Introduction
Gait disorders are commonly caused by peripheral
nerve and muscle weakness due to the external environment or
hereditary factors (1). Skeletal
muscles are connected to the motor axons of peripheral nerves
through neuromuscular junctions that control body movement
(2). Peripheral neuropathy causes
damage to nerves and muscles, leading to gait disturbances
(3–5).
Previous studies have reported that hereditary
peripheral neuropathy causes proximal and distal muscle atrophy due
to axonal loss in motor nerves in murine models (6–8).
However, the majority of current neuromuscular disease therapies
are focused on regenerating axon nerves and myelination, not on
muscle regeneration (3,9–11).
For instance, a candidate drug for Schwann cell myelination
improves the phenotype of peripheral neuropathy, motor nerve
electrophysiology and locomotor coordination (10,11).
However, the drug does not directly improve muscle regeneration or
gait pattern.
In vivo gene targeting therapies provide
stronger therapeutic effects in the treatment of hereditary
diseases compared with drug therapies (12,13).
A clustered regularly interspaced short palindromic repeats
(CRISPR)/caspase 9-based therapy demonstrated a significant
improvement in peripheral neuropathy following direct intraneural
CRISPR-ribonucleoprotein injection into the sciatic nerve
immediately distal to the sciatic notch in a C2C12 murine model
with Charcot-Marie-Tooth disease type 1A (CMT1A) (13). The peripheral myelin protein 22
(PMP22)-overexpressing murine model C22 presents with severe
demyelinating neuropathy, including muscle weakness and sensory
depression, similar to symptoms exhibited by patients with CMT 1A.
These mice usually develop CMT symptoms at 3 weeks of age (14). However, in the previous study,
treatment for neuropathy did not improve gait regeneration
(12).
Myostatin (Mstn) serves crucial roles in muscle
regeneration and embryonic skeletal myogenesis (15,16).
Previous studies have demonstrated that Mstn inhibition is a strong
potential candidate for the treatment of neuromuscular disease,
including Duchenne muscular dystrophy (17–19).
These results strongly indicated that adeno-associated
virus-mediated Mstn inhibition improved muscle regeneration in
terms of muscle mass and size. Another previous study reported that
insulin-like growth factor-1 stimulates muscle hypertrophy by
suppressing the Mstn signaling pathway (20). Therefore, it has been hypothesized
that regulating Mstn expression may treat neuromuscular diseases,
including amyotrophic lateral sclerosis (ALS), neuromuscular
junction disorders and peripheral neuropathy.
In murine models of diseases, conventional
behavioral tests, including the rotarod performance test, hind-limb
grip strength test and footprint analysis, are useful tools for
examining therapeutic effects on the central nervous system and
peripheral nervous system (PNS) (7–11).
Animal studies have attempted to analyze walking patterns using
digital recording systems (21–23).
For instance, in an ALS disease mouse model (23), the DigiGait™ Imaging System
measures step length and >30 gait-related traits, including
stance duration, swing duration and stance width (21,22).
Therefore, the DigiGait™ Imaging System may be a valuable tool for
developing novel therapies to treat abnormal gait diseases.
The present study aimed to improve gait disorders
using short hairpin (sh)RNA shLenti-Mstn therapy and evaluate
effectiveness in the CMT1A mouse model using a digital gait system.
RNA-based gene silencing, specifically using shRNAs, is a valuable
tool and has clinical applications for target validation and
therapeutics (24,25).
Materials and methods
Cell culture
Mouse myoblast C2C12 (cat. no. CRL-1772) and 293T
(cat. no. CRL-11268) cell lines were purchased from the American
Type Culture Collection. C2C12 and 293T cells were cultured in DMEM
with high glucose supplemented with 10% FBS, 100 U/ml penicillin
and 100 µg/ml streptomycin (all from Thermo Fisher Scientific,
Inc.). Cultures were maintained at 37°C at 5% CO2. C2C12
cells were differentiated into myotubes by replacing the growth
medium with a medium containing 2% horse serum (Thermo Fisher
Scientific, Inc.) with 1% penicillin-streptomycin (Thermo Fisher
Scientific, Inc.) and 1X insulin-transferrin-selenium supplement
(Sigma-Aldrich; Merck KGaA).
In vitro transfection and selection of
allele-specific shRNAs. Allele-specific shRNAs and control shRNAs
were purchased from OriGene Technologies, Inc. A total of five
vectors (one p-green fluorescence protein (GFP)-V-mock and four
different shMstn sequences; 4 µg/well) were transfected into C2C12
myoblasts using Lipofectamine® 3000 (Invitrogen; Thermo
Fisher Scientific, Inc.), according to the manufacturer's protocol.
The four different shRNAs contained the following sequences:
shRNA-Mstn A, 5′-TGGCTCTTGAAGATGACGATTATCACG−3′; B,
5′-ACAATCATTACCATGCCTACAGAGTCTGA-3′; C,
5′-CAACAGTGTTTGTGCAAATCCTGAGACTC-3′; and D,
5′-AATTCCAGCCATGGTAGTAGACCGCTGTG-3′. To differentiate from myoblast
cell to nascent myotubes, growth medium was replaced the following
day and for five additional days with fresh medium containing 2%
horse serum and 1% penicillin-streptomycin in DMEM with high
glucose. At 7 days post-transfection, allelic specificity of the
shRNAs was determined by comparing the expression of mstn
mRNA in each transfected group using reverse
transcription-quantitative PCR (RT-qPCR).
RNA isolation and cDNA synthesis
To quantify mRNA levels in the cell lines and
lentivirus injected tissues, total mRNA was prepared for RT using
an RNeasy Mini kit (Qiagen GmbH), according to the manufacturer's
protocol. mRNA concentrations were determined by optical density as
measured by a spectrophotometer (NanoDrop 2000; Thermo Fisher
Scientific, Inc.). Total mRNA (2 µg) was used for cDNA synthesis
using SuperScript™ II reverse transcriptase (Invitrogen; Thermo
Fisher Scientific, Inc.).
PCR
A total of 1 µl template cDNA, 45 µl PCR SuperMix
(Invitrogen; Thermo Fisher Scientific, Inc.) and 10 pmol (total
volume, 50 µl) of each of the following primers: mstn
forward, 5′-AGTGGATCTAAATGAGGGCAGT-3′ and reverse,
5′-GTTTCCAGGCGCAGCTTAC-3′; GAPDH forward, 5′-TCACCATGGAGAAGGC-3′
and reverse, 5′-GCTAAGCAGTTGGTGCA-3′ were processed using a TP600
thermocycler (Takara Biotechnology Co., Ltd.). PCR products were
confirmed by ethidium bromide staining following electrophoresis on
1% agarose gels. Quantitative analyses of band densities were
performed using a Gel Doc XR imaging system (Bio-Rad Laboratories,
Inc.).
RT-qPCR
A total of 1 µl template cDNA, 5 µl SYBR-Green PCR
master mix (Applied Biosystems; Thermo Fisher Scientific, Inc.) and
10 pmol (total volume, 10 µl) of each primer aforementioned was
amplified were performed using an ABI 7900 PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The following
thermocycling conditions were used for qPCR: 3 min at 95°C,
followed by two-step PCR at 95°C for 15 sec and 60°C for 60 sec for
40 cycles, with fluorescence monitoring at the end of each
elongation step. Relative mRNA expression of target genes was
calculated using the 2−ΔΔCq method (26). GAPDH was utilized as the endogenous
control.
In vitro transduction
Viral particles of shLenti-Mstn GFP were purchased
from Sirion Biotech GmbH based on the shRNA-Mstn A. To identify
shLenti-Mstn GFP expression, 0.5 µl viral particles (VPs;
8×108 VP/ml) were transduced into 293T cells. At 3 days
post-transduction, GFP expression in transfected cells was
confirmed using a fluorescence microscope (Ti2U FL; Nikon
Corporation) and photographed using NIS-Element software (version
5.01.00; Nikon Corporation).
Western blotting
Proteins were collected from differentiated C2C12
cells using RIPA lysis buffer (Biosesang) supplemented with a 1X
protease/phosphatase inhibitor cocktail (cat. no. 5871; Cell
Signaling Technology, Inc.). Cell lysates were centrifuged at
13,000 × g for 20 min at 4°C and the supernatants were used for
quantification using the BCA method. A total of 15 µg/lane of each
sample was separated on a 12% SDS-PAGE gel and transferred to PVDF
membranes. The membranes were blocked with 5% bovine serum albumin
(cat. no. 0219989680; MP Biomedicals, LLC) in TBST [20 mM Tris-HCl
(pH 8.0), 150 mM NaCl, 0.1% (v/v) Tween-20; all Sigma-Aldrich;
Merck KGaA] for 1 h at room temperature. Then, membranes were
incubated overnight at 4°C with the following primary antibodies:
Anti-GDF8 (cat. no. ab71808; Abcam; 1:1,000) and anti-β-actin (cat.
no. ab8227; Abcam; 1:1,000). Subsequently, bound antibodies were
incubated with horseradish peroxidase-conjugated anti-rabbit
immunoglobulin G secondary antibodies (cat. no. 7074; Cell
Signaling Technology, Inc.; 1:2,000) for 1 h at room temperature.
Western blotting luminol reagent (Santa Cruz Biotechnology, Inc.)
was used to detect proteins. Integrated optical densities of the
immunoreactive protein bands were measured using ImageJ software
(version 1.8.0_112; National Institutes of Health).
Prepared mouse model
All animals were tested in a blinded manner. After
administration, rotarod test, grip strength, MRI, NCS data
acquisition, gait analysis and muscle isolation were performed
separately by different researchers without information regarding
the treatments. Animals were managed with strict adherence to
protocols approved by the Institutional Animal Care and Use
Committee of the Samsung Medical Center (approval no.
SMC-20160507001). The mice were maintained in an environment of an
illuminance of 300 lux, 30–70% humidity and at 21°C (±2°C). The
mice were fed a normal diet, and access to food and water was free.
The dark/light cycle was performed through dimming control at 08:00
am and 06:00 pm. C22 mice [B6;CBACa-Tg(PMP22)C22Clh/H] were
purchased from MRC Harwell Institute. A total of 24 C22 and its
wild-type littermate (WT) mice were used. According to a previous
study, myelination in rodents begins on postnatal day 6, and thus
treatment for demyelinating neuropathy was conducted on postnatal
day 6 (7). Therefore,
administration of shLenti-Mstn or shLenti-mock was initiated on
postnatal day 90 (P90) in C22 mice, which represented the
phenotypic expression of CMT. At this day, WT (male, 30.0±2.2 g;
female, 25.50±1.6 g) and C22 (male, 29.6±1.1 g; female, 24.0±1.2 g)
were weighed. Thereafter, rotarod test, grip strength,
electrophysiological study, magnetic resonance imaging and gait
analysis were performed in all groups of mice at postnatal day 120
(P120). Subsequently, mice were sacrificed for muscle isolation.
The mice were divided into the following groups: Mock (male, n=4;
female, n=3) and shLenti-Mstn A (male, n=7; female, n=3).
Mouse genotyping
Tail biopsies (0.5 cm) of mice at P24 were incubated
at 55°C for 5–6 h in 150 µl Direct PCR Reagent (cat. no. 102; FIAT
International Ltd.) containing 200 µg/ml proteinase K (cat. no.
p6556; Sigma-Aldrich; Merck KGaA). Following centrifugation at 300
× g for 1 min at room temperature, the supernatant was inactivated
at 85°C for 45 min. Subsequently, the supernatant was diluted with
deionized water at 1/10, and 1 µl supernatant was used as a
template gDNA for PCR. Genotyping of C22 mice was performed using a
pair of primers that amplify mouse and human cDNA. The typical PCR
sample consisted of a 25 µl volume containing 5 pmol primers for
Yeast artificial chromosome (YAC;
forward,5′-GCTACTTGGAGCCACTATCGAGACGCGAT-3′ and reverse,
TGATAAATTAAAGTCTTGCGCCTTAAACC-3′) and a control pair of primers for
amplifying CD79b as a wild-type allele [IC1250; forward,
5′-GAGACTCTGGCTACTGATCC-3′ and reverse, 5′-CCTTCAGCAAGAGCTGGGGAC−3′
(27)]. The reaction also
contained Hifi mixture (cat. no. 12532016; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. The
following PCR conditions were applied: Initial denaturation for 5
min at 94°C; cyclic denaturation for 30 sec at 94°C, annealing for
30 sec at 58°C and elongation for 1 min at 72°C for 35 cycles; and
elongation for 5 min at 72°C. PCR amplification products were
analyzed by agarose gel electrophoresis as aforementioned.
In vivo transduction
Mice were injected with 120 µl (8×108
VP/ml) shLenti-Mstn or 120 µl PBS at the gastrocnemius muscle (GA)
and rectus femoris (RF) sites into hind muscles using 1 ml insulin
syringe (cat. no. 328820; Becton, Dickinson and Company). As 120 µl
is a large quantity compared with muscle volume, administration was
divided into three injections of 40 µl with 10 min intervals. Doses
were calculated using an in vitro assay (20–100 pmol/ml;
cat. no. 631235; Clonetech Laboratories, Inc.) according to the
manufacturer's protocol.
Muscle isolation
To evaluate muscle weight and evaluate mRNA
expression levels at injection sites (GA and RF) of C22 transgenic
mice administered with lentivirus, mice were euthanized at P120 at
the end of the experiment. All mice euthanasia was performed by
appropriately trained personnel approved by the Institutional
Animal Care and Use Committee of the Samsung Medical Center
(approval no. SMC-20160507001). Mice were euthanized by slow (30%
per minute) displacement of chamber air with compressed
CO2 gas for 5 min in a CO2 chamber, and
decapitation was performed to confirm complete irreversible
euthanasia. Subsequently, each tissue of all groups was separated
in a blinded manner by an experienced researcher. The isolated
muscle was weighed using a microbalance and mRNA was isolated from
the tissues as aforementioned.
Rotarod performance test
To evaluate the motor coordination and balance of
all groups of mice, mice performed the rotarod performance test by
gradual acceleration on a 3-cm horizontal rotating rod at a maximum
speed of 2 m/min. Prior to the experiment, mice were pretrained for
3 days for acclimation. Performance was measured for a maximum of 5
min.
Grip strength test
Grip strength of hind limbs in all groups of mice
was measured every other day using a Grip Strength Meter (Ugo
Basile S.R.L). Maximal forces of three trials were averaged and
averaged values were divided by body weight.
Electrophysiological study
To assess electrophysiological status in mice groups
of mock and shMstn-A, a nerve conduction study (NCS) was performed
as previously described (13).
Briefly, mice were initially anesthetized with CO2 gas
and maintained with 1.5% isoflurane supplied using a nose cone for
the duration of the procedure. This is a common and mandatory
procedure to examine electrophysiological functions in NCS
(12,13). The present study administered
10–30% chamber vol/min of CO2 gas used for the
initiation of anesthesia and then changed to isoflurane (2–3%; ≥1
l/min) to reduce any potential health risk and obtain accurate
results. Previous studies have reported that an increased
catecholamine concentration due to CO2 exposure can
cause a higher stress response (increased heart rate and blood
pressure), compared with isoflurane exposure (28–31).
This method is frequently required by the Institutional Animal Care
and Use Committee of the Samsung Medical Center (approval no.
SMC-20160507001) for any repeated measurements of
electrophysiological experiments to reduce potential stress.
Fur from the distal back to the hind limbs was
completely removed. NCS was performed using a Nicolet Viking Quest
device (Natus Medical, Inc.). For sciatic nerve motor NCS, an
active recording needle electrode was placed into the gastrocnemius
muscle with the reference electrode on the tendon. The stimulating
cathode was placed proximal to the recording electrode in the
midline of the posterior thigh to obtain the distal response and 6
mm proximally in the medial gluteal region to obtain the proximal
response. Compound motor action potentials amplitudes (CMAP) and
motor nerve conduction velocities (MNCV) were determined.
Lower limb magnetic resonance imaging
(MRI) studies
To evaluate the muscle size of all groups of mice
including WT, mock and shMstn A, MRI was performed using a 1.5T
system (Siemens AG). Axial, mid-sagittal and coronal scout rapid
acquisition with fast low angle shot imaging was used to localize
the legs. Area analysis using MRI images was measured using
Paravision software (version 6.0; Bruker Corporation).
Gait analysis
Mouse gait was analyzed in all groups of mice using
a TSE system MotoRater 303030 instrument (TSE Systems). Prior to
measurement, mice were trained to spontaneously walk through the
passage of the experimental apparatus. Mice were allowed to make a
free round trip, for about 10 min, by going back and forth from the
beginning to the end of the equipment passage (width, 4 cm; length,
80 cm), at the same location for 4 days. At the end of each mouse's
training, the inside of the passage was disinfected using 70%
ethanol and dried. Video recordings were made to compare the
changes in mouse pace throughout the daily training process. Prior
to and to facilitate joint observation, the hair on the right hind
flank of the mouse was flattened and white points were used to
display each point of the iliac crest-hip-knee-ankle-foot. Video
recordings began following confirmation that walking was performed
normally compared with the 4-day training records. After the mouse
walk measurements were completed, the interior of the aisle was
disinfected using 70% ethanol and dried. The collected video
recordings were analyzed to measure the time-dependent distance and
joint angle changes using MotoRater and TSE System software
(version 8.5.12; TSE Systems). Three indicators were selected as
evaluation items: i) Stride length, which was the distance between
the right front foot and the right rear foot with all feet touching
the ground; ii) duty factor, which was the % of time the feet
stayed in the air and on the ground; and iii) base of support,
which was the gap with both forefeet touching the ground.
Statistical analysis. All data are presented as mean
± SEM from at least three independent experiments. The statistical
significance of the data presented in all figures was evaluated
using one-way ANOVA with Tukey's post hoc multiple comparisons
test. P<0.05 was considered to indicate a statistically
significant difference. Additionally, all animals were randomly
assigned to each treatment and tested in a blinded manner.
Results
Determination of an optimal shRNA-Mstn
system to suppress endogenous Mstn expression
Mstn expression regulates myoblast differentiation
and proliferation by serving as a crucial negative activator during
embryonic myogenesis (15–17).
The downregulation of Mstn expression using a
shRNA system in the myogenic mouse C2C12 line was investigated.
Four different types of shRNA-Mstn (A, B, C and D) and mock
(control shRNA) were transfected into C2C12 cells. When examined by
PCR, shRNA-Mstn A exhibited the highest significant Mstn
suppressive effect (>60%; P<0.01) compared with the other
shRNA-Mstns and shRNA-Mstn A exhibited a higher significant
suppressive effect compared with the mock group (Fig. 1A). Furthermore, shRNA-Mstn A
decreased expression the endogenous protein level of Mstn (≥50%;
P<0.001) compared with controls.
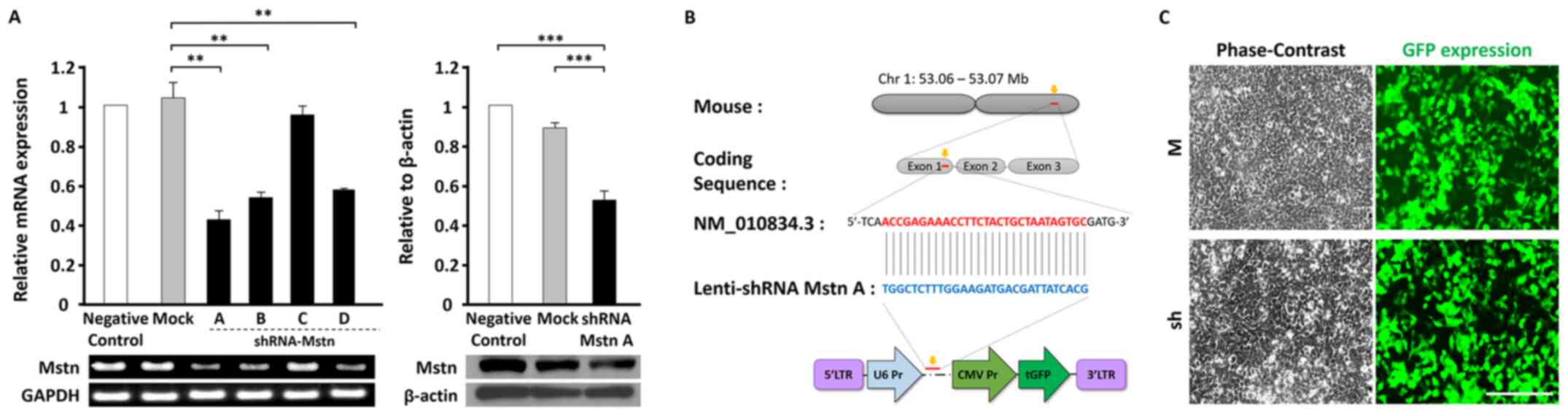 | Figure 1.The optimal design of shRNA
mstn and construction of the shLenti-system. (A) PCR
analysis of the inhibition levels of mstn gene expression by
four different shRNAs (shRNA-Mstn A, B, C and D) compared with the
negative control and mock groups (left). Western blotting following
transfection of shRNA-Mstn A to analyze the expression of
endogenous Mstn (right). (B) Construction of the Lenti-shRNA system
with GFP expression. Co-expression of shRNA-Mstn A and GFP
following transduction genomic integration. (C) Expression of
shLenti-Mstn A-GFP in 293T cells post-transfection. Magnification,
×200. Scale bar, 40 µm. **P<0.01. ***P<0.001. Mstn,
myostatin; GFP, green fluorescent protein; Chr, chromosome; Pr,
promoter; CMV, cytomegalovirus; LTR, long terminal repeat; W,
wild-type; M, mock; sh, shLenti-Mstn A. |
Following this, a Lenti-associated viral vector was
constructed, which included shRNA-Mstn A and GFP (shLenti-Mstn A)
for the in vivo study (Fig.
1B). Fluorescence imaging demonstrated GFP-expressing cells in
293T cells following shLenti-Mstn transduction (Fig. 1C). GFP expression was observed in
most cells (~80%) and levels were similar to those in the mock
group (Lenti-positive GFP). These results indicated that
shLenti-Mstn-GFP may decrease Mstn expression in an in vivo
model.
Improvement of muscle regeneration
using shLenti-Mstn in C22 mice
The regulation of Mstn expression serves an
important role in recovering skeletal muscle volume and mass
(16–19). Whether treatment with shLenti-Mstn
increased muscle volume and mass in C22 mice with hind-limb muscle
atrophy was investigated. The current study attempted to increase
skeletal muscle volume and mass by injecting shLenti-Mstn A into
two different sites, GA and RF of the hind limb, in C22 mice on
postnatal day 90 (Fig. 2A).
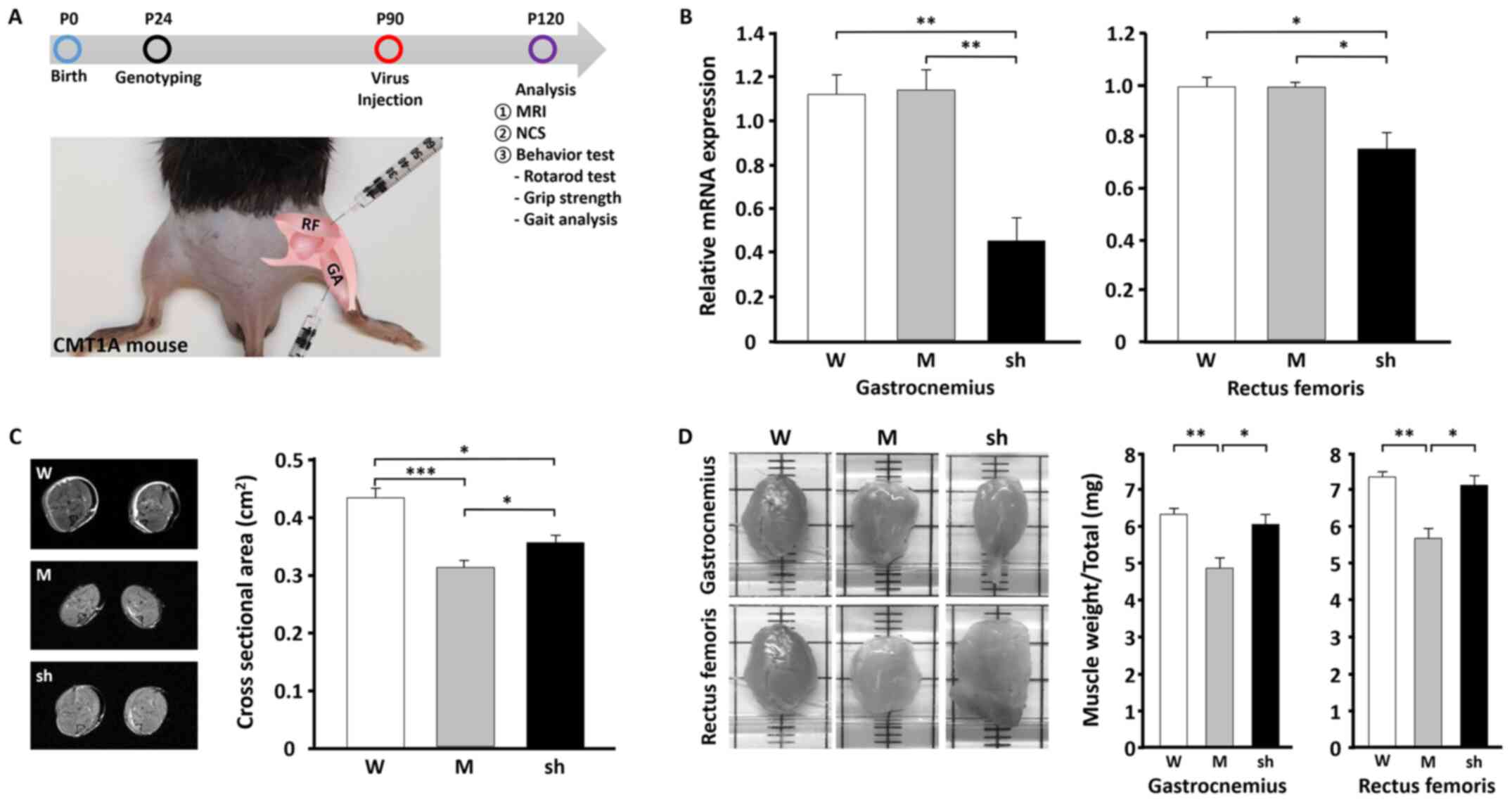 | Figure 2.In vivo efficacy of
shLenti-Mstn A treatment in CMT1A mice. (A) Schematic diagram of
the in vivo study using the CMT1A mouse (C22) model and
sites of infection by shLenti-Mstn in the hind limb of mice. (B)
Reverse transcription-quantitative PCR of mstn expression
levels at day 30 post-infection at two different sites, GA and RF,
in the hind limb. (C) MRI images of the hind limb muscle area
(left) in three different mice and quantification (right)(n=3). (D)
Images of the GA and RF muscle mass (left) and quantification
(right). WT (male n=3, female n=4), mock (male n=4, female n=3) and
shLenti-Mstn A (male n=6, female n=3) groups were analyzed.
*P<0.05, **P<0.01 and ***P<0.001. Mstn, myostatin; GA,
gastrocnemius; RF, rectus femoris; MRI, magnetic resonance imaging;
TG, transgenic; W, wild-type; M, mock; sh, shLenti-Mstn A; P,
postnatal; NCS, nerve conduction study; CMT1A, Charcot-Marie-Tooth
disease type 1A. |
Previous studies have demonstrated that C22 mice
exhibit demyelinating neuropathy on P6 and muscle atrophy on P30
(3,4,8).
mRNA of the muscles from GA and RF of the hind limbs were isolated
on P120. The results of RT-qPCR demonstrated that Mstn expression
in the hind limb was significantly reduced (by 46.9%, P<0.01) by
shLenti-Mstn compared with the wild-type (WT) and mock groups
(Fig. 2B). Furthermore,
shLenti-Mstn decreased the level of endogenous Mstn in the C22 mice
following long-term transgene expression (Fig. 2B). MRI revealed that shLenti-mock
(muscle atrophy, 0.312±0.012 cm2) caused a ~27% decrease
(P<0.001) in the cross sectional areas of hind limb volume in
C22 mice compared with WT mice (muscle atrophy, 0.429±0.018
cm2; Fig. 2C). In
contrast, shLenti-Mstn A (muscle atrophy, 0.355±0.014
cm2) exhibited a significantly increased cross sectional
area of the hind limb compared with the shLenti-mock group
(P<0.05). Additionally, whether muscle mass was altered in
shLenti-Mstn A-treated C22 mice was investigated by measuring the
masses of GA and RF in each group. Following excision, muscle
weight was significantly higher in the shLenti-Mstn A group (GA,
6.09±0.25 mg; RF, 7.11±0.48 mg) mice compared with mock (GA,
4.89±0.27 mg; RF, 5.51±0.47 mg) and WT (GA, 6.38±0.14 mg; RF,
7.35±0.29 mg) groups (Fig. 2D).
These results demonstrated that shLenti-Mstn increased the muscle
mass of both the GA and RF of the hind limb in C22 mice.
Enhancement of the electrophysiology
in hind limbs following muscle regeneration
Nerve electrophysiology changes in the hind limb
following shLenti-Mstn treatment were examined. C22 mice exhibiting
demyelinating neuropathies, including abnormal MNCV and CMAP caused
by human PMP22 gene overexpression (14,32–34).
Following shLenti-Mstn A injections into the hind limbs of C22
mice, electrophysiological patterns were improved in the
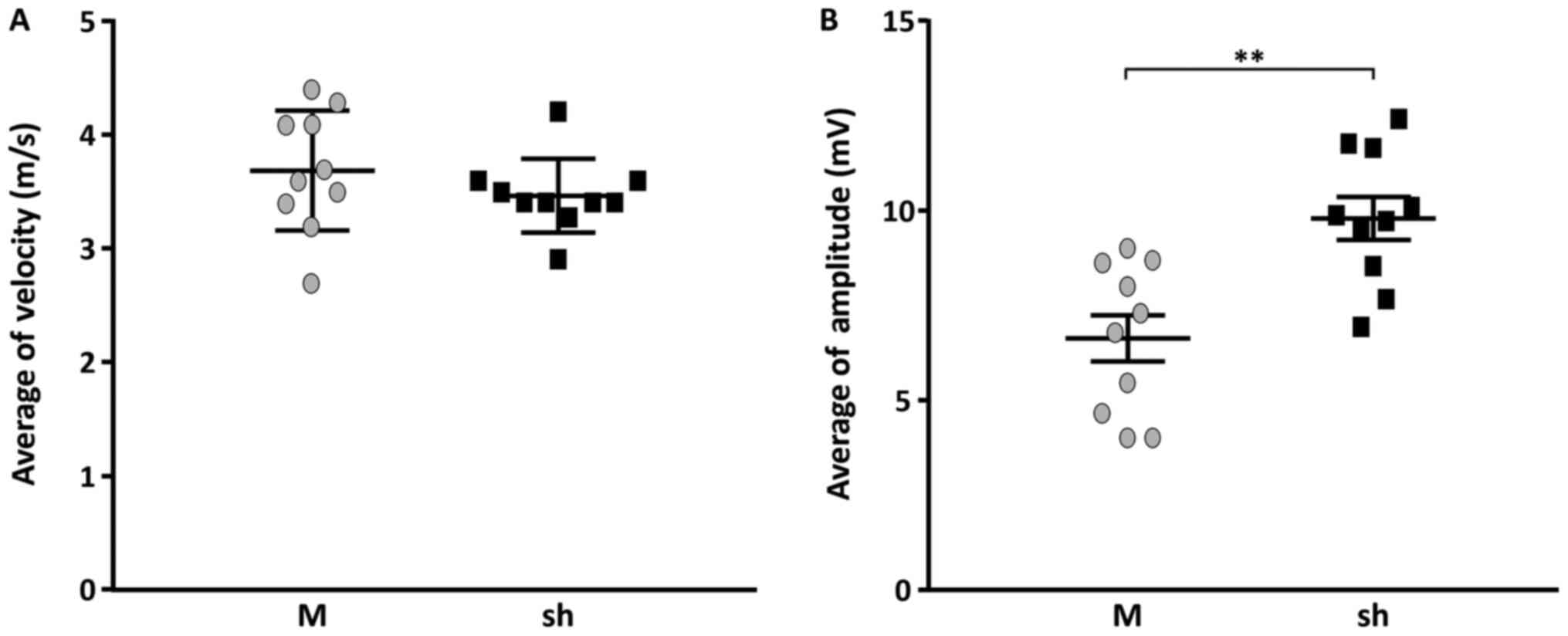
shLenti-Mstn A group compared with the mock group (Fig. 3). However, the NCS demonstrated
that MNCV in the shLenti-Mstn A group (3.4±0.4 m/sec) was not
significantly different compared with the mock group (3.7±0.5
m/sec) (Fig. 3A). In contrast,
CMAP was significantly increased by ~23% in the shLenti-Mstn A
group (9.9±0.7 mv) compared with the mock group (6.7±0.6 mv)
(P<0.01; Fig. 3B). Thus, these
results indicated that the increased muscle mass improved CMAP in
the hind limb of C22 mice injected with shLenti-Mstn A.
Regenerated hind-limb muscle improves
locomotor coordination in C22 mice
Whether shLenti-Mstn A injections led to the
improvement of locomotor coordination of C22 mice due to an
increase in muscle mass was investigated. Behavioral test results,
including those of the rotarod, grip strength and gait analysis
were analyzed. This evaluation was used to determine locomotor
coordination during mouse movement (13,14).
C22 mice are reported to have unsteady gaits and
muscle weakness caused by demyelinating peripheral neuropathies
(32). In the current study,
behavioral evaluations were measured on day 30 following injections
of shLenti-Mstn A and a mock viral particle. As determined by the
rotarod test (shLenti-mock, 2.8±0.49 sec; shLenti-Mstn A,
14.17±2.71 sec; Fig. 4A) and grip
strength test [shLenti-mock, 0.25±0.11 gram force (gf);
shLenti-Mstn A, 1.12±0.17 gf; Fig.
4B], mobility in the hind limb (>5-fold, P<0.01) and grip
strength (>1.7-fold, P<0.01) were significantly increased in
shLenti-Mstn A-injected group compared with the mock group.
However, increased behavioral values in the shLenti-Mstn A group
were significantly lower compared with the WT group (P<0.001).
Following this, gait improvement was examined using a MotoRater
system, which analyzes walking in C22 mice. Numerous previous
studies on ALS have used walking analysis with the DigiGait system
in mice (21–23). In these studies, mice exhibiting
muscle atrophy demonstrated shortened stride and increased swing of
the hind legs.
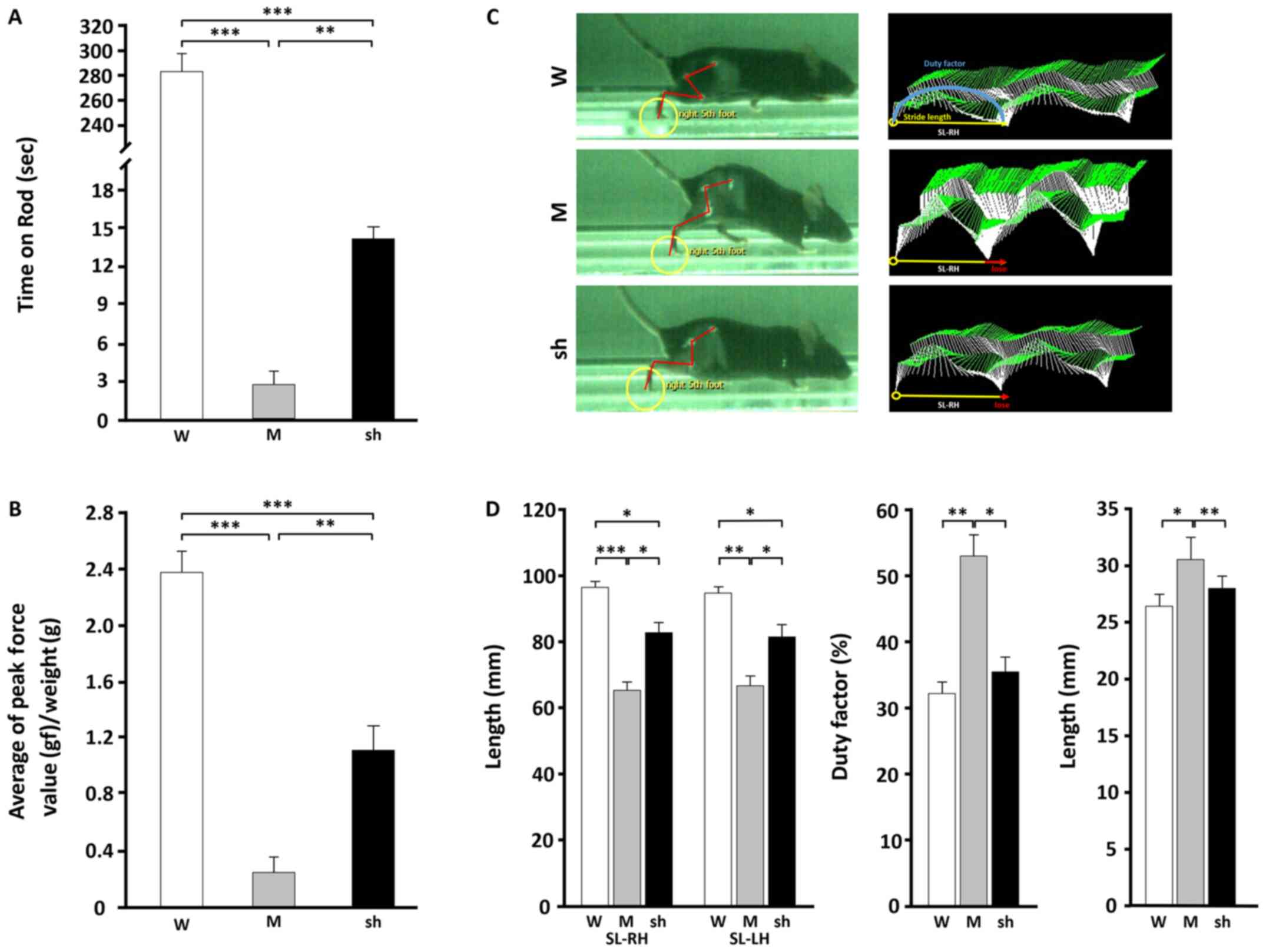 | Figure 4.Effects of shLenti-Mstn A treatment
on locomotion in C22 mice. (A) Rotarod test and (B) hind-limb grip
strength following muscular injections of shLenti-Mstn A in the
mock (n=5) and shLenti-Mstn A (n=6) groups. (C) Real-time gait
analysis of WT, mock and shLenti-Mstn A-treated mice (n=3/group).
Footprint colors were assigned by the software (foot, yellow
circles; yellow line, distance between each footstep; red line,
line connecting the pelvis, hip, knee, ankle and foot points
together for analysis; red arrow, decreased step length compared
with WT). (D) Quantitative analysis of SL (left panel), duty factor
(middle panel) and base of support (right panel). n=3/group.
*P<0.05. **P<0.01. ***P<0.001. Mstn, myostatin; W,
wild-type; M, mock; sh, shLenti-Mstn A; SL, stride length; RH,
right hind limb; LH, left hindlimb. |
In the current study, swinging and walking in three
different mice was measured using the MotoRater system. The results
demonstrated that mock mice exhibited significantly lower
stride-to-stride variability in both legs compared with the WT
(SL-RH, P<0.001; SL-LH, P<0.01) and shLenti-Mstn A groups
(SL-RH, P<0.05; SL-LH, P<0.05) (indicated by red arrows;
Fig. 4C and D). Furthermore,
shLenti-Mstn A mice demonstrated improved swinging and walking
compared with mock mice (Fig. 4D).
These results indicated that shLenti-Mstn A injections resulted in
improved swinging and walking in the hind limbs of C22 mice.
Discussion
The current study investigated shRNA-based gene
therapy using shLenti-Mstn in a mouse model of CMT1A. The results
demonstrated that gait improved following therapy. shLenti-Mstn
therapy reported a >60% reduction of mstn mRNA in the
myogenic C2C12 mouse cell line. Furthermore, a single dose [240 µl
(8×108 VP/ml)/site] of shLenti-Mstn by intramuscular
injection to C22 mice was accompanied with a >40% reduction in
mstn mRNA expression, which was observed on day 30.
To support the results of mstn silencing, MRI
imaging demonstrated that cross sectional area and muscle mass in
GA significantly increased alongside the levels of CMAP.
Consequently, the locomotion and gait pattern of C22 mice were
improved. The shLenti-Mstn system used in the present study may be
an important approach in the treatment of gait disorders and may
increase the quality of life in patients with PNS conditions.
RNA-based gene-silencing methodologies represent an
effective therapeutic strategy for the inhibition of specific
target molecules in disease pathways. Due to the potential for
increased specificity and reduced toxicity and side effects, RNA
interference has the potential to surpass treatment with standard
pharmacological drugs (12,35).
Previous studies have demonstrated that non-coding
RNAs, including microRNA and small interfering RNA, are potent
therapeutic strategies for the treatment of neuropathies (12,35).
However, few studies have reported improvement in gait disorders
due to muscle regeneration. Therefore, using the shMstn system, the
current study attempted to increase the recovery of neurological
disease-induced muscle atrophy. Among the four designed shMstn
sequences targeting Mstn exon 1, shMstn A exhibited the highest
suppression of Mstn in vitro with high transfection
efficiency.
The regulation of Mstn expression is essential
during myogenesis and skeletal muscle regeneration (15,16).
Following damage to the skeletal muscle, the regeneration process
is activated and myoblasts sequentially differentiate into
myocytes, myotubes and myofibers (15–20).
In adulthood, MSTN is produced by skeletal muscles, circulated by
the blood and acts to limit muscle fiber growth and it is a
well-established endogenous inhibitor of myogenesis (15,16,36).
Furthermore, several studies have demonstrated that skeletal muscle
satellite cells differentiate into myofibers during the
regeneration mechanism in adult muscles (15–20,36).
This has led to speculation about the potential physiological roles
of Mstn in peripheral neuropathies. Other previous studies have
established animal models of muscle hypertrophy in various species
through mstn gene knockouts or mutations (37–40).
Notably, Lin et al (41)
reported increased myogenesis and decreased adipogenesis through
examination of intramuscular mechanisms in an mstn-knockdown
mouse model.
PNS diseases, including Guillain-Barrè syndrome and
CMT, are accompanied by the demyelination of Schwann cells and
axonal damage to sensory and motor nerve cells (1–5). In
particular, motor nerve weakness gradually leads to muscle atrophy
and gait disorders (1,4,5).
Following the onset of neuropathy, patients with CMT develop gait
disorders due to the degeneration of motor nerves and skeletal
muscle (1,5). Treatments for skeletal muscle and the
PNS are required to overcome these challenges. To the best of our
knowledge, a direct correlation between CMT and Mstn muscle
regeneration has not yet been reported. A recent study demonstrated
that follistatin (a protein secreted by skeletal muscle)-based
fusion protein binds and potently neutralizes Mstn (42). Furthermore, to the best of our
knowledge, the current study is the first to use shLenti-Mstn
therapy to treat gait disorder in a CMT disease mouse model.
Prepared shLenti-Mstn A was injected into two
locations in the CMT1A disease mouse model. The two injection
points, at the GA and RF, are the major muscles of the hind limb
and are indispensable for walking (43). The results confirmed that
shLenti-Mstn A significantly suppressed mstn gene expression
and increased muscle volume and mass in the hind limbs in the CMT1A
disease model compared with the mock group. Furthermore, the
results of electrophysiological analysis revealed a statistically
significant increase in CMAP compared with the mock group. However,
there was no significant difference in MNCV between the
shLenti-Mstn A group and the mock group. This indicated that in the
neuropathy model, improved muscle mass ameliorated poor motor
coordination, even if nerve function was not regenerated by shMstn
therapy. The results suggested that shLenti-Mstn A treatment lead
to enhanced muscle hypertrophy and functionality.
Furthermore, the present results demonstrated that
increased muscle mass of the hind limb supported gait improvement
in the C22 mouse model. The C22 mouse develops abnormal behavior at
~2 weeks of age and gradually exhibits abnormal gait (10–13).
Previous studies focused on behavior assessments of locomotor
coordination in CMT mouse models (12,13,35).
MotoRater analysis provides more detailed
information about mouse gait through real-time recording compared
with footprint analysis (21,22).
Generally, footprint analysis can demonstrate the ability of mouse
locomotion, including walking stability and body balance, which is
based on the distance between footprints (44). Mancuso et al (23) focused on gait analysis using the
DigiGait system for measuring the walking pattern in a mouse model
of ALS.
The present study investigated the footprints and
complete walking patterns in a mouse model of CMT1. Accordingly,
shLenti-Mstn A treatment improved the unstable walking patterns in
terms of stride length, duty factor and base of support.
Additionally, during measurement of stride length in the right hind
limb, stride was significantly decreased in the mock group compared
with the WT group. The shLenti-Mstn A-injected mice demonstrated a
significantly improved stride length and duty factor, and these
values were increased in WT mice, although not significant for
stride length. These results indicated that an increase in muscle
mass may be able to mitigate walking patterns in the neuropathy
model. Therefore, it was hypothesized that the regulation of Mstn
activity improves gait problems caused by CMT disease.
In conclusion, it was hypothesized that shLenti-Mstn
A-inducted Mstn suppression is associated with the growth and
differentiation underlying CMT disease, which suppresses muscle
contractions and induces muscle changes. As a result, Mstn
suppression served an important role in CMT disease, thereby
directly or indirectly affecting gait improvement. The regulation
of muscle activity in neurological diseases alleviated the
condition, which is associated with Mstn suppression. Further
studies are required to identify the potential mechanism of action
between the muscle and Mstn. Although lentivirus was used to
knockdown Mstn expression in the present study, using lentivirus
has limitations in clinical drug development as neuropathy-induced
DNA integration, muscle loss and gait are regulated by regulating
the expression of genes involved in muscle formation and
development. Therefore, it is necessary to develop symptomatic
therapeutic strategies for improving gait of CMT 1A by further
investigating the mechanisms underlying neuromuscular
development.
Acknowledgements
Not applicable.
Funding
The current study was supported by the National
Research Foundation of Korea grants funded by the Korean government
(grant nos. 2016 R1A5A 2007009, 2017 R1A2B 2004699, 2018 R1A4A
1024506 and 2018 R1C1B 6002586) and by the Korean Health Technology
R&D Project, Ministry of Health and Welfare (grant nos. HI14C
3484 and HI16C 0426).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YBH, JHK and BOC designed and managed the current
study. HMD, GK and HH designed the current study and collected
data. HMD, SHN, HWM, SBK, JH and KWC analyzed and interpreted data.
HMD and JH drafted and critically revised the manuscript for
important intellectual content. YBH, JHK and BOC supervised the
study. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The current study was approved by the Institutional
Animal Care and Use Committees of Samsung Medical Center (Seoul,
Korea; approval no. SMC-20160507001).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Skre H: Genetic and clinical aspects of
Charcot-Marie-Tooth's disease. Clin Genet. 6:98–118. 1974.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Levitan IB and Kaczmarek LK: Intercellular
Communication. The Neuron: Cell and Molecular Biology. 4th edition.
Oxford University Press; New York, NY: pp. 153–328. 2015,
View Article : Google Scholar
|
|
3
|
Rotthier A, Baets J, Timmerman V and
Janssens K: Mechanisms of disease in hereditary sensory and
autonomic neuropathies. Nat Rev Neurol. 8:73–85. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rossor AM, Kalmar B, Greensmith L and
Reilly MM: The distal hereditary motor neuropathies. J Neurol
Neurosurg Psychiatr. 83:6–14. 2012. View Article : Google Scholar
|
|
5
|
Harding AE and Thomas PK: The clinical
features of hereditary motor and sensory neuropathy types I and II.
Brain. 103:259–280. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim JY, Woo SY, Hong YB, Choi H, Kim J,
Choi H, Mook-Jung I, Ha N, Kyung J, Koo SK, et al: HDAC6 inhibitors
rescued the defective axonal mitochondrial movement in motor
neurons derived from the induced pluripotent stem cells of
peripheral neuropathy patients with HSPB1 mutation. Stem Cells Int.
2016:94759812016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fledrich R, Stassart RM and Sereda MW:
Murine therapeutic models for Charcot-Marie-Tooth (CMT) disease. Br
Med Bull. 102:89–113. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Robertson AM, Perea J, McGuigan A, King
RH, Muddle JR, Gabreels-Festen AA, Thomas PK and Huxley C:
Comparison of a new pmp22 transgenic mouse line with other mouse
models and human patients with CMT1A. J Anat. 200:377–390. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Meyer zu Horste G, Prukop T, Liebetanz D,
Mobius W, Nave KA and Sereda MW: Antiprogesterone therapy uncouples
axonal loss from demyelination in a transgenic rat model of CMT1A
neuropathy. Ann Neurol. 61:61–72. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sereda MW, Meyer zu Horste G, Suter U,
Uzma N and Nave KA: Therapeutic administration of progesterone
antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A). Nat
Med. 9:1533–1537. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Passage E, Norreel JC, Noack-Fraissignes
P, Sanguedolce V, Pizant J, Thirion X, Robaglia-Schlupp A,
Pellissier JF and Fontes M: Ascorbic acid treatment corrects the
phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat Med.
10:396–401. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee JS, Kwak G, Kim HJ, Park HT, Choi BO
and Hong YB: MiR-381 attenuates peripheral neuropathic phenotype
caused by overexpression of PMP22. Exp Neurobiol. 28:279–288. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee JS, Lee JY, Song DW, Bae HS, Doo HM,
Yu HS, Lee KJ, Kim HK, Hwang H, Kwak G, et al: Targeted PMP22
TATA-box editing by CRISPR/Cas9 reduces demyelinating neuropathy of
Charcot-Marie-Tooth disease type 1A in mice. Nucleic Acids Res.
48:130–140. 2020.PubMed/NCBI
|
|
14
|
Robaglia-Schlupp A, Pizant J, Norreel JC,
Passage E, Saberan-Djoneidi D, Ansaldi JL, Vinay L,
Figarella-Branger D, Levy N, Clarac F, et al: PMP22 overexpression
causes dysmyelination in mice. Brain. 125:2213–2221. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McPherron AC, Lawler AM and Lee SJ:
Regulation of skeletal muscle mass in mice by a new TGF-beta
superfamily member. Nature. 387:83–90. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee SJ and McPherron AC: Regulation of
myostatin activity and muscle growth. Proc Natl Acad Sci USA.
98:9306–9311. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mariot V, Joubert R, Hourde C, Feasson L,
Hanna M, Muntoni F, Maisonobe T, Servais L, Bogni C, Le Panse R, et
al: Downregulation of myostatin pathway in neuromuscular diseases
may explain challenges of anti-myostatin therapeutic approaches.
Nat Commun. 8:18592017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wagner KR, McPherron AC, Winik N and Lee
SJ: Loss of myostatin attenuates severity of muscular dystrophy in
mdx mice. Ann Neurol. 52:832–836. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Holzbaur EL, Howland DS, Weber N, Wallace
KE, She Y, Kwak S, Tchistiakova LA, Murphy E, Hinson J, Karim R, et
al: Myostatin inhibition slows muscle atrophy in rodent models of
amyotrophic lateral sclerosis. Neurobiol Dis. 23:697–707. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hennebry A, Oldham J, Shavlakadze T,
Grounds MD, Sheard P, Fiorotto ML, Falconer S, Smith HK, Berry C,
Jeanplong F, et al: IGF1 stimulates greater muscle hypertrophy in
the absence of myostatin in male mice. J Endocrinol. 234:187–200.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dorman CW, Krug HE, Frizelle SP,
Funkenbusch S and Mahowald ML: A comparison of DigiGait™ and
TreadScan™ imaging systems: Assessment of pain using gait analysis
in murine monoarthritis. J Pain Res. 7:25–35. 2013.PubMed/NCBI
|
|
22
|
Xu Y, Tian N, Bai Q, Chen Q, Sun XH and
Wang Y: Gait assessment of pain and analgesics: Comparison of the
DigiGait™ and CatWalk™ gait imaging systems. Neurosci Bull.
35:401–418. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mancuso R, Olivan S, Osta R and Navarro X:
Evolution of gait abnormalities in SOD1(G93A) transgenic mice.
Brain Res. 1406:65–73. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
DiGiusto DL, Krishnan A, Li L, Li H, Li S,
Rao A, Mi S, Yam P, Stinson S, Kalos M, et al: RNA-based gene
therapy for HIV with lentiviral vector-modified CD34(+) cells in
patients undergoing transplantation for AIDS-related lymphoma. Sci
Transl Med. 2:36ra432010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maples PB, Kumar P, Yu Y, Wang Z, Jay C,
Pappen BO, Rao DD, Kuhn J, Nemunaitis J and Senzer N: FANG Vaccine:
Autologous tumor cell vaccine genitically modified to express
GM-CSF and block production of furin. BioProcess J. 8:4–14. 2009.
View Article : Google Scholar
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-tie quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hasenberg A, Hasenberg M, Männ L, Neumann
F, Borkenstein L, Stecher M, Kraus A, Engel DR, Klingberg A,
Seddigh P, et al: Catchup: A mouse model for imaging-based tracking
and modulation of neutrophil granulocytes. Nat Methods. 12:445–452.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Boivin GP, Hickman DL, Creamer-Hente MA,
Pritchett-Corning KR and Bratcher NA: Review of CO2 as a
euthanasia agent for laboratory rats and mice. J Am Assoc Lab Anim
Sci. 56:491–499. 2017.PubMed/NCBI
|
|
29
|
Marquardt N, Feja M, Hünigen H, Plendl J,
Menken L, Fink H and Bert B: Euthanasia of laboratory mice: Are
isoflurane and sevoflurane real alternatives to carbon dioxide?
PLoS One. 13:e02037932018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Makowska IJ and Weary DM: Rat aversion to
induction with inhalant anaesthetics. Appl Anim Behav Sci.
119:229–235. 2009. View Article : Google Scholar
|
|
31
|
Leach MC, Bowell VA, Allan TF and Morton
DB: Degrees of aversion shown by rats and mice to different
concentrations of inhalational anaesthetics. Vet Rec. 150:808–815.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Verhamme C, King RH, ten Asbroek AL,
Muddle JR, Nourallah M, Wolterman R, Baas F and van Schaik IN:
Myelin and axon pathology in a long-term study of
PMP22-overexpressing mice. J Neuropathol Exp Neurol. 70:386–398.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Komyathy K, Neal S, Feely S, Miller LJ,
Lewis RA, Trigge G, Siskind CE, Shy ME and Ramchandren S: Anterior
tibialis CMAP amplitude correlations with impairment in CMT1A.
Muscle Nerve. 47:493–496. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stojkovic T: Hereditary neuropathies: An
update. Rev Neurol (Paris). 172:775–778. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee JS, Chang EH, Koo OJ, Jwa DH, Mo WM,
Kwak G, Moon HW, Park HT, Hong YB and Choi BO: Pmp22 mutant
allele-specific siRNA alleviates demyelinating neuropathic
phenotype in vivo. Neurobiol Dis. 100:99–107. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chal J and Pourquie O: Making muscle:
Skeletal myogenesis in vivo and in vitro. Development.
144:2104–2122. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
McPherron AC and Lee SJ: Double muscling
in cattle due to mutations in the myostatin gene. Proc Natl Acad
Sci USA. 94:12457–12461. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Grobet L, Martin LJ, Poncelet D, Pirottin
D, Brouwers B, Riquet J, Schoeberlein A, Dunner S, Menissier F,
Massabanda J, et al: A deletion in the bovine myostatin gene causes
the double-muscled phenotype in cattle. Nat Genet. 17:71–74. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kambadur R, Sharma M, Smith TP and Bass
JJ: Mutations in myostatin (GDF8) in double-muscled Belgian blue
and piedmontese cattle. Genome Res. 7:910–916. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Grobet L, Poncelet D, Royo LJ, Brouwers B,
Pirottin D, Michaux C, Menissier F, Zanotti M, Dunner S and Georges
M: Molecular definition of an allelic series of mutations
disrupting the myostatin function and causing double-muscling in
cattle. Mamm Genome. 9:210–213. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lin J, Arnold HB, Della-Fera MA, Azain MJ,
Hartzell DL and Baile CA: Myostatin knockout in mice increases
myogenesis and decreases adipogenesis. Biochem Biophys Res Commun.
291:701–706. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pearsall RS, Davies MV, Cannell M, Li J,
Widrick J, Mulivor AW, Wallner S, Troy ME, Spaits M, Liharska K, et
al: Follistatin-based ligand trap ACE-083 induces localized
hypertrophy of skeletal muscle with functional improvement in
models of neuromuscular disease. Sci Rep. 9:113922019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bonnefoy A and Armand S: Normal Gait.
Orthopedic Management of Children With Cerebral Palsy: A
Comprehensive Approach. Nova Science Publishers Inc.; Hauppauge,
NY: pp. 5672015
|
|
44
|
Chester VL, Biden EN and Tingley M: Gait
analysis. Biomed Instrum Technol. 39:64–74. 2005.PubMed/NCBI
|