RJ, Grayburn PA, Eichhorn EJ,
Myers SI and Clagett GP: Coronary artery disease is highly
prevalent among patients with premature peripheral vascular
disease. J Vasc Surg. 19:668–674. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McDivitt JD, Braun M and Kassop D:
Cardiovascular disease: Lower extremity peripheral artery disease.
FP Essent. 479:11–15. 2019.PubMed/NCBI
|
|
3
|
Asakura Y, Suzuki M, Nonogi H, Haze K,
Sato A, Inada H, Okuda Y, Yamashita K and Harano Y: Restenosis
after percutaneous transluminal coronary angioplasty in patients
with non-insulin-dependent diabetes mellitus (NIDDM). J Cardiovasc
Risk. 5:331–334. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Goel SA, Guo LW, Liu B and Kent KC:
Mechanisms of post- intervention arterial remodelling. Cardiovasc
Res. 96:363–371. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Weintraub WS: The pathophysiology and
burden of restenosis. Am J Cardiol. 100:3K–9K. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Broos K, Feys HB, De Meyer SF,
Vanhoorelbeke K and Deckmyn H: Platelets at work in primary
hemostasis. Blood Rev. 25:155–167. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hoel AW, Yu P, Nguyen KP, Sui X, Plescia
J, Altieri DC and Conte MS: Mitochondrial heat shock protein-90
modulates vascular smooth muscle cell survival and the vascular
injury response in vivo. Am J Pathol. 181:1151–1157. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lianos GD, Alexiou GA, Mangano A, Mangano
A, Rausei S, Boni L, Dionigi G and Roukos DH: The role of heat
shock proteins in cancer. Cancer Lett. 360:114–118. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim J, Jang SW, Park E, Oh M, Park S and
Ko J: The role of heat shock protein 90 in migration and
proliferation of vascular smooth muscle cells in the development of
atherosclerosis. J Mol Cell Cardiol. 72:157–167. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lang SA, Klein D, Moser C, Gaumann A,
Glockzin G, Dahlke MH, Dietmaier W, Bolder U, Schlitt HJ, Geissler
EK and Stoeltzing O: Inhibition of heat shock protein 90 impairs
epidermal growth factor-mediated signaling in gastric cancer cells
and reduces tumor growth and vascularization in vivo. Mol Cancer
Ther. 6:1123–1132. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hermane J, Eichner S, Mancuso L, Schröder
B, Sasse F, Zeilinger C and Kirschning A: New geldanamycin
derivatives with anti Hsp properties by mutasynthesis. Org Biomol
Chem. 17:5269–5278. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mellatyar H, Talaei S,
Pilehvar-Soltanahmadi Y, Barzegar A, Akbarzadeh A, Shahabi A,
Barekati-Mowahed M and Zarghami N: Targeted cancer therapy through
17-DMAG as an Hsp90 inhibitor: Overview and current state of the
art. Biomed Pharmacother. 102:608–617. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sausville EA, Tomaszewski JE and Ivy P:
Clinical development of 17-allylamino, 17-demethoxygeldanamycin.
Curr Cancer Drug Targets. 3:377–383. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang D, Zhuang J, Peng W, Lu Y, Liu H,
Zhao Q, Chi C, Li X, Zhu G, Xu X, et al: Phospholipase Cγ1 mediates
intima formation through Akt-Notch1 signaling independent of the
phospholipase activity. J Am Heart Assoc. 6:2017. View Article : Google Scholar
|
|
15
|
Collins TC, Beyth RJ, Nelson DB, Petersen
NJ, Suarez- Almazor ME, Bush RL, Hirsch AT and Ashton CM: Process
of care and outcomes in patients with peripheral arterial disease.
J Gen Intern Med. 22:942–948. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schillinger M and Minar E: Restenosis
after percutaneous angioplasty: The role of vascular inflammation.
Vasc Health Risk Manag. 1:73–78. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu Q, Metzler B, Jahangiri M and Mandal K:
Molecular chaperones and heat shock proteins in atherosclerosis. Am
J Physiol Heart Circ Physiol. 302:H506–H514. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Madrigal-Matute J, López-Franco O,
Blanco-Colio LM, Muñoz-García B, Ramos-Mozo P, Ortega L, Egido J
and Martín-Ventura JL: Heat shock protein 90 inhibitors attenuate
inflammatory responses in atherosclerosis. Cardiovasc Res.
86:330–337. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mayer MP: Gymnastics of molecular
chaperones. Mol Cell. 39:321–331. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pearl LH and Prodromou C: Structure and
mechanism of the Hsp90 molecular chaperone machinery. Annu Rev
Biochem. 75:271–294. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mjahed H, Girodon F, Fontenay M and
Garrido C: Heat shock proteins in hematopoietic malignancies. Exp
Cell Res. 318:1946–1958. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Helkin A, Maier KG and Gahtan V:
Thrombospondin-1, −2 and −5 have differential effects on vascular
smooth muscle cell physiology. Biochem Biophys Res Commun.
464:1022–1027. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Doran AC, Meller N and McNamara CA: Role
of smooth muscle cells in the initiation and early progression of
atherosclerosis. Arterioscler Thromb Vasc Biol. 28:812–819. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu DH, Yuan HY, Cao CY, Gao ZP, Zhu BY,
Huang HL and Liao DF: Heat shock protein 90 acts as a molecular
chaperone in late-phase activation of extracellular
signal-regulated kinase 1/2 stimulated by oxidative stress in
vascular smooth muscle cells. Acta Pharmacol Sin. 28:1907–1913.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Boucherat O, Peterlini T, Bourgeois A,
Nadeau V, Breuils- Bonnet S, Boilet-Molez S, Potus F, Meloche J,
Chabot S, Lambert C, et al: Mitochondrial HSP90 accumulation
promotes vascular remodeling in pulmonary arterial hypertension. Am
J Respir Crit Care Med. 198:90–103. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang GK, Li SH, Zhao ZM, Liu SX, Zhang GX,
Yang F, Wang Y, Wu F, Zhao XX and Xu ZY: Inhibition of heat shock
protein 90 improves pulmonary arteriole remodeling in pulmonary
arterial hypertension. Oncotarget. 7:54263–54273. 2016.PubMed/NCBI
|
|
27
|
Li YP, Chen JJ, Shen JJ, Cui J, Wu LZ,
Wang Z and Li ZR: Synthesis and biological evaluation of
geldanamycin analogs against human cancer cells. Cancer Chemother
Pharmacol. 75:773–782. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Supko JG, Hickman RL, Grever MR and
Malspeis L: Preclinical pharmacologic evaluation of geldanamycin as
an antitumor agent. Cancer Chemother Pharmacol. 36:305–315. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Egorin MJ, Lagattuta TF, Hamburger DR,
Covey JM, White KD, Musser SM and Eiseman JL: Pharmacokinetics,
tissue distribution, and metabolism of
17-(dimethylaminoethylamino)-17-demethoxygeldanamycin (NSC 707545)
in CD2F1 mice and Fischer 344 rats. Cancer Chemother Pharmacol.
49:7–19. 2002. View Article : Google Scholar : PubMed/NCBI
|