Introduction
Congenital heart diseases (CHDs), which may have a
significant impact on cardiac structure, function, or both, are the
most common defects in liveborn children (1). Affecting the heart and/or great
vessels, CHDs can be isolated or identified within a syndromic
presentation; they are highly diverse, ranging from asymptomatic to
fatal. Multiple genetic factors have been implicated in playing a
role in CHDs' complex etiology (2).
Most of the known CHD causative genes encode
transcription factors (TF) that assist in regulating the cardiac
embryogenesis, such as NKX2-5, GATA5, TBX20, TBX1, GATA4, GATA6,
TBX5, MEF2C, HAND1, NR2F2, and HAND2 (3). Among the identified TF genes are the
T-box gene family, which encodes proteins notably harboring a
well-conserved T-box domain which assists in DNA binding (4). One of the T-box genes is TBX5
(OMIM ID: 601620) that is predominantly expressed in the heart and
the forelimbs (5). Pathogenic
variants in TBX5 have been associated with Holt-Oram
syndrome (HOS; OMIM ID: 142900), which is characterized by
congenital anomalies in both the heart and the upper limbs, in line
with the TBX5 protein expression (6). Interestingly, variants in
TBX5 have also been reported to cause non-syndromic CHD
(7,8). Previous observations showed that the
malformations of HOS were distinctively heterogenous even among
individuals sharing the same genetic variant (9–13).
A wide phenotypic spectrum of septal defects, conduction
abnormalities, and tetralogy of Fallot has been frequently linked
to the bulk of the TBX5-associated heart disorders (14). However, the clinical association
between HOS and total anomalous pulmonary venous return (TAPVR) has
rarely been described (15).
TAPVR makes up 1–3% of the total CHD cases, with an
incidence of around 7 per 100,000 live births (16). TAPVR describes the improper
connection of the pulmonary veins to the right atrium or to the
systemic venous system rather than the normal connection to the
left atrium (17). It can also be
anatomically sub-divided based on the level of the venous
connection anomaly into 4 subtypes (supracardiac, infracardiac,
cardiac, and mixed types) (18).
To our knowledge, all the studies that claimed the association
between HOS and TAPVR were only based on clinical diagnosis-no
genetic testing was performed in the previous literature to confirm
that TBX5 was the implicated gene (15,19,20). Furthermore, to date, no case of
mixed-type TAPVR has been reported in patients with HOS.
Over the past few years, whole-exome sequencing
(WES) has been successfully implemented to uncover the underlying
molecular etiology of multiple diseases. including CHD (21,22). Consequently, an accurate diagnosis
of certain cases was achieved based on the identified genetic
findings, which may inform families and influence patient care
(23).
We present a multi-generation family affected by
autosomal dominant (AD)-CHD with intrafamilial variability of
specific manifestations and severity. Interestingly, the proband
had a rare lethal mixed-type TAPVR. Our genetic investigation
identified a hereditary disease-causing variant (DCV) in the T-box
domain of TBX5. Consequently, the identified genetic
findings prompted additional phenotyping which modified the
diagnosis from non-syndromic CHD to HOS. Noteworthy, this is the
first time TBX5 has been associated with TAPVR,
specifically, mixed-type TAPVR, in patients with or without HOS.
Also, we aimed to augment the previously reported corpus of genetic
knowledge related to null-variants in the T-box domain of
TBX5 to underpin the phenotypic intra- and interfamilial
variable expressivity. Finally, we intended to illustrate the
impact of harboring the variant on the protein's structure and
function.
Materials and methods
Ethical compliance
This study was conducted in concordance with the
tents of the Declaration of Helsinki and was approved by the
Institutional Review Board (IRB) of Jordan University Hospital
(protocol code 2018/198, 26 June 2018). Before enrollment, written
informed consents were secured from the participating individuals
and the legal guardian (for the newborn patient).
Clinical assessment
Cardiac clinical evaluation was initially done using
echocardiography (Philips HD 11 XE ultrasound system; Philips
Healthcare), while the confirmation of diagnosis and detailed
anatomy of the pulmonary venous drainage was achieved using
contrast computed tomography (CT) scanning (Somatom Definition
VA44, 2012; Siemens AG Healthcare).
Study subjects and sample
collection
Blood samples were collected using EDTA tubes from
the following individuals: III-1, III-2, and the proband (IV-1)
(Fig. 1A). DNA isolation was
conducted on the collected samples by using Wizard Genomic DNA
Purification Kit (Promega) according to the manufacturer's
protocol.
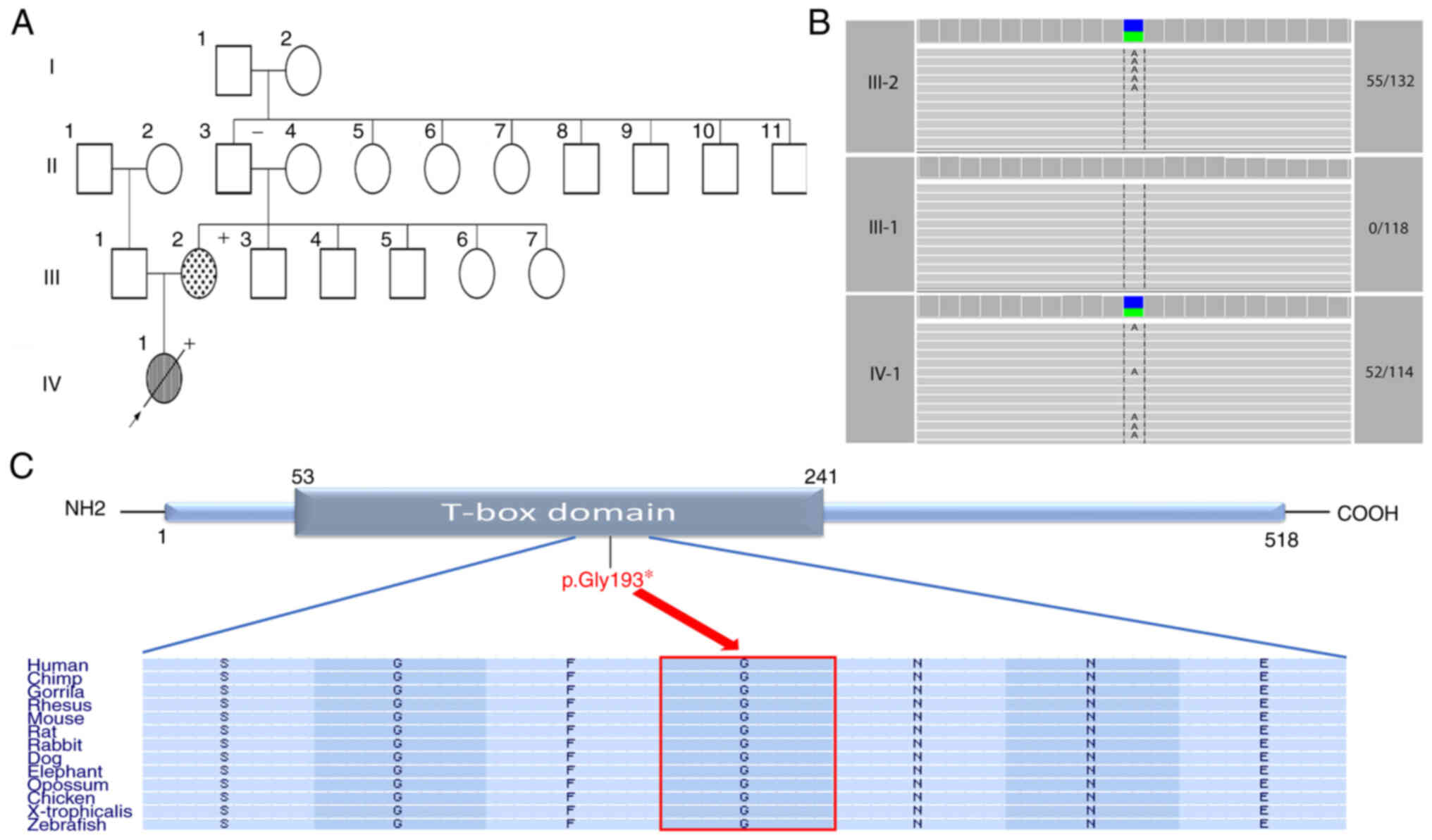 | Figure 1.Description of the participating
family and the identified variant. (A) Pedigree of the
participating family shows the affected and unaffected family
members across two generations (III–IV). The striped symbol
indicates that the individual is affected by mixed-type total
anomalous pulmonary venous return, while the dot-filled symbol
represents the affected member with atrial septal defect. The +
signs, which are located on the upper right corner of the affected
individuals, indicate the presence of triphalangeal thumb. Arrow,
proband; empty symbol, unaffected member; circles, females;
squares, males; diagonal line, deceased member. (B) Cropped IGV
screenshots showing the trio-coverage of the identified DCV in the
parents and proband. The numbers on the right represent the number
of reads capturing the variant over the total number of reads. (C)
Schematic representation of the TBX5 protein. The identified
nonsense DCV (p.Gly193*) is located within the T-box domain. The
horizontal rectangle at the bottom represents the amino acid
alignment around the variated residue and their corresponding
conservation among selected species, created by the UCSC browser
(https://genome.ucsc.edu). The arrow points
towards the altered glycine residue which is highly conserved
across various species. NH2 and COOH represent amino- and
carboxyl-terminus, respectively. |
WES and data analysis
WES was performed on samples from parents (III-1,
and III-2) and proband (IV-1). De-identified genomic DNA samples
were provided to Yale under its IRB-approved protocol with a waiver
of consent. Trio-exome sequencing was conducted at the Yale Center
for Genomic Analysis (YCGA) through the Pediatric Genomics
Discovery Program (PGDP, http://www.yalemedicine.org/departments/pediatric-genomics),
a program that is freely available to coordinate sequencing,
analysis, and additional testing with pediatric critical care
researchers caring for children with diseases of suspected genetic
etiology. Capture was performed on IDT xGen capture kit followed by
Illumina DNA sequencing (HiSeq 4000) using YCGA whole-exome
sequencing (WES) protocol. Paired-end sequence reads (101 bases)
were converted to FASTQ format and were aligned to the reference
human genome (hg19). Genetic variants were called by GATK (24), and they were annotated by ANNOVAR
(25) and a custom pipeline that
includes population allele frequencies, OMIM and ClinVar citations,
and numerous in silico attributes.
The samples were sequenced to a mean depth of at
least 110× independent reads per targeted base, with at least 20×
independent reads in 98% of targeted bases, or 50× in 90% of
targeted bases (Table SI). We
filtered exonic or splice-site rare variants (MAF ≤0.01 from
publicly available population databases e.g., 1000 Genomes,
NHLBI-EVS, gnomAD, and our institutional database) that exhibited
high quality sequence reads. De novo variants that were only
present in the parents' samples (≥20% alternate allele ratio in the
proband, alternate allele ratio <3% in parents) were called.
Homozygous or compound heterozygous variants in the proband, or
very rare inherited heterozygous variants from target genes were
recorded (Table SII,Table SIII,Table SIV,Table SV). All the recorded variants
were then visualized and verified manually by Integrative Genomics
Viewer (IGV). The visualization of the conserved amino acids
flanking the variants was conducted by the help of UCSC browser,
GRCh37/hg19 (https://genome.ucsc.edu).
Protein modeling of TBX5 (mutant and
wild type)
A template search with BLAST and HHBlits has been
performed against the SWISS-MODEL template library (26,27). A total of 30 templates were found.
Models are built based on the target-template alignment using
ProMod3 (28). Coordinates that
are conserved between the target and the template are copied from
the template to the model. Insertions and deletions are remodeled
using a fragment library. The sidechains are then rebuilt. Finally,
the geometry of the resulting model is regularized by using a force
field. In case loop modeling with ProMod3 fails, an alternative
model is built with PROMOD-II (29). The global and per-residue model
quality has been assessed using tools implemented in SWISSS-MODEL,
including QMEAN (Qualitative Model Energy Analysis) score and
MolProbity score (30). The
former is a composite of 6 energy values within the homology model
matrix related to protein nativeness, with score values ≤-4.0
indicating poor quality homology models. The latter, on the other
hand, combines several protein parameters, including clash score,
Ramachandran Plot criteria (Ramachandran Favored and Ramachandran
Outliers) (31).
Results
Initial clinical history
The proband (IV-1; Fig. 1A) was a 12-day-old female infant
who presented to the emergency room with cyanosis and respiratory
distress. Physical examination showed a cyanotic infant with
tachypnea and tachycardia. She had no apparent dysmorphic facial
features. Evaluation by chest radiography showed a normal-sized
heart with bilateral pulmonary congestion. The echocardiographic
evaluation showed mixed-type TAPVR, with obstruction of pulmonary
venous drainage. There was a drainage of the pulmonary vein to the
posterior aspect of the superior vena cava (SVC), with significant
obstruction at its entry. In addition, pulmonary venous flow was
also seen draining below the diaphragm to the inferior caval vein.
The right ventricle (RV) was dilated and hypertrophic with moderate
tricuspid regurgitation. The left atrium (LA) was small with right
to left shunt at the atrial septum. Left ventricle appeared
adequate in size with normal function. The ventricular septum was
intact.
The infant was admitted to the pediatric intensive
care unit on supplementary oxygen, and a CT angiogram was done the
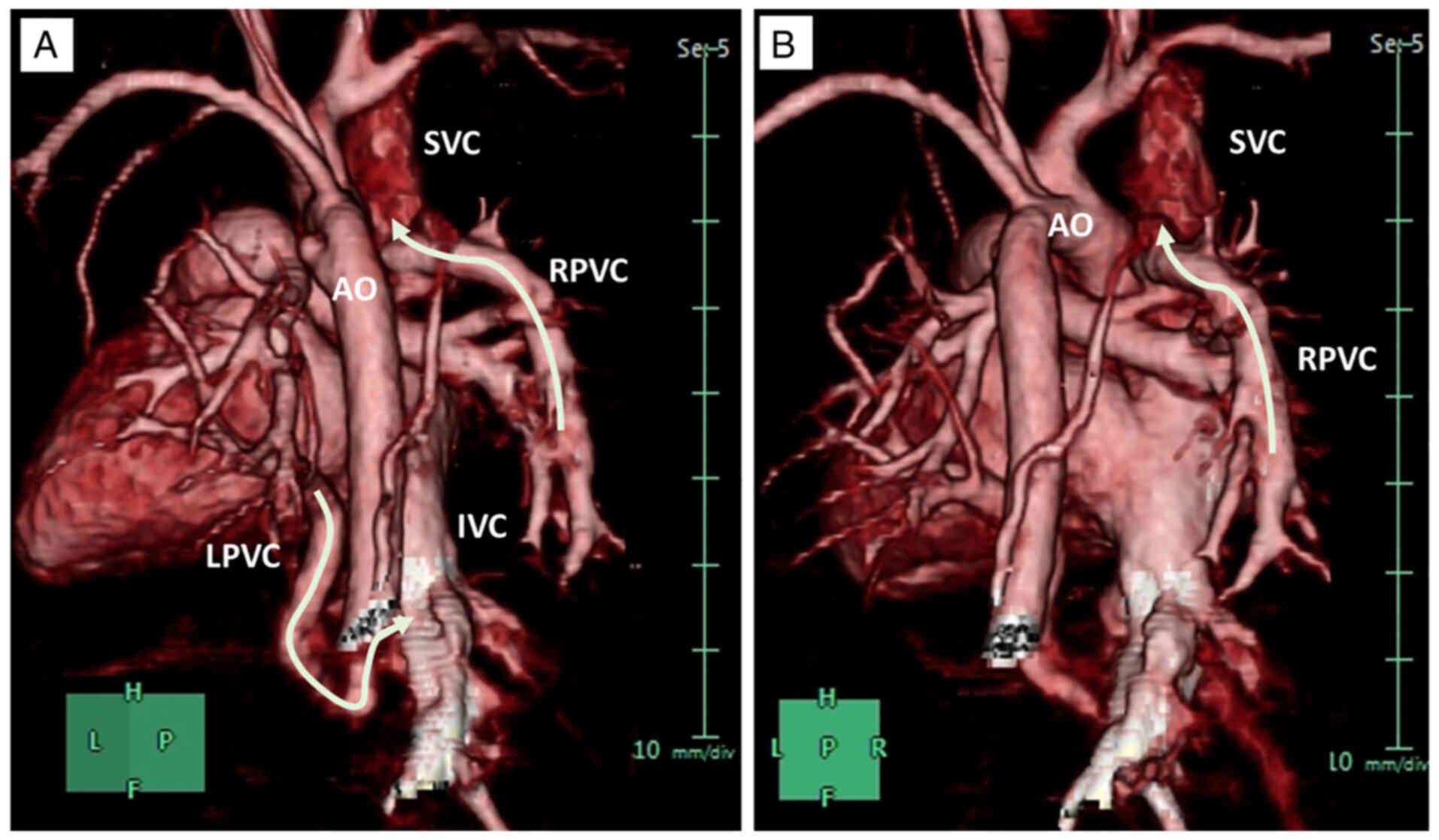
following day to confirm the complex pulmonary venous anatomy
(Fig. 2). CT scan showed that the
right pulmonary venous confluence (RPVC) drained to the
postero-rightward aspect of the superior caval vein, and the
left-sided venous confluence (LPVC) coursed behind the left atrium
and descended through the diaphragm and drained to the inferior
caval vein via a tortuous route (Fig.
2). The patient was transferred to the cardiac center for
urgent surgical repair, but unfortunately, at the age of one month,
she died before surgery from respiratory failure.
The proband (IV-1), who is the only child in this
family, had a family history of CHD from the maternal lineage as it
was revealed that her mother (III-2) had ostium secundum atrial
septal defect (ASD) for which she underwent surgical repair during
childhood (Fig. 1A). Noteworthy,
no clinical examinations were done for the distant family members,
and the data of their clinical status were based on the provided
history.
Genetic analysis
As a rare cardiac anomaly, the finding of TAPVR
triggered a genetic workup to better understand this abnormal
clinical presentation on the molecular level. The affected proband
(IV-1) along with her parents (III-1 and III-2) (Fig. 1A) underwent WES. The trio-based
WES analysis revealed a maternally inherited heterozygous nonsense
DCV in the TBX5 gene (Table
I and Fig. 1B). This DCV
(c.577G>T; p.Gly193*) is located in exon 6 at an evolutionarily
highly conserved position (GERP score=5.75). Also, this variant is
located within the T-box domain of the TBX5 protein (Fig. 1C).
 | Table I.Details of the identified
disease-causing variant. |
Table I.
Details of the identified
disease-causing variant.
| Gene | Variant
coordinate | RefSeq
transcript | Exon | HGVS cDNA | HGVS aa | Consequence | Zygosity | Highest MAF in
gnomAD V2, V3 | ClinVar | ACMG classification
(Richards et al 2015) (33) |
|---|
| TBX5 | GRCh37 (hg19)
chr12:114832632 | GRCh38 (hg38)
chr12:114394827 | NM_181486.4 | 6/9 | c.577G>T | p.Gly193* | Nonsense | HET | Not found | Not reported | Pathogenic |
The characteristics and classification
of the identified DCV
This DCV in TBX5 (c.577G>T) introduces a
stop codon at residue number 193 (p.Gly193*; Table I) of the resultant protein. This
premature termination codon (PTC) can lead to the translation of an
aberrant short protein-the last 325 amino acids (35% of the protein
length) might be lost. Potential nonsense-mediated mRNA decay (NMD)
is also a predicted mechanism as the distance between the mRNA's
stop codon and the nearest 3′-end of the exon-exon junction is more
than 55 nucleotides long. This DCV is absent from the population
database (gnomAD; http://gnomad.broadinstitute.org). It has been
previously reported in a Chinese Han family with multiple affected
individuals with CHDs (32).
ClinVar has no prior entry for this variant. Multiple
loss-of-function (LoF) variants in TBX5 have been already
reported to be disease-causing. Given together, according to the
ACMG guidelines (Richards et al 2015), we classify this DCV
as pathogenic (Table I) (33). This is the first finding for
TBX5 to be associated with the complex cardiac manifestation
of mixed-type TAPVR.
Protein modeling of the wild type and
mutant (p.Gly193*) versions of TBX5
The resulting homology structure was evaluated
employing structure assessment tools within SWISS-MODEL. The
results are as follows (in brackets): QMEAN (−0.30), MolProbity
score (1.5), Clash score (1.17), Ramachandran Favored (96.42%). The
truncated protein was prepared using Discovery Studio (version
4.5). Both wild type (WT) and mutated proteins (in dimer forms)
were minimized for 10 steps using Discovery Studio (version 4.5),
and then the non-bond interactions between the two units were
identified using the implemented tool in Discovery Studio (version
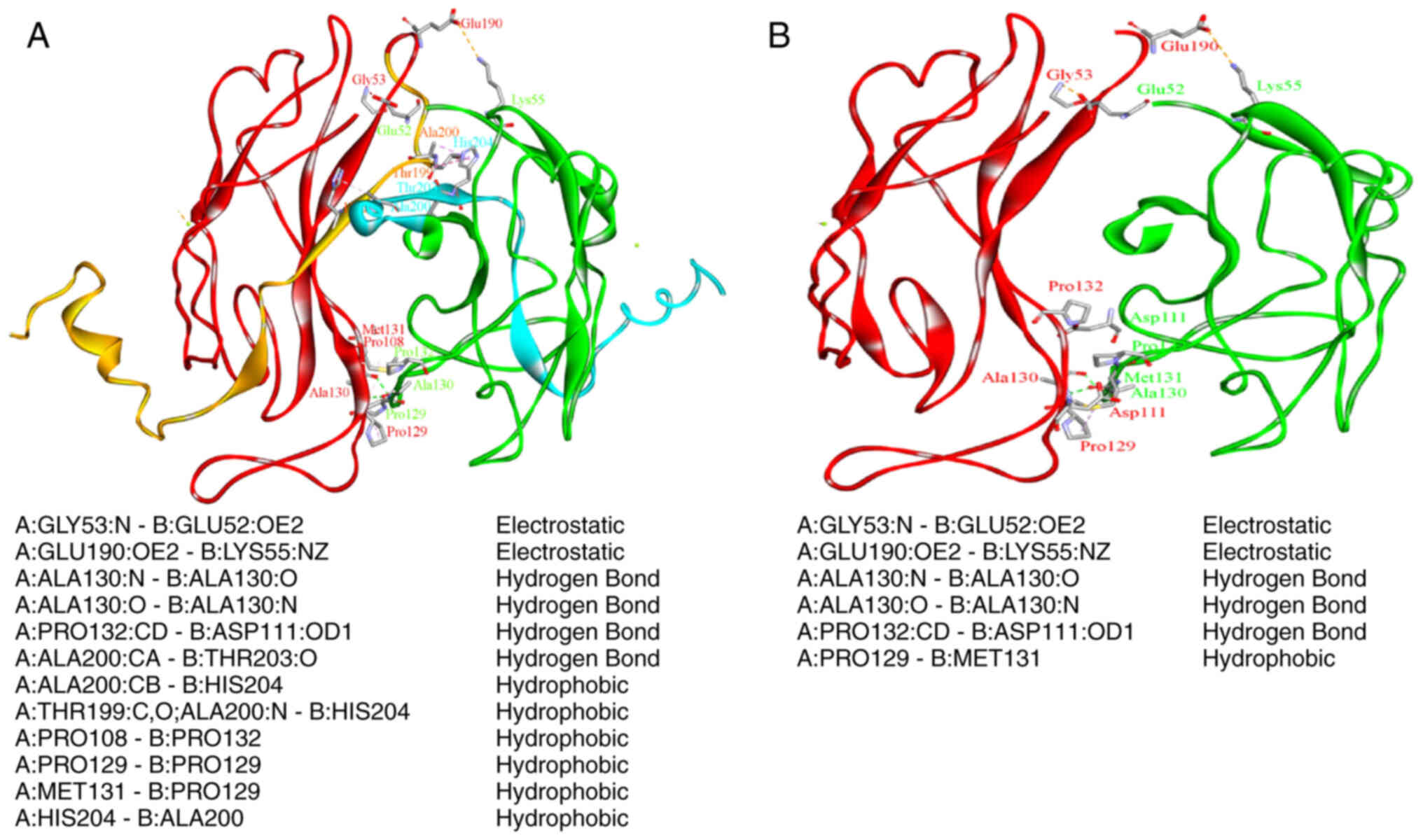
4.5). Fig. 3 shows the
significant reduction in the number of non-covalent bond
interactions, which clarifies the less stabile state of the mutated
dimer form. A significant reduction in the number of non-covalent
bonds (interactions) may affect the stability of the dimer
formation and thus its binding to DNA.
Outcome of identifying the DCV
Our genetic analysis and interpretation revealed
TBX5 as the underlying genetic etiology of the proband's
disease. This finding granted re-evaluating the clinical diagnosis
of the proband's non-syndromic cardiac anomaly to be part of a
manifestation of HOS. A thorough assessment of the family's medical
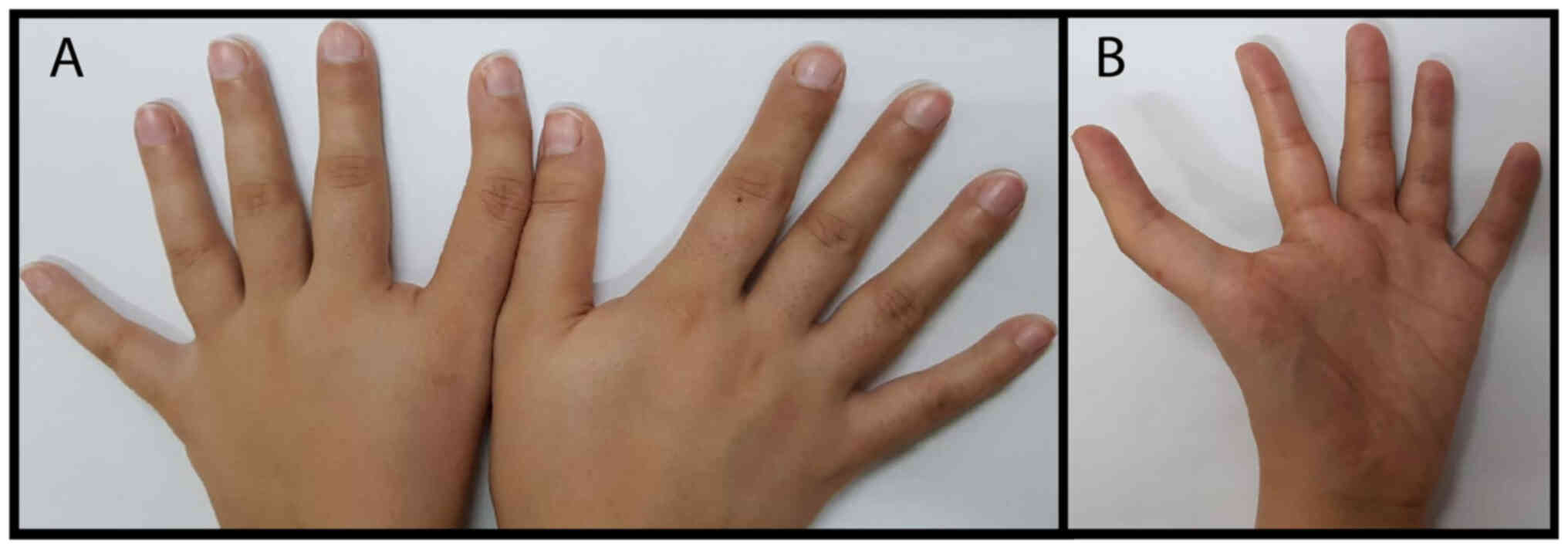
history revealed that the mother (III-2; Fig. 1), carrying the same variant, does
indeed have a triphalangeal thumb in her left hand (Fig. 4). In addition, the mother declared
the same bilateral thumb anomaly in her deceased child (IV-1).
The finding of triphalangeal thumb in the proband
(IV-1) and her affected mother (III-2), along with their respective
CHDs, suggested manifestations of HOS in the setting of a DCV in
TBX5. This is the first documented HOS patient with the
cardiac anomaly of mixed-type TAPVR (Fig. 2).
Discussion
The TBX5 gene encodes for a transcriptional
factor protein belonging to the T-box protein family and plays a
major role in regulating the early cardiac and upper limb
developmental processes (34).
Interestingly, TBX5 interacts with other ‘cardiac-critical’
transcriptional factors, such as NKX2-5 and GATA4, which both also
help in the early stages of the heart development pathways
(35). Thus, genetic variations
in the TBX5 can lead to both cardiac and/or skeletal defects
(36). Variants in TBX5
cause HOS, also known as the heart-hand syndrome, which is
inherited as an AD trait (6).
In this study, we identified the underlying genetic
etiology behind a family with AD-CHDs in which the index case
presented with mixed-type TAPVR while her mother had ASD. After
conducting a trio-based WES analysis, we identified a maternally
inherited pathogenic DCV in TBX5. Notably, prior to our
investigation, the proband's mother (III-2), who suffered from ASD
along with triphalangeal thumb, had never been diagnosed with HOS.
The genetic finding of the DCV in TBX5 triggered us to
perform a thorough review of her medical records and consequently
reached her proper diagnosis with HOS.
LoF variants in TBX5, such as the pathogenic
nonsense variant (c.577G>T; p.Gly193*) identified here, are
established to be disease-causing in HOS. The resultant protein is
predicted to harbor a PTC and probably would yield an aberrantly
truncated protein (only 193 out of 518 TBX5′s amino acid residues
might be translated, representing nearly 35% of the total protein's
length). The transcribed TBX5 mRNA harboring the PTC might
instead undergo degradation through the NMD process, resulting in
TBX5 haploinsufficiency (37).
Previous functional in vitro analysis for this DCV showed
that the mutated TBX5 protein failed to activate its targeted genes
(MYH6, and NPPA), and no synergistic transactivation
between the mutated protein and other transcription factors
(NKX2-5, and GATA4) was detected. The designated pathways, which
are considered essential for cardiac development, could be
nullified in embryos harboring this DCV and probably lead to the
consequent heart defect (32). By
utilizing protein modeling tools, we studied the impact of
harboring this variant (p.Gly193*) on the TBX5′s structure and
function. The resultant TBX5 structure formed a destabilized dimer
due to a significant reduction in the number of the non-covalent
bonds, and its ability to bind DNA might be subsequently
deteriorated.
Our findings illustrate that at the intrafamilial
level, the elucidated CHD ranged from a simple cardiac
manifestation (ASD) to a complex, ultimately lethal one (mixed-type
TAPVR). Furthermore, the same DCV (c.577G>T; p.Gly193*) has been
previously reported in a Chinese Han family variably affected by
ASD and/or ventricular septal defect (VSD), bicuspid aortic valve
(BA), and atrial fibrillation (AF) (32). Hence, variable expressivity of the
cardiac defects accompanied by this specific DCV can be observed at
both the intrafamilial and interfamilial levels, and it is widely
ranging in severity, structural deficits, and functional
compromise.
The nonsense DCV (c.577G>T; p.Gly193*) is located
in the TBX5′s T-box domain. Although pathogenic DCVs have been
identified throughout the entire gene, they appear to cluster
within this domain in particular (4). The T-box domain is a highly
conserved domain across various species; it is also essential for
interactions with other transcription factors and DNA binding
(38). Therefore, we reviewed the
reported clinical effects of the T-box's null variants on the
expressivity of cardiac and skeletal malformations (Table II). We noticed that the cardiac
defects were mainly associated with limb defects (and thus HOS) and
ranged from isolated cases of septal defects (most common) to
complex cases presenting with a combination of multiple cardiac
manifestations, left ventricular noncompaction cardiomyopathy,
conductive heart failure, and even valve anomalies. Nevertheless,
this is the first time TBX5 has been discovered to cause
mixed-type TAPVR.
| Table II.A summary of the T-box's
null-variants that are reported in the HGMD Pro (Version 2020.4)
database and their associated cardiac manifestations. |
Table II.
A summary of the T-box's
null-variants that are reported in the HGMD Pro (Version 2020.4)
database and their associated cardiac manifestations.
|
|
|
|
|
| Associated skeletal
anomalies |
|
|
|---|
|
|
|
|
|
|
|
|
|
|---|
| TBX5-null
variant | Age at last
examination, years | Relationship with
proband | Sex | Reported
phenotype |
Hands//deformity |
Forearms//deformity | Arm and shoulder
complex//deformity | Lower
extremity//deformity | Other | Cardiac
features | (Refs.) |
|---|
| Lys88* | 23 years | Proband | F | HOS | -R-TB//HP in 1st
MCB and phalanges -L-TB//HP −2ndand 5th HF//middle phalanges
HP | -Forearm//Pro. and
sup. | None | None | None | ASD-II, and MVP
with slight regurgitation | (10) |
|
| 13 years | Sister | F | HOS | -TB//Bi-proximally
set −5th HF//HP and clinodactyly −2nd and 5th HF//Bi-HP of middle
phalanges (R>L) -CB//Scaphoid and trapezium fusions |
-Forearm//Bi-abnormal pro. and sup.
(L>R) | None | None | None | MR | (10) |
|
| 42 years | Mother | F | HOS | -TB//Bi-Abs.
-MCB//Bi-Abs. | -Forearm//Abnormal
pro. and sup. | R-shoulder//Limited
rotation | Various anomalies
in 2nd, 3rd, and 4th toes | None | Atrial septal
hypermobility, but no shunt | (10) |
| Tyr136* | 46 years | Proband of Family
A | F | HOS | -TB//Bi-HP
(L>R) | None | -Deltoid//
Bi-HP | None | None | ASD | (43) |
|
| 44 years | Family A
Brother | M | HOS | -TBs//Bi-HP | None |
-Deltoid//Bi-HP | None | None | ASD | (43) |
|
| 18 years | Family A Niece | F | HOS | -TB//Bi-HP | None |
-Deltoid//Bi-HP | None | None | ASD | (43) |
|
| 40 years | Proband of family
B | M | HOS | -L-TB//Abs. | -L-Radius// Abs.
-R-Radius//HP | -Deltoid//HP | None | None | ASD | (43) |
|
| 55 years | Family B
Brother | M | HOS | -TBs//Bi-abs. | None | -Deltoid//HP | None | None | ASD | (43) |
|
| 53 years | Family B
Brother | M | HOS | -L-TB//Abs.
-R-TB//TF | None | -Deltoid//HP | None | None | ASD | (43) |
| Gln151* | ND | Proband | M | HOS | -L-TB//TF with
distal phalanx ulnar deviation | None | None | None | None | ASD-II, MVI,
AVB | (44) |
| c.243-2A>G | 31 years | Proband | F | HOS |
-TBs//Bi-anomaly | None | None | None | None | CHF | (45) |
| c.243-1G>C | 2 years | Proband | F | HOS | -L 2nd and 3rd
HF//Syndactyly -TBs//Bilateral agenesis -MCB//Bi-abs −1st HF//Abs
phalanges | -L-arm//Phocomelia
(L<R) -R-arm//Phocomelia -Radioulnar joint// Bi-HP -L-ulna//Abs.
-Superior limbs//Spasticity (L>R) -Upper limbs//Hypotonia | None | -Hip//HP
-Femoral-tibial angle//genu varum | -Nasal bones//HP
-Trunk/Hypotonia | ASD-II, muscular
VSD, and pulmonary hypertension | (46) |
| c.242+1G>A | ND | ND | ND | HOS | Preaxial radial
ray | None | None | None | None | VSD | (36) |
| c.363-1G>A | 23 years | Proband | F | HOS | Present (ND) | Present (ND) | None | None | None | ASD | (42) |
|
| ND | Mother | F | ND | ND | ND | ND | ND | ND | ASD and DCM but was
not genetically tested | (42) |
| c.362+1G>T | ND | ND | ND | HOS | -TBs//Abs.
-Wrist//Malformation -CB//disarticulation | -Radius
bone//disarticulation | None | None | None | Conduction
abnormality | (36) |
| c.510+1G>T along
with another path missense variant | ND | ND | ND | HOS | ND | ND | ND | ND | ND | ASD, VSD, CoA | (36) |
| c.510+5G>T | 50 years | Proband
(Mother) | F | HOS | -TB anomaly | None | None | None | None | ASD, LVNC | (47) |
|
| ND | Daughter | F | HOS | -TBs//TF -CB//HP
and extra CB | Radius//HP | None | None | None | ASD | (47) |
| c.664-1G>A | 36 years | Proband | M | HOS | TBs//Bi-HP | L-radius//Deviated
with HP | -Clavicles//HP
-Shoulders//Narrow | None | Hemithorax//HP | ASD-II and
anomalous right coronary artery | (48) |
| c.663+1G>C | ND | Proband
(Father) | M | HOS | TB//Abs. | None | None | None | None | ASD | (49) |
|
| 3 years | Son | M | HOS | TB//TF | None | None | None | None | ASD | (49) |
| p.(Leu65
Glnfs*10) | 8 years | Proband | M | HOS | -TBs//Bi-abs.
-Bi-2nd and 5th HF//HP 2nd phalanges | -L-radius and
ulna//HP -L-elbow//abnormal with subluxation | -Scapula glenoid
fossae//insufficient development -Shoulders//Bi-subluxation
-Humeri//Proximal epiphyseal dysplasia | None | None | VSD | (50) |
| p.(Asn
95Ilefs*29) | 14 years | Proband | F | HOS | -L-TB//Aplastic
-R-TB//TF | None | None | None | None | VSD | (45) |
|
| 16 years | Proband | F | HOS | TBs//Bi-TF | L-ulna//HP |
Claviculae//Bi-HP | None | None | ASD-II, several
VSDs | (45) |
|
| 42 years | Proband | F | HOS | None | None | None | None | None | VSD | (45) |
| p.(Asp 118del) | ND | ND | ND | Atrial
fibrillation | ND | ND | ND | ND | ND | AF | (51) |
| p.(Pro139
Glnfs*11) | ND | Proband
(Mother) | F | HOS | -Lhand//connected
to the shoulder joint -Rt hand// Connected to the elbow joint
-TBs//Bi-abs | -L-arm//Abs.
-R-forearm//Abs. | None | None | None | ASD | (52) |
|
| ND | Son | M | HOS | TBs//Bi-abs | -Upper
limbs//Maldevelopment -Radii and ulnae//Curved | None | None | None | ASD | (52) |
| p.(Val153
Serfs*21) | ND | ND | F | HOS | 2nd HF//Bi-abs |
Bi-radius//Abs. | None | None | None | ASD | (40) |
| p.(Phe168
Leufs*6) | ND | Proband (Son) | M | HOS | -R-TB//TFl with
ulnar deviation of the distal phalanx -L-TB//HP-TF | None | Shoulder
gridle//Abnormal with limited motion | None | None | ASD-II, VSD,
PDA | (44) |
|
| ND | Mother | F | HOS | -R-TB//Abs
-L-TB//HP | Forearms//Bi
shortening with limited pro. and sup. | Shoulder
gridle//Abnormal with limited motion | None | None | MVP, TVP
regurgitation | (44) |
| p.(Val214
Aspfs*14) | ND | Proband
(Mother) | F | HOS | TBs//Bi-HP | Bi-radial
deviation | None | None | None | Normal | (44) |
|
| ND | Son | M | HOS | TBs//Bi-HP | Bi-radial
deviation | None | None | None | AVSD | (44) |
| p.(His 220del) | 11 months | Proband | F | HOS | TBs//Bi-TF | -R-radius//Aplasia
-L-radius and ulna//Shortened | None | None | Ribs//11 pairs of
ribs | VSDs, AVSD, HPRV,
AV-valve insufficiency, PVS | (35) |
| p.(Met
242Ilefs*10) | ND | ND | ND | HOS | Radial club
hand | None | None | None | None | ASD-II | (53) |
| p.(Arg134
Profs*49) | ND | Proband | F | HOS |
TBs//Bi-digitalized | None | None | None | None | ASD | (40) |
|
| ND | Uncle | M | HOS | TBs//Bilateral
abs | R-radius//HP | None | None | None | VSD | (40) |
|
| ND | Mother | F | HOS | TBs//Bilateral
digitalized | R-radius//HP | None | None | None | ASD or VSD | (40) |
|
| ND | Maternal
grandmother | F | HOS | ND | ND | ND | ND | ND | VSD | (40) |
|
| ND | Brother | M | HOS | -L-TB//HP
-R-TB//TF | None | None | None | None | Multiple VSDs | (40) |
| p.(Ala143
Argfs*40) | ND | Proband | M | HOS | -L-TB// Abs. digit
-R-TB// HP digit | L-radius//HP | None | None | None | ASD-II, muscular
VSDs | (40) |
|
| ND | Mother | F | HOS | TB//Bi-abs.
digit | -L-ulna and
radius//Abs. -R-ulna and radius//HP | L-humerus//HP | None | None | VSD | (40) |
|
| ND | Brother | M | HOS | -R hand//Extra
digit (R1) -L-TB//TF | None | None | None | None | Muscular-VSD | (40) |
|
| ND | Maternal
grandfather | M | HOS | -TB//HP | Radioulnar
synostosis | None | None | None | ASD or VSD | (40) |
|
p.(Lys126_Arg134del) | ND | Extended
family | ND | HOS | ND (Can be with or
without skeletal defect) | ND | ND | ND | ND | ASDs VSDs, MVP, and
others | (54) |
The associated skeletal deformities accompanied by
the CHDs in HOS were mainly described in the hand and predominantly
in the preaxial radial ray (Table
II). Similarly, both the deceased proband (IV-1) and her mother
(III-2) presented with a triphalangeal thumb, which has been
frequently reported in the literature as a manifestation of HOS
(Table II). Other skeletal
malformations were reported to a lesser extent in the postaxial
region and the other upper extremities (forearms, arms, and
shoulder complex), but were rarely ascribed to the lower
extremities (feet, hip, etc.), as shown in Table II.
While HOS typically features both cardiac and limb
deformities, in certain instances, the severity of the accompanying
malformations tends to be more pronounced either in the heart or in
the limbs of the same patient. Moreover, a wide range of
accompanying anomalies (whether they were cardiac or skeletal) seem
to be variably expressed even among patients harboring the same DCV
(Table II). Several models
propose that the disease-causing mechanism and the severity levels
can be attributed to disturbances in TBX5′s binding to downstream
protein-partners or recognizing targeted motifs that are vital to
the embryo's heart and/or skeletal development, such as NKX2-5,
GATA4, GATA6, TBX20, and MEF2C (38).
Although Table II
shows a spectrum of the composite cardiac defects, we did not
notice any correlation between the type or the location of the
null-variant within the T-box domain and the consequent phenotypic
severity. Broadly speaking, early studies suggested that type
(missense vs. null variants) and/or the location of the DCVs could
influence the severity of HOS's cardiac and skeletal manifestations
(39). Nonetheless, these
observations were based on a small number of cases, and
inconsistencies were observed in the later-described ones (36,40). A recent study by Vanlerberghe
et al, conducted a phenotype-genotype correlation on the
largest molecular investigation of 78 HOS cases and found out that
the null-variants were associated with less-severe cardiac
manifestations when compared with the missense variants (9). Additionally, no relation was
established between the type of the molecular variation
(null-variants vs. missense) and the severity of the skeletal
anomalies (9). Ultimately, as
demonstrated in the family we report, variability is such to
prevent predictions of phenotypic consequences based on genotype
(40).
The TAPVR subtype identified here revealed both
supracardiac and infracardiac venous anomalies, resulting in a
mixed-typed classification of the TAPVR phenotype. The 3D CT scan
of the proband's heart (IV-) showed that both the RPVC and LPVC
failed to connect to the left atrium and instead drained directly
into the SVC and IVC, respectively. The mixed-type TAPVR is rare
amongst TAPVR cases-accounting for around 5% of the total
occurrences (41). Cardiac,
supracardiac, and infracardiac subtypes of TAPVR have been
previously reported with HOS (15,19,20,42). To the best of our knowledge,
mixed-type TAPVR has never been reported in HOS. Hence, this is the
first genetic investigation to identify an HOS case with mixed-type
TAPVR.
In summary, we utilized the trio-based WES approach
to identify a pathogenic nonsense DCV in a family with hereditary
CHDs. Also, we showed the importance of genetic testing in reaching
the proper diagnosis-our findings aided the correct diagnosis of
the proband's and her mother's cases with HOS. We explored the
broad phenotypic spectrum of the reported null-variants within the
T-box domain of TBX5. Importantly, we reported a patient presenting
with mixed-type TAPVR; to our knowledge, this is the first study
documenting the connection between the mixed-type TAPVR and HOS.
Additionally, this is the first molecular investigation to ever
associate TAPVR with HOS.
Supplementary Material
Supporting Data
Acknowledgements
We thank Ms Monica Konstantino (Pediatric Genomics
Discovery Program, Yale University Department of Pediatrics, New
Haven, USA) for her work in coordinating the samples logistics.
Funding
This work was supported by the Deanship of Academic Research at
the University of Jordan (grant no. 2205).
Availability of data and materials
The proband's high-throughput data generated or
analyzed during this study are available in the NCBI SRA repository
(accession number: PRJNA822821; http://www.ncbi.nlm.nih.gov/biosample/27279246).
Authors' contributions
BA, IAA and SL conceptualized the study. DA
interpreted the data. WJ, LJ, NJI, ASAA, HM, YAO, MAS, ZD and MMH
performed the data analysis. BA and IAA acquired funding. DA, WJ,
NJI, ASAA, HM, YAO, MAS, ZD and MMH designed the methodology. BA,
IAA and SL were project administrators and supervised the study.
BA, WJ, LJ, NJI, ASAA, HM, YAO, MAS, ZD, IAA and SL conducted the
experiments and generated data. DA prepared the figures and tables.
BA and DA wrote the original draft. BA, WJ, LJ, IAA and SL wrote,
reviewed and edited the manuscript. BA and DA confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
This study was conducted in concordance with the
tents of the Declaration of Helsinki and was approved by the
Institutional Review Board of Jordan University Hospital (approval
no. 2018/198; 26 June 2018). Before enrollment, written informed
consents were secured from the participating individuals and the
legal guardian (for the newborn patient).
Patient consent for publication
The proband's parents provided their consent for the
publication of data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Triedman JK and Newburger JW: Trends in
congenital heart disease: The next decade. Circulation.
133:2716–2733. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Diab NS, Barish S, Dong W, Zhao S,
Allington G, Yu X, Kahle KT, Brueckner M and Jin SC: Molecular
genetics and complex inheritance of congenital heart disease. Genes
(Basel). 12:10202021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pierpont ME, Brueckner M, Chung WK, Garg
V, Lacro RV, McGuire AL, Mital S, Priest JR, Pu WT, Roberts A, et
al: Genetic basis for congenital heart disease: Revisited: A
scientific statement from the american heart association.
Circulation. 138:e653–e711. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Isphording D, Leylek AM, Yeung J, Mischel
A and Simon HG: T-box genes and congenital heart/limb
malformations. Clin Genet. 66:253–264. 2004. View Article : Google Scholar
|
|
5
|
Bruneau BG, Logan M, Davis N, Levi T,
Tabin CJ, Seidman JG and Seidman CE: Chamber-specific cardiac
expression of Tbx5 and heart defects in Holt-Oram syndrome. Dev
Biol. 211:100–108. 1999. View Article : Google Scholar
|
|
6
|
Basson CT, Bachinsky DR, Lin RC, Levi T,
Elkins JA, Soults J, Grayzel D, Kroumpouzou E, Traill TA,
Leblanc-Straceski J, et al: Mutations in human TBX5 [corrected]
cause limb and cardiac malformation in Holt-Oram syndrome. Nat
Genet. 15:30–35. 1997. View Article : Google Scholar
|
|
7
|
Marie Reamon-Buettner S and Borlak J: TBX5
mutations in non-Holt-Oram syndrome (HOS) malformed hearts. Hum
Mutat. 24:1042004. View Article : Google Scholar
|
|
8
|
Yoshida A, Morisaki H, Nakaji M, Kitano M,
Kim KS, Sagawa K, Ishikawa S, Satokata I, Mitani Y, Kato H, et al:
Genetic mutation analysis in Japanese patients with non-syndromic
congenital heart disease. J Hum Genet. 61:157–162. 2016. View Article : Google Scholar
|
|
9
|
Vanlerberghe C, Jourdain AS, Ghoumid J,
Frenois F, Mezel A, Vaksmann G, Lenne B, Delobel B, Porchet N,
Cormier-Daire V, et al: Holt-Oram syndrome: Clinical and molecular
description of 78 patients with TBX5 variants. Eur J Hum Genet.
27:360–368. 2019. View Article : Google Scholar
|
|
10
|
Garavelli L, De Brasi D, Verri R,
Guareschi E, Cariola F, Melis D, Calcagno G, Salvatore F, Unger S,
Sebastio G, et al: Holt-Oram syndrome associated with anomalies of
the feet. Am J Med Genet Part A. 146A:1185–1189. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guo Q, Shen J, Liu Y, Pu T, Sun K and Chen
S: Exome sequencing identifies a c.148-1G>C mutation of TBX5 in
a Holt-Oram family with unusual genotype-phenotype correlations.
Cell Physiol Biochem. 37:1066–1074. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guo DF, Li RG, Yuan F, Shi HY, Hou XM, Qu
XK, Xu YJ, Zhang M, Liu X, Jiang JQ, et al: TBX5 loss-of-function
mutation contributes to atrial fibrillation and atypical Holt-Oram
syndrome. Mol Med Rep. 13:4349–4356. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhou W, Zhao L, Jiang JQ, Jiang WF, Yang
YQ and Qiu XB: A novel TBX5 loss-of-function mutation associated
with sporadic dilated cardiomyopathy. Int J Mol Med. 36:282–288.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Goldmuntz E: The genetic contribution to
congenital heart disease. Pediatr Clin North Am. 511721–1737.
(x)2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Correa-Villaseñor A, Ferencz C, Boughman
JA and Neill CA: Total anomalous pulmonary venous return: Familial
and environmental factors. The Baltimore-Washington infant study
group. Teratology. 44:415–428. 1991. View Article : Google Scholar
|
|
16
|
Karamlou T, Gurofsky R, Al Sukhni E, Coles
JG, Williams WG, Caldarone CA, Van Arsdell GS and McCrindle BW:
Factors associated with mortality and reoperation in 377 children
with total anomalous pulmonary venous connection. Circulation.
115:1591–1598. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shi X, Cheng L, Jiao X, Chen B, Li Z,
Liang Y, Liu W, Wang J, Liu G, Xu Y, et al: Rare copy number
variants identify novel genes in sporadic total anomalous pulmonary
vein connection. Front Genet. 9:5592018. View Article : Google Scholar
|
|
18
|
Konduri A and Aggarwal S: Partial and
total anomalous pulmonary venous connection. StatPearls [Internet]
Treasure Island (FL): StatPearls Publishing; 2021
|
|
19
|
Marcus RH, Marcus BD and Levin SE: The
upper limb-cardiovascular syndrome (Holt-Oram syndrome) in a South
African family. S Afr Med J. 67:1013–1014. 1985.PubMed/NCBI
|
|
20
|
Sahn DJ, Goldberg SJ, Allen HD and Canale
JM: Cross-sectional echocardiographic imaging of supracardiac total
anomalous pulmonary venous drainage to a vertical vein in a patient
with Holt-Oram syndrome. Chest. 79:113–115. 1981. View Article : Google Scholar
|
|
21
|
Szot JO, Cuny H, Blue GM, Humphreys DT, Ip
E, Harrison K, Sholler GF, Giannoulatou E, Leo P, Duncan EL, et al:
A screening approach to identify clinically actionable variants
causing congenital heart disease in exome data. Circ Genomic Precis
Med. 11:e0019782018. View Article : Google Scholar
|
|
22
|
Yang Y, Muzny DM, Reid JG, Bainbridge MN,
Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al:
Clinical whole-exome sequencing for the diagnosis of mendelian
disorders. N Engl J Med. 369:1502–1511. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Volk A, Conboy E, Wical B, Patterson M and
Kirmani S: Whole-exome sequencing in the clinic: Lessons from six
consecutive cases from the clinician's perspective. Mol Syndromol.
6:23–31. 2015. View Article : Google Scholar
|
|
24
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Camacho C, Coulouris G, Avagyan V, Ma N,
Papadopoulos J, Bealer K and Madden TL: BLAST+: Architecture and
applications. BMC Bioinformatics. 10:4212009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Remmert M, Biegert A, Hauser A and Söding
J: HHblits: Lightning-fast iterative protein sequence searching by
HMM-HMM alignment. Nat Methods. 9:173–175. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Waterhouse A, Bertoni M, Bienert S, Studer
G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C,
Bordoli L, et al: SWISS-MODEL: Homology modelling of protein
structures and complexes. Nucleic Acids Res. 46:W296–W303. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guex N, Peitsch MC and Schwede T:
Automated comparative protein structure modeling with SWISS-MODEL
and Swiss-PdbViewer: A historical perspective. Electrophoresis. 30
(Suppl 1):S162–S173. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Williams CJ, Headd JJ, Moriarty NW,
Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen
VB, et al: MolProbity: More and better reference data for improved
all-atom structure validation. Protein Sci. 27:293–315. 2018.
View Article : Google Scholar
|
|
31
|
Lovell SC, Davis IW, Arendall WB III, de
Bakker PI, Word JM, Prisant MG, Richardson JS and Richardson DC:
Structure validation by Calpha geometry: Phi, psi and Cbeta
deviation. Proteins. 50:437–450. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jiang WF, Xu YJ, Zhao CM, Wang XH, Qiu XB,
Liu X, Wu SH and Yang YQ: A novel TBX5 mutation predisposes to
familial cardiac septal defects and atrial fibrillation as well as
bicuspid aortic valve. Genet Mol Biol. 43:e202001422020. View Article : Google Scholar
|
|
33
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li QY, Newbury-Ecob RA, Terrett JA, Wilson
DI, Curtis AR, Yi CH, Gebuhr T, Bullen PJ, Robson SC, Strachan T,
et al: Holt-Oram syndrome is caused by mutations in TBX5, a member
of the Brachyury (T) gene family. Nat Genet. 15:21–29. 1997.
View Article : Google Scholar
|
|
35
|
Boogerd CJ, Dooijes D, Ilgun A, Mathijssen
IB, Hordijk R, van de Laar IM, Rump P, Veenstra-Knol HE, Moorman
AF, Barnett P and Postma AV: Functional analysis of novel TBX5
T-box mutations associated with Holt-Oram syndrome. Cardiovasc Res.
88:130–139. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
McDermott DA, Bressan MC, He J, Lee JS,
Aftimos S, Brueckner M, Gilbert F, Graham GE, Hannibal MC, Innis
JW, et al: TBX5 genetic testing validates strict clinical criteria
for Holt-Oram syndrome. Pediatr Res. 58:981–986. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nogueira G, Fernandes R, García-Moreno JF
and Romão L: Nonsense-mediated RNA decay and its bipolar function
in cancer. Mol Cancer. 20:722021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Steimle JD and Moskowitz IP: TBX5: A key
regulator of heart development. Current Topics in Developmental
Biology. Vol. 122. Academic Press Inc.; Cambridge, MA, USA: pp.
195–221. 2017, View Article : Google Scholar
|
|
39
|
Basson CT, Huang T, Lin RC, Bachinsky DR,
Weremowicz S, Vaglio A, Bruzzone R, Quadrelli R, Lerone M, Romeo G,
et al: Different TBX5 interactions in heart and limb defined by
Holt-Oram syndrome mutations. Proc Natl Acad Sci USA. 96:2919–2924.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Brassington AM, Sung SS, Toydemir RM, Le
T, Roeder AD, Rutherford AE, Whitby FG, Jorde LB and Bamshad MJ:
Expressivity of Holt-Oram syndrome is not predicted by TBX5
genotype. Am J Hum Genet. 73:74–85. 2003. View Article : Google Scholar
|
|
41
|
Tarniceriu CC, Hurjui LL, Tanase DM,
Nedelcu AH, Gradinaru I, Ursaru M, Stefan Rudeanu A, Delianu C and
Lozneanu L: The pulmonary venous return from normal to
pathological-clinical correlations and review of literature.
Medicina (Kaunas). 57:2932021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Patterson J, Coats C and McGowan R:
Familial dilated cardiomyopathy associated with pathogenic TBX5
variants: Expanding the cardiac phenotype associated with Holt-Oram
syndrome. Am J Med Genet Part A. 182:1725–1734. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gruenauer-Kloevekorn C and Froster UG:
Holt-Oram syndrome: A new mutation in the TBX5 gene in two
unrelated families. Ann Genet. 46:19–23. 2003. View Article : Google Scholar
|
|
44
|
Borozdin W, Bravo Ferrer Acosta AM,
Bamshad MJ, Botzenhart EM, Froster UG, Lemke J, Schinzel A,
Spranger S, McGaughran J, Wand D, et al: Expanding the spectrum of
TBX5 mutations in Holt-Oram syndrome: Detection of two intragenic
deletions by quantitative real time PCR, and report of eight novel
point mutations. Hum Mutat. 27:975–976. 2006. View Article : Google Scholar
|
|
45
|
Heinritz W, Moschik A, Kujat A, Spranger
S, Heilbronner H, Demuth S, Bier A, Tihanyi M, Mundlos S,
Gruenauer-Kloevekorn C and Froster UG: Identification of new
mutations in the TBX5 gene in patients with Holt-Oram syndrome.
Heart. 91:383–384. 2005. View Article : Google Scholar
|
|
46
|
Ríos-Serna LJ, Díaz-Ordoñez L, Candelo E
and Pachajoa H: A novel de novo TBX5 mutation in a patient with
Holt-oram syndrome. Appl Clin Genet. 11:157–162. 2018. View Article : Google Scholar
|
|
47
|
Ross SB, Bagnall RD, Yeates L, Sy RW and
Semsarian C: Holt-Oram syndrome in two families diagnosed with left
ventricular noncompaction and conduction disease. HeartRhythm Case
Rep. 4:146–151. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Vianna CB, Miura N, Pereira AC and Jatene
MB: Holt-Oram syndrome: Novel TBX5 mutation and associated
anomalous right coronary artery. Cardiol Young. 21:351–353. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Qin L, Lou G, Guo L, Zhang Y, Wang H, Wang
L, Hou Q, Liu H, Li X and Liao S: Targeted next-generation
sequencing-based molecular diagnosis of congenital hand
malformations in Chinese population. Sci Rep. 8:127212018.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Atik T, Dervisoglu H, Onay H, Ozkinay F
and Cogulu O: A new mutation in the TBX5 gene in Holt-Oram
syndrome: Two cases in the same family and prenatal diagnosis. J
Trop Pediatr. 60:257–259. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ma JF, Yang F, Mahida SN, Zhao L, Chen X,
Zhang ML, Sun Z, Yao Y, Zhang YX, Zheng GY, et al: TBX5 mutations
contribute to early-onset atrial fibrillation in Chinese and
Caucasians. Cardiovasc Res. 109:442–450. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yang J, Hu D, Xia J, Yang Y, Ying B, Hu J
and Zhou X: Three novel TBX5 mutations in Chinese patients with
Holt-Oram syndrome. Am J Med Genet. 92:237–240. 2000. View Article : Google Scholar
|
|
53
|
Debeer P, Race V, Gewillig M, Devriendt K
and Frijns JP: Novel TBX5 mutations in patients with Holt-Oram
syndrome. Clin Orthop Relat Res. 462:20–26. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Fan C, Duhagon MA, Oberti C, Chen S, Hiroi
Y, Komuro I, Duhagon PI, Canessa R and Wang Q: Novel TBX5 mutations
and molecular mechanism for Holt-Oram syndrome. J Med Genet.
40:e292003. View Article : Google Scholar
|