Introduction
Myocardial infarction (MI) is an ischemic heart
condition that poses a significant burden to society and may lead
to death or disability (1–3).
Myocardial ischemia-reperfusion (I/R) injury is considered as the
main pathophysiology of ischemic heart diseases (4). It has been reported that acute and
chronic cell death following MI affects cardiac function and
patient prognosis (5,6). A previous study has reported that
the apoptotic cascade in myocardial I/R is initiated either by
mitochondrial damage and activation of caspase-9 or by death
receptor ligation and activation of caspase-8 (7). Myocardial ischemia may induce
inflammatory responses, while reperfusion may promote cell
apoptosis, thus resulting in impaired myocardial structure and
function (8–10). Therefore, inhibiting inflammatory
responses and apoptosis following I/R could be considered as an
effective strategy for preventing myocardial I/R injury.
C1q/TNF-related proteins (CTRPs) are a family of
adipokines, similar to adiponectin, which exert numerous biological
activities, including anti-atherosclerotic, insulin sensitivity,
anti-inflammatory and vascular functions (11). CTRP12, also known as adipolin, is
a member of the CTRP family, which is abundantly expressed in the
fat tissue (12). It has
previously been reported that CTRP12 is involved in the protection
against metabolic disorders such as type 2 diabetes,
atherosclerosis and obesity (13–15). Furthermore, it has been suggested
that CTRP12 may serve a critical role in cardiovascular injury
(16). Another study has
demonstrated that CTRP12 could alleviate lipopolysaccharide
(LPS)-induced cardiomyocyte injury by preventing inflammation and
apoptosis (17), which suggests
that CTRP12 may be tightly linked with the cardiovascular system.
However, its role in myocardial I/R injury has not been previously
reported.
Krueppel-like factor 15 (KLF15), a member of the
zinc-finger family of transcription factors, is tightly associated
with several disorders, including inflammation, obesity and
metabolic dysfunction (18).
Emerging evidence has indicated that KLF15 enhances the activity of
the CTRP12 promoter and regulates CTRP12 expression in adipocytes
(19). Furthermore, another study
reported that KLF15 attenuates hypoxia-induced apoptosis and
oxidative stress in myocardial cells (20). Therefore, the aim of the present
study was to evaluate the functional role of CTRP12 in myocardial
I/R injury and explore the possible underlying molecular mechanisms
of CTRP12 and KLF15.
Materials and methods
Animals
The present study was approved by the Animal Care
and Use Committee of the Shenzhen Peking University, The Hong Kong
University of Science and Technology Medical Center (Shenzhen,
China) and conducted in accordance with Chinese legislation
regarding animal experiments (21). A total of 30 c57BL6/J male mice
(age, 8 weeks; weight, 18–22 g) were purchased from the Comparative
Medicine Centre of Yangzhou University and housed under standard
conditions of 25°C, with a relative humidity of 60%, in a 12-h
light/dark cycle with free access to standard laboratory food and
water.
Establishment of the myocardial I/R
model
To establish the I/R mouse model, the left anterior
descending coronary artery (LAD) was ligated (22). Mice were anesthetized using
isoflurane (4% for induction and 2% for maintenance). Subsequently,
a 6–0 silk suture slipknot was placed around the LAD and was
released after 30 min. During the operation, mice were
subcutaneously injected with 0.05 mg/kg buprenorphine hydrochloride
as an intraoperative analgesic. Mice in the sham group underwent
the same surgical procedure but without LAD ligation. Following the
operation, 0.1 ml 5% glucose was subcutaneously injected into the
mice to prevent dehydration. Following recovery from anesthesia,
mice received buprenorphine hydrochloride (0.05 mg/kg
subcutaneously) for post-operative analgesia (23–26). Animals were sacrificed by cervical
dislocation following anesthesia at 24 h following reperfusion and
myocardial tissue samples were subsequently collected and stored at
−80°C for further analysis.
Histopathological examination
Myocardial tissue specimens derived from mice who
underwent myocardial I/R and sham surgery were collected and fixed
using 4% paraformaldehyde for 24 h at room temperature.
Subsequently, the tissues were dehydrated in different
concentrations of ethanol, embedded in paraffin and sliced into
4-µm-thick sections. The tissue sections were then stained with
hematoxylin (0.4%) and eosin (0.1%) (H&E) solution at 37°C for
5 min and the pathological changes after myocardial I/R injury were
observed using a light microscope.
Cell culture and hypoxia-reoxygenation
(H/R) treatment
The rat myoblast H9c2 cell line was obtained from
the American Type Culture Collection. Cells were maintained in DMEM
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS
(Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin solution (Gibco; Thermo Fisher Scientific,
Inc.) in a humidified atmosphere with 5% CO2 at 37°C. To
establish an in vitro cardiomyocyte I/R model, H9c2 cells
were cultured in glucose-free/serum-free DMEM and maintained in an
incubator containing a gaseous mixture of 94% N2, 5%
CO2 and 1% O2 at 37°C for 4 h. Following
incubation, cells were transferred to 30 mM high-glucose DMEM,
containing 10% FBS, was cultured under normoxic conditions for 4 h
at 37°C for reoxygenation. H9c2 cells were not previously cultured
under hypoxic conditions and these cells were cultured in high
glucose DMEM containing 10% FBS under normoxic conditions and
served as the control group.
Cell transfection
CTRP12- or KLF15-specific pcDNA overexpression
plasmids [overexpressed (Ov)-CTRP12 or Ov-KLF15] and the
corresponding negative control (NC; Ov-NC), CTRP12-specific small
interfering (si) RNA-CTRP12 (siRNA-CTRP12) and the corresponding NC
(siRNA-NC), were synthesized by Shanghai GenePharma Co., Ltd. The
following siRNA sequences were used: CTRP12 forward (F),
5′-CUGUGACGGUAGACAAGAAGA-3′ and reverse (R),
5′-UUCUUGUCUACCGUCACAGGG-3′; and siRNA-NC F,
5′-GAUCAUACGUGCGAUCAGA-3′ and R, 5′-UCUGAUCGCACGUAUGAUC-3′. H9c2
cells (1×105 cells/well) were transfected with the
recombinants (1 µg) using Lipofectamine® 2000 reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions for 6 h at room temperature. After 48 h
cells underwent H/R injury.
Cell viability assay
The Cell Counting Kit-8 (CCK-8) assay was utilized
to detect cell viability (27).
Briefly, at 48 h following transfection, H9c2 cells were seeded
into 96-well plates at a density of 5×103 cells/well and
treated with H/R for 8 h (4 h hypoxia followed by 4 h reperfusion)
at 37°C. Subsequently, each well was supplemented with 10 µl CCK-8
solution (Beyotime Institute of Biotechnology) and cells were
incubated at 37°C for a further 2 h. The absorbance of each well
was measured at a wavelength of 450 nm using a microplate reader
(Bio-Rad Laboratories, Inc.).
Detection of lactate dehydrogenase
(LDH) activity
H9c2 cells were seeded into 96-well plates at a
density of 5×103 cells/well and cultured for 4 h of
hypoxia and 4 h of reperfusion at 37°C. The cell supernatants were
harvested using centrifugation at 1,000 × g for 15 min at 4°C to
quantify the secretory levels of LDH using the LDH Assay Kit (cat.
no. A020-2-2; Nanjing Jiancheng Bioengineering Institute) according
to the manufacturer's protocol.
ELISA
The secretory levels of TNF-α and IL-6 were measured
using the TNF-α Assay Kit (cat. no. H052-1; Nanjing Jiancheng
Bioengineering Institute) and IL-6 Kit (cat. no. H007-1-1; Nanjing
Jiancheng Bioengineering Institute) according to the manufacturer's
protocol. IL-1β levels were detected using the Human IL-1β ELISA
Kit (cat. no. ab214025; Abcam) according to the manufacturer's
protocol. Culture supernatants from H9c2 cells (2×104
cells/well) were cultured at 37°C under 4 h of hypoxia and 4 h of
reperfusion and were collected. Subsequently, 50 µl supernatant was
added to each well. The optical density was detected at 450 nm
using a microplate reader (BioTek Instruments, Inc.). A total of
eight parallel wells were used for each ELISA.
Cell apoptosis
The TUNEL assay was performed to evaluate cell
apoptosis (28). Briefly,
following treatment, H9c2 cells (2×106 cells/well) were
washed twice with PBS. Following fixation with 4% paraformaldehyde
for 15 min at room temperature, cells were incubated with 15 µg/ml
proteinase K for 15 min at 37°C and endogenous peroxidase activity
was blocked using 3% H2O2 for 15 min at room
temperature. After washing, cells were treated with TUNEL working
solution for 60 min at 37°C, followed by co-labeling with DAPI (0.5
µg/ml) for 5 min at room temperature. The cells were mounted in
Antifade Mounting Medium (Beyotime Institute of Biotechnology) and
cells from five randomly selected fields of view were captured
using a fluorescence microscope (Leica Microsystems GmbH).
Apoptosis was quantified using ImageJ v1.8.0 software (National
Institutes of Health).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from murine myocardial
tissues and H9c2 cells using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). Subsequently, RNA was
reverse transcribed into first-strand cDNA using the PrimeScript RT
Master Mix (Takara Bio, Inc.) according to the manufacturer's
instructions. qPCR was performed using the SYBR Premix ExTaq Kit
(Takara Bio, Inc.) using the ABI PRISM 7900 Real-Time System
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The
thermocycling conditions were as follows: initial denaturation at
95°C for 30 sec; 40 cycles of denaturation at 95°C for 5 sec,
annealing at 60°C for 30 sec and extension at 72°C for 60 sec. The
program was terminated via final extension at 72°C for 1 min and
cooling at 40°C. The primer sequences used for qPCR were as
follows: CTRP12 F, 5′-CCTGTCCTTGGGCCGATTTA-3′ and R,
5′-CAGGGACGTATGACGGTGAC-3′; KLF15 F, 5′-GGGATCGTGGAGGAGAGCCT-3′ and
R, 5′-CCAGCTGAGAGCTGGCTACA-3′; and GAPDH F,
5′-GTCAAGGCTGAGAACGGGAA-3′ and R 5′-AAATGAGCCCCAGCCTTCTC-3′. The
mRNA expression levels were quantified using the 2−ΔΔCq
method and were normalized using the internal reference gene GAPDH
(29).
Western blotting
Total protein was extracted from myocardial tissues
and H9c2 cells using RIPA buffer (Auragene; Hunan Aijia
Biotechnology Co., Ltd.). Total protein (30 µg protein/lane) was
separated via SDS-PAGE (Bio-Rad Laboratories, Inc.) on a 10% gel.
Separated protein was transferred onto a PVDF membrane
(MilliporeSigma). Following blocking with 5% skimmed milk for 1 h
at room temperature, the membranes were incubated overnight at 4°C
with primary antibodies targeting CTRP12 (1:1,000; cat. no.
ab177991), KLF15 (1:1,000; cat. no. ab2647), Bax (1:2,000; cat. no.
ab182733), cleaved caspase-3 (1:500; cat. no. ab32042), cleaved
poly (ADP-ribose) polymerase (PARP; 1:1,000; cat. no. ab32064),
Bcl-2 (1:1,000; cat. no. ab194583) and GAPDH (1:3,000; cat. no.
ab125247) (all purchased from Abcam). Subsequently, the membranes
were washed four times with PBS and incubated with horseradish
peroxidase-labeled goat anti-rabbit IgG secondary antibodies
(1:3,000; cat. no. 7074S; Cell Signaling Technology, Inc.) for 1 h
at room temperature. The protein bands were visualized using an
Amersham™ ECL™ Prime Western Blotting
Detection Reagent (Amersham; Cytiva) and analyzed using ImageJ
software v1.8.0 (National Institutes of Health).
Bioinformatic analysis
The binding site between KLF15 and the CTRP12
promoter was analyzed using the JASPAR database 2018 (https://jaspar.genereg.net/). The JASPAR database is
an open-access database containing manually curated, non-redundant
transcription factor (TF) binding profiles for TFs across six
taxonomic groups.
Dual-luciferase reporter assay
The activity of the CTRP12 promoter was evaluated
using a dual-luciferase reporter assay (30). Briefly, the CTRP12 promoter
covering predicted binding sites was cloned into the pGL3 basic
plasmid (Shanghai Genomeditech Co., Ltd.) with the
luciferase/Renilla reporter gene. The Ov-KLF15 vector or the
empty vector and luciferase/Renilla plasmids were
co-transfected into H9c2 cells (1×105 cells/well) using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) for
24 h at room temperature. Each well was then supplemented with 100
µl Renilla Luciferase Reporter Gene Assay Cell Lysis Buffer
(Beyotime Institute of Biotechnology) and the lysed cells were
centrifuged at 5,000 × g for 1 min at 4°C to collect the
supernatants. Subsequently, the cells were supplemented with 20 µl
lysis buffer and then with 100 µl 1X firefly and 1X Renilla
luciferase reaction solutions (Wuhan Scithera Microbiological
Technologies, Inc.) to detect firefly and Renilla luciferase
activity, respectively. After 48 h, luciferase activity was
measured using the Dual Luciferase Reporter System, purchased from
Promega Corporation, according to the manufacturer's instructions.
The results are presented as the ratio of luciferase to
Renilla activity.
Statistical analysis
All data are presented as the mean ± SD. All
statistical analyses were carried out using GraphPad Prism 6
software (GraphPad Software, Inc.) using unpaired two-tailed
Student's t-tests or a one-way ANOVA followed by a Bonferroni's
post hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
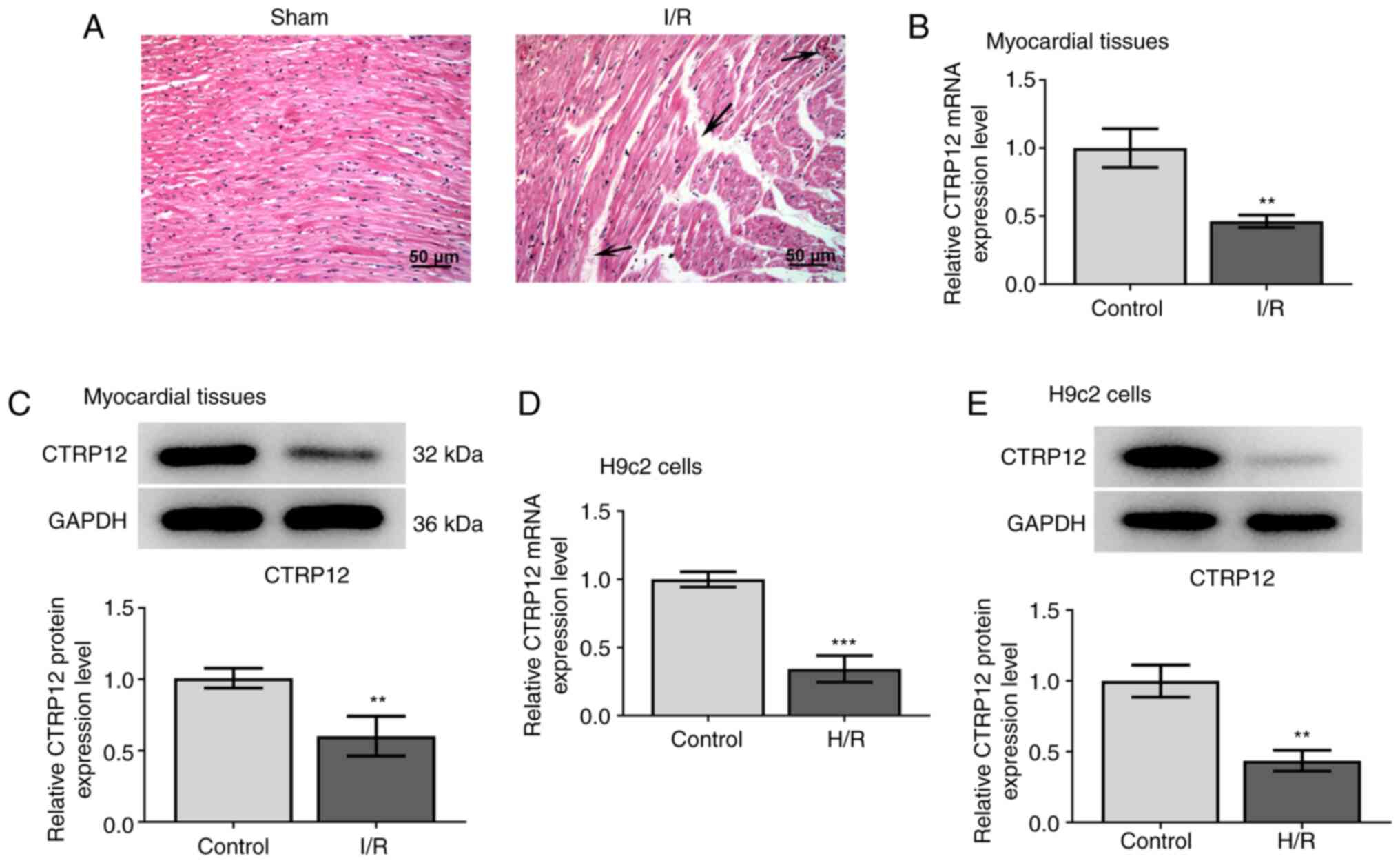
CTRP12 is downregulated in I/R-induced
myocardial tissues and H/R-induced cardiomyocytes
To explore the role of CTRP12 in myocardial I/R
injury, a myocardial I/R mouse model was established. H&E
staining demonstrated that mice who underwent the sham operation
exhibited normal myocardial tissue structure, whereas those in the
I/R group displayed damaged myocardial fiber structure, broken
vascular walls and hemocyte and inflammatory molecule infiltration
in the myocardial tissue (Fig.
1A), indicating that the model was successfully established.
Subsequently, the mRNA expression levels of CTRP12 were determined
in myocardial tissue samples and H9c2 cardiomyocytes using RT-qPCR
and western blotting. The mRNA and protein expression levels of
CTRP12 were significantly decreased in both I/R-induced myocardial
tissues and H/R-induced H9c2 cells compared with the corresponding
control groups (Fig. 1B-E). These
results suggested that the decreased expression of CTRP12 was
associated with myocardial I/R injury.
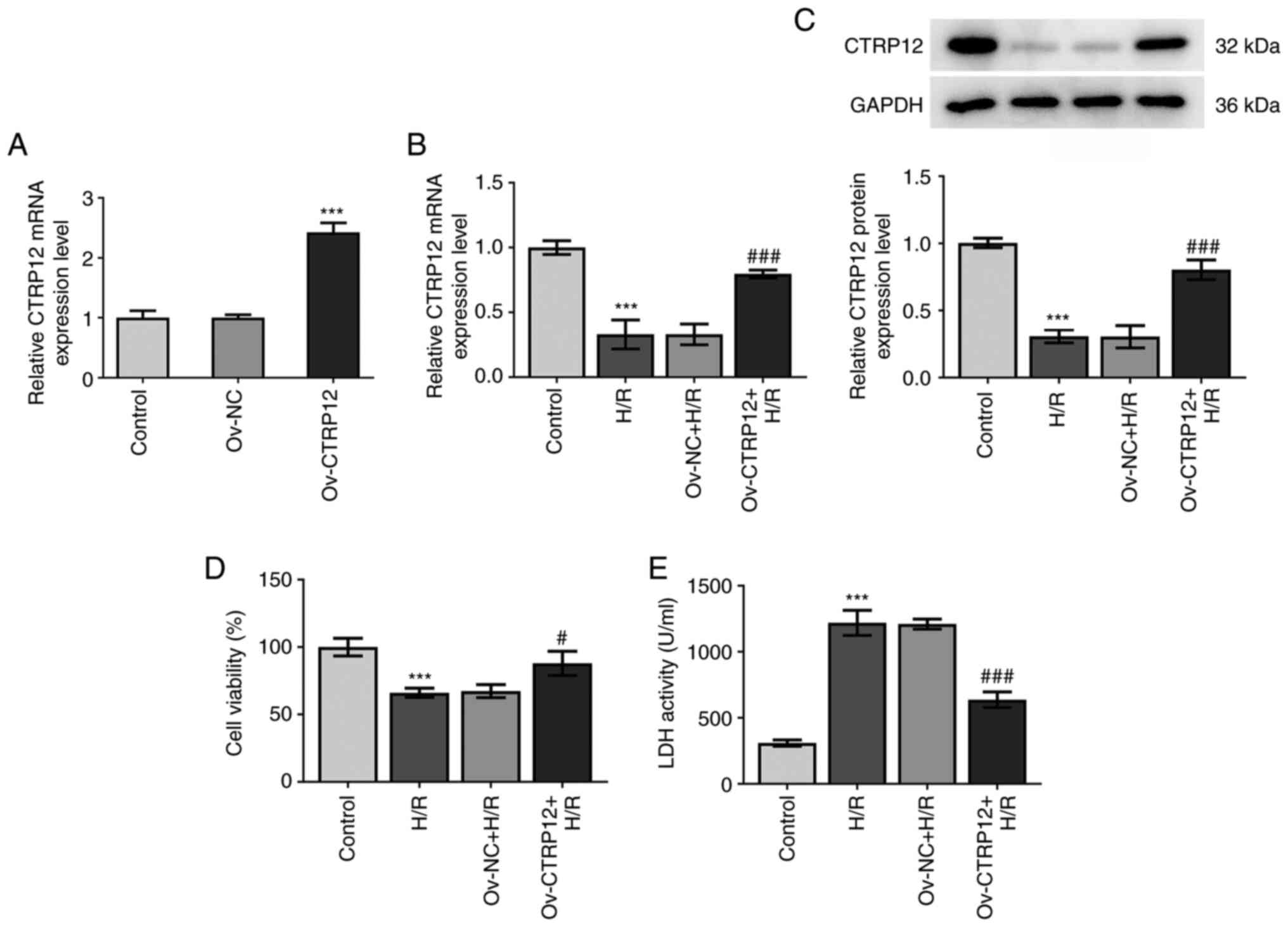
CTRP12 overexpression alleviates
H/R-induced cell injury in H9c2 cells
To uncover the biological role of CTRP12 in
hypoxia-induced cardiomyocytes, H9c2 cells were transfected with
Ov-CTRP12 to enhance CTRP12 expression. The transfection efficiency
of Ov-CTRP12 was evaluated using RT-qPCR and it was demonstrated
that the mRNA expression levels of CTRP12 were significantly
elevated compared with the Ov-NC group (Fig. 2A). The results demonstrated that
CTRP12 mRNA and protein expression levels were significantly
downregulated under H/R conditions compared with the control group,
which was significantly reversed following transfection with
Ov-CTRP12 in H/R cells compared with the H/R only group (Fig. 2B and C). The CCK-8 assay was
performed to measure the effects of CTRP12 overexpression on cell
viability and the results demonstrated that H/R injury
significantly reduced cell viability compared with the control.
However, CTRP12 overexpression significantly restored the
H/R-mediated reduced cell viability compared with the H/R group
(Fig. 2D). Furthermore, the
levels of LDH were measured to determine the severity of
cardiomyocyte injury. The results demonstrated that LDH activity
was significantly enhanced in hypoxic cardiomyocytes compared with
the control, whereas CTRP12 overexpression significantly reversed
this effect compared with the H/R group (Fig. 2E). The aforementioned findings
indicated that CTRP12 overexpression exerted a protective role
against H/R-induced myocardial cell injury.
CTRP12 overexpression relieves the
secretion of inflammatory factors and apoptosis in H/R-induced
cells
Subsequently, the present study aimed to investigate
whether CTRP12 overexpression could potentially protect H9c2 cells
from H/R-induced inflammation and apoptosis. The secretory levels
of TNF-α, IL-1β and IL-6 were significantly elevated by H/R injury
compared with the control, whereas their levels were significantly
reduced following CTRP12 overexpression compared with the H/R group
(Fig. 3A-C). Furthermore, the
TUNEL assay results demonstrated that H/R treatment significantly
promoted cardiomyocyte apoptosis compared with the control, whereas
CTRP12 overexpression significantly prevented the effects of H/R
induction on cardiomyocyte apoptosis compared with the H/R group
(Fig. 3D and E). Moreover,
western blotting demonstrated that compared to the control the
protein expression levels of Bax, cleaved caspase-3 and cleaved
PARP were significantly increased, whereas those of Bcl-2 were
significantly decreased in H/R-treated cells. However, CTRP12
overexpression significantly reversed the effects of H/R exposure
on the expression profile of the aforementioned proteins, compared
with the H/R group (Fig. 3F).
These results suggested that CTRP12 overexpression may ameliorate
hypoxia-induced H9c2 cell injury via attenuating inflammation and
apoptosis.
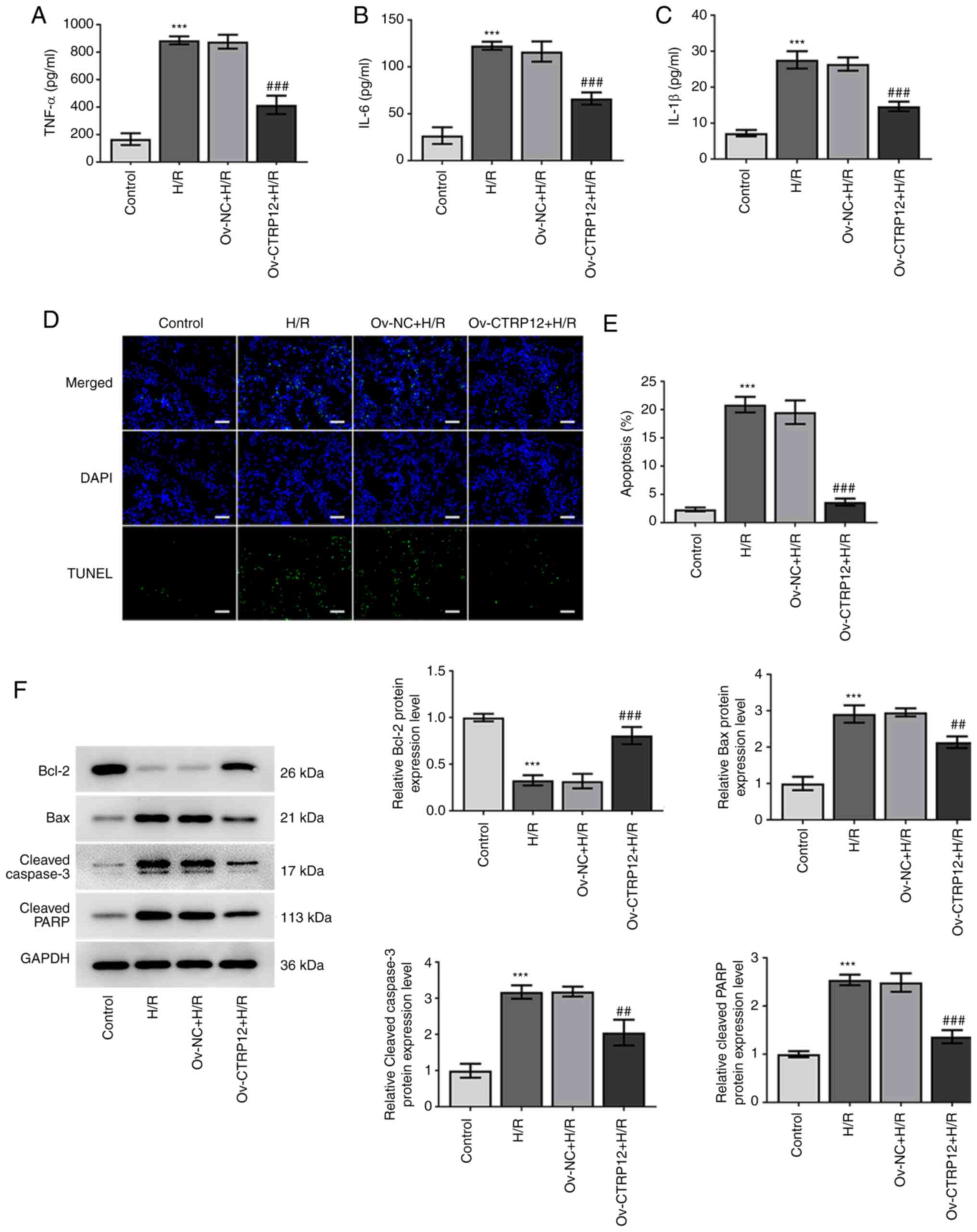 | Figure 3.Overexpression of CTRP12 inhibits the
release of inflammatory factors and apoptosis in H/R-induced H9c2
cells. (A) TNF-α, (B) IL-1β and (C) IL-6 levels were quantified
using ELISA. (D and E) Cell apoptosis was detected using the TUNEL
assay. Scale bar, 100 µm. (F) Western blotting was performed to
assess protein expression levels of Bcl-2, Bax, cleaved caspase-3
and cleaved PARP. Results are presented as the mean ± SD analyzed
using three independent experiments. ***P<0.001 vs. control; and
##P<0.01, ###P<0.001 vs. H/R group.
CTRP12, C1q/TNF-related protein 12; H/R, hypoxic/reoxygenated;
PARP, poly(ADP-ribose) polymerase; Ov, overexpressed; NC, negative
control. |
KLF15 enhances the activity of the
CTRP12 promoter and regulates CTRP12 expression levels
To elucidate whether KLF15 was involved in the
regulation of CTRP12 expression in myocardial I/R injury, the
expression levels of KLF15 were determined in I/R-induced
myocardial tissues and H/R-induced cardiomyocytes. The results
demonstrated that the mRNA and protein expression levels of KLF15
were significantly reduced in both myocardial tissues and H9c2
cells compared with those in the untreated control groups (Fig. 4A-D). The transfection efficiency
of Ov-KLF15 was assessed using RT-qPCR and the results demonstrated
that that the KLF5 mRNA expression levels were significantly
elevated in the Ov-KLF15 group (Fig.
4E). Subsequently, H/R-treated H9c2 cells were transfected with
Ov-KLF15 to overexpress KLF15 and the results demonstrated that
KLF15 mRNA expression in the Ov-KLF15 + H/R group was significantly
increased compared with the Ov-NC + H/R group (Fig. 4F and G). The mRNA and protein
expression levels of CTRP12 were significantly increased in
KLF15-overexpressing H9c2 cells compared with the H/R group
(Fig. 4H and I). The binding site
of the KLF15 and CTRP12 promoter was predicted using the JASPAR
database (Fig. 4J). The results
of the dual-luciferase reporter assay demonstrated that the
luciferase activity was significantly enhanced in cells transfected
with Ov-KLF15 after exposure to H/R compared with the H/R group
(Fig. 4K). These results
therefore suggested that KLF15 overexpression may increase the
promoter activity of CTRP12.
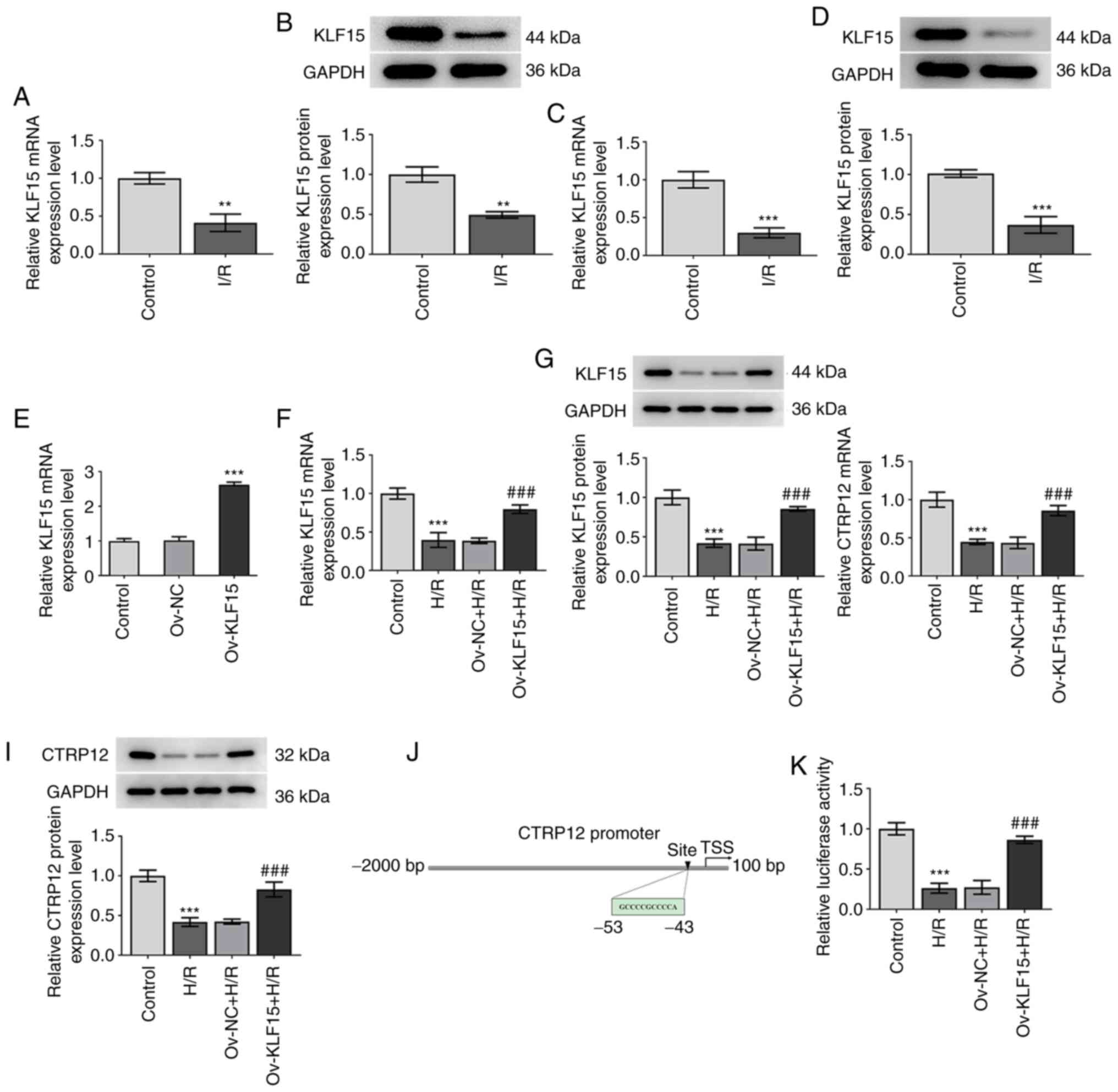 | Figure 4.KLF15 regulates CTRP12 expression
levels in H9c2 cells. (A) mRNA and (B) protein expression levels of
KLF15 were determined via RT-qPCR and western blotting,
respectively, in myocardial tissue in I/R-induced mice. (C) mRNA
and (D) protein expression levels of KLF15 were determined using
RT-qPCR and western blotting, respectively, in H/R-induced H9c2
cells following exposure to H/R compared with the control. (E)
Transfection efficiency of Ov-KLF15 was assessed using reverse
transcription-quantitative PCR. (F) mRNA and (G) protein expression
levels of KLF15 in H/R cells were detected following transfection
with Ov-KLF15. (H) mRNA and (I) protein expression levels of CTRP12
were assessed following transfection with Ov-KLF15. (J) Binding
site of KLF15 and CTRP12 promoter was predicted using the JASPAR
database. (K) CTRP12 promoter activity was determined using the
dual-luciferase reporter assay. Results are presented as the mean ±
SD analyzed using three independent experiments. **P<0.01,
***P<0.001 vs. control; and ###P<0.001 vs. H/R
group. KLF15, Krueppel-like factor 15; CTRP12, C1q/TNF-related
protein 12; I/R, ischemic/reperfused; H/R, hypoxic/reoxygenated;
RT-qPCR, reverse transcription-quantitative PCR; Ov, overexpressed;
NC, negative control; TSS, transcriptional start site. |
KLF15 overexpression attenuates
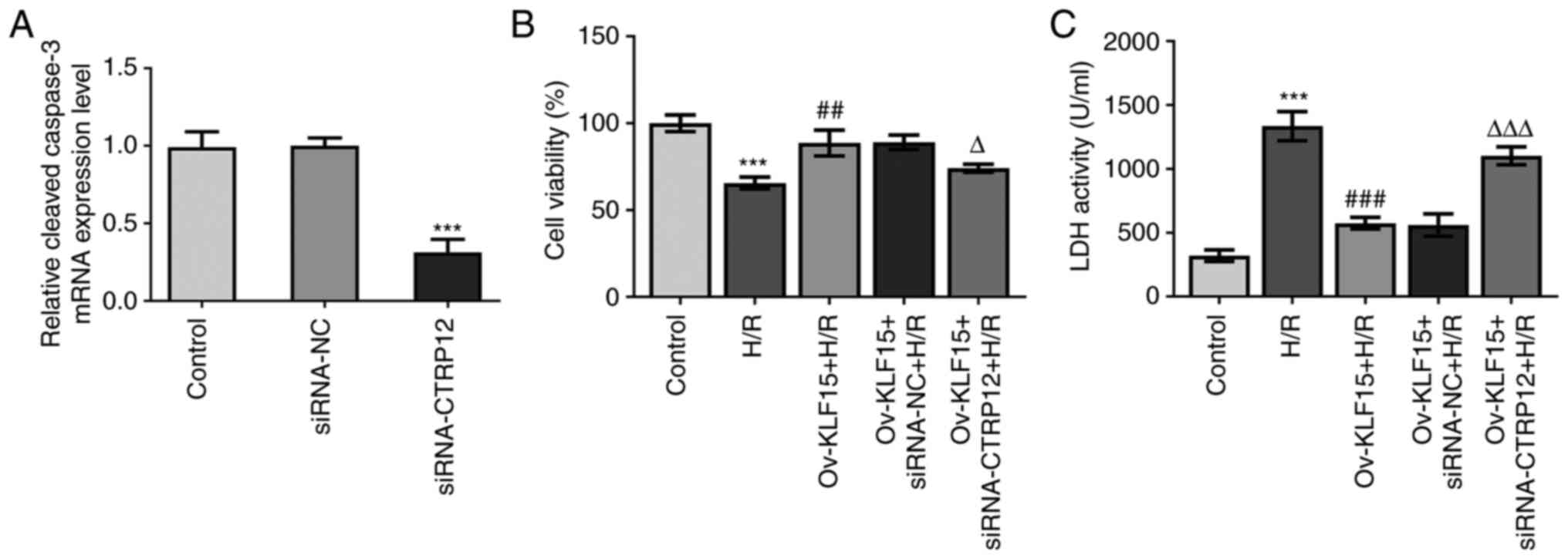
H/R-induced H9c2 cell injury via regulation of CTRP12
Based on the aforementioned findings, the present
study further investigated the effect of KLF15 on regulating CTRP12
expression levels in cardiomyocytes. The transfection efficiency of
siRNA-CTRP12 was assessed using RT-qPCR and the results
demonstrated that CTRP12 mRNA expression were significantly reduced
in the siRNA-CTRP12 group compared with the siRNA-NC (Fig. 5A). Compared with the H/R group,
KLF15 overexpression significantly rescued H/R-mediated reduced
cell viability in H/R-treated H9c2 cells (Fig. 5B). Furthermore, CTRP12 silencing
significantly reduced the effects of H/R on cell viability compared
with the Ov-KLF15 + siRNA-NC + H/R group. LDH activity was also
assessed to determine the degree of H9c2 cell injury and the
results demonstrated that CTRP12 silencing significantly elevated
LDH activity in H/R-treated H9c2 cells transfected with Ov-KLF15
compared with the Ov-KLF15 + siRNA-NC + H/R group (Fig. 5C). These results therefore
supported the involvement of KLF15 in CTRP12-mediated H/R-induced
cardiomyocyte injury.
KLF15 overexpression inhibits
inflammation and apoptosis in H/R-induced H9c2 cells via regulation
of CTRP12
The effects of KLF15 overexpression on H/R-induced
inflammation and apoptosis were evaluated in CTRP12-silenced H9c2
cells. The secretory levels of TNF-α, IL-1β and IL-6 were
significantly reduced by KLF15 overexpression compared with the H/R
group, whereas this effect was significantly reversed following
CTRP12 knockdown compared with the Ov-KLF15 + siRNA-NC + H/R group
(Fig. 6A-C). Furthermore, the
apoptotic rate of H/R-induced cells was significantly decreased
following KLF15 overexpression compared with the H/R group, whereas
CTRP12 silencing resulted in a significant increase in the
apoptotic rate in H/R-treated H9c2 cells transfected with Ov-KLF15
compared with the Ov-KLF15 + siRNA-NC + H/R group (Fig. 6D and E). Furthermore, western
blotting demonstrated that the protein expression levels of Bax,
cleaved caspase-3 and cleaved PARP were significantly decreased,
whereas Bcl-2 protein expression levels were significantly
increased in the Ov-KLF15 + H/R group compared with the H/R group.
However, transfection of H9c2 cells with si-CTRP12 significantly
upregulated Bax, cleaved caspase-3 and cleaved PARP, and
significantly downregulated Bcl-2 compared with the Ov-KLF15 +
siRNA-NC + H/R group (Fig. 6F).
These data suggested that KLF15 overexpression may attenuate
H/R-induced inflammation and apoptosis in H9c2 cells via the
regulation of CTRP12 expression.
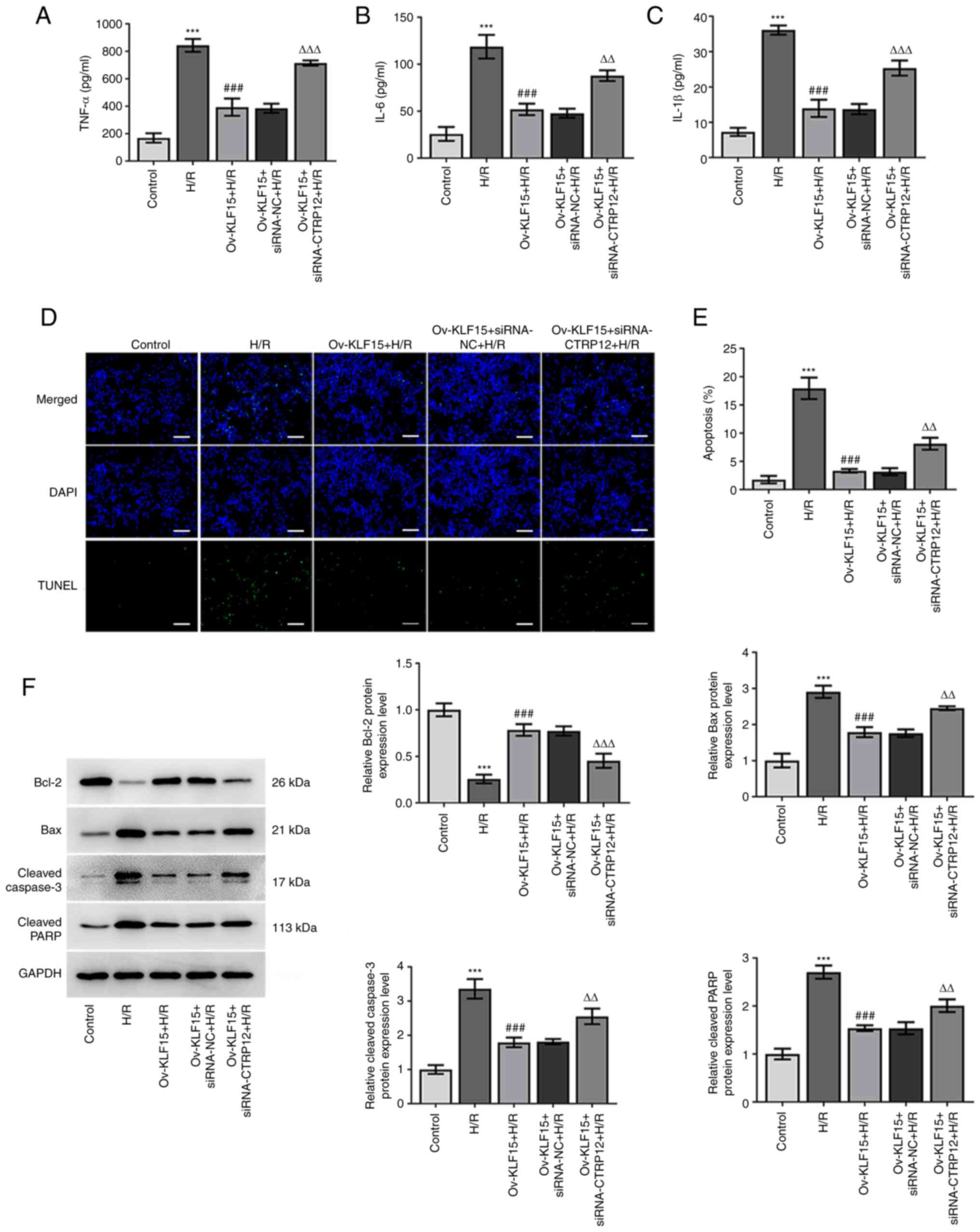 | Figure 6.KLF15 overexpression suppresses H/R
induced inflammation and apoptosis in H9c2 cells by regulating
CTRP12. (A) TNF-α, (B) IL-1β and (C) IL-6 levels were quantified
using ELISA. (D and E) Cell apoptosis was detected using the TUNEL
assay. Scale bar, 100 µm. (F) Western blotting was performed to
assess protein expression levels of Bcl-2, Bax, cleaved caspase-3
and cleaved PARP. Results are presented as the mean ± SD analyzed
using three independent experiments. ***P<0.001 vs. control;
###P<0.001 vs. H/R group; and ∆∆P<0.01,
∆∆∆P<0.001 vs. Ov-KLF15 + siRNA-NC + H/R. KLF15,
Krueppel-like factor 15; CTRP12, C1q/TNF-related protein 12; H/R,
hypoxic/reoxygenated; PARP, poly (ADP-ribose) polymerase; Ov,
overexpressed; siRNA, small interfering RNA; NC, negative
control. |
Discussion
The present study demonstrated that the expression
levels of CTRP12 could be modulated by cardiac I/R, while its
overexpression exerted a protective effect against I/R injury via
alleviating reperfusion-induced inflammation and apoptosis.
Furthermore, mechanistic investigations demonstrated that CTRP12
exerted its therapeutic benefits via its KLF15-regulated enhanced
promotor activity.
Myocardial I/R injury is involved in complicated
pathophysiological mechanisms and is considered to be a significant
cause of heart diseases, which makes it a severe global burden
(31). Myocardial ischemia may
lead to hypoxia, thus resulting in cell injury and increased
cytotoxicity, which in turn can promote cell apoptosis and
oxidative stress (32,33). In the present study, a myocardial
I/R mouse model was established after LAD ligation and reperfusion
(34). H&E staining
demonstrated typical myocardial injury, characterized by damaged
myocardial fiber structure, broken vascular walls and hemocyte
infiltration. Furthermore, H9c2 cells were treated with H/R to
construct an in vitro myocardial I/R model. Subsequently,
the mRNA and protein expression levels of CTRP12 in I/R-exposed
myocardial tissues and H/R-induced cardiomyocytes was determined.
The results demonstrated that CTRP12 was significantly
downregulated both in vivo and in vitro models. CCK-8
assays and the determination of LDH activity are sensitive tools
for evaluating myocardial cell viability and injury, respectively
(35). In the present study,
following CTRP12 overexpression, cell viability was significantly
improved and LDH activity was significantly reduced, supporting the
hypothesis that CTRP12 has a protective effect against I/R-induced
cardiomyocyte injury, which is consistent with previous studies
(17,36).
It has also been reported that CTRP12 regulates
inflammation, glucose metabolism, vascular remodeling and cardiac
fibrosis (15,37,38). Zhou et al (17) demonstrated that CTRP12 ameliorated
LPS-induced inflammatory responses and cell apoptosis in
cardiomyocytes in a nuclear factor erythroid 2-related factor 2
(Nrf2)-dependent manner. Nrf2, a critical antioxidant gene in
scavenging reactive oxygen species (ROS) and maintaining redox
homeostasis, could mediate redox balance (39–41). Moreover, Fadaei et al
(16) reported that the serum
levels of CTRP12 were decreased and negatively associated with
TNF-α and IL-6 in patients with coronary artery disease. Consistent
with previously published findings (36), the results of the present study
demonstrated that the secretory levels of TNF-α, IL-1β and IL-6
were significantly increased in cell culture supernatants from
H/R-treated cells, whereas CTRP12 overexpression significantly
abrogated this effect. These findings indicated that CTRP12
overexpression may protect cardiomyocytes against H/R-induced
inflammation.
Myocardial cell apoptosis contributes to
cardiomyocyte loss in ischemic heart diseases (42). A previous study suggested that
cell apoptosis is a crucial ‘blasting fuse’ for hypoxia-induced
cardiac ischemic injury (43).
Therefore, in the present study a TUNEL staining assay and western
blotting were carried out to assess the effects of CTRP12
overexpression on H/R-induced cardiomyocyte apoptosis. The results
demonstrated that CTRP12 significantly attenuated cardiomyocyte
apoptosis induced by H/R, accompanied by the significant
upregulation of the anti-apoptotic protein Bcl-2 and significantly
decreased expression of the pro-apoptotic proteins, Bax, cleaved
caspase-3 and cleaved PARP. The aforementioned findings suggested
that CTRP12 exhibited an inhibitory effect on the apoptosis of
cardiomyocytes with H/R injury. However, it was also demonstrated
that cell apoptosis is not completely reversed by CTRP12 silencing.
These results therefore suggested that CTRP12 silencing may reverse
cell apoptosis in part and the degree of reversal is associated
with numerous factors such as concentration of transfection
reagent, transfection efficiency, cell type and culture time.
It has previously been reported that the zinc finger
transcription factor KLF15 serves a significant role in heart
diseases, including pathological forms of left ventricular
hypertrophy and heart failure (44,45). Furthermore, it has been proposed
that KLF15 protects cardiomyocytes via attenuating hypertrophic
remodeling, regulating cardiomyocyte gene expression and lipid
oxidation and inhibiting cardiomyocyte apoptosis and oxidative
stress (20,45,46). In the present study the mRNA and
protein expression levels of KLF15 were significantly downregulated
in both I/R-exposed myocardial tissues ad H/R-induced
cardiomyocytes. Moreover, the results determined that KLF15
overexpression significantly promoted the mRNA and protein
expression of CTRP12, which therefore indicated that KLF15 may have
exerted a regulatory role on CTRP12 expression. Furthermore, the
dual-luciferase reporter assay was performed and demonstrated KLF15
overexpression increased CTRP12 promoter activity. It was therefore
hypothesized that KLF15 regulated CTRP12 expression at the
transcriptional level by transcriptionally activating the CTRP12
promoter. Rescue experiments demonstrated that KLF15 overexpression
significantly relieved cardiomyocyte cell viability and ameliorated
H/R-induced cardiomyocyte inflammation and apoptosis. However,
CTRP12 silencing significantly reversed the effects of KLF15
overexpression on H/R-mediated cardiomyocyte injury. These data
provided direct evidence to suggest that KLF15 may be involved in
the CTRP12-mediated protection of cardiomyocytes against H/R
injury.
There are several limitations of the present study.
First, due to the aim of the present study investigating the
biological role of CTRP12 in myocardial I/R injury, the expression
levels of other CTRPs were not explored in the in vivo and
in vitro models. Furthermore, although the relationship
between CTRP12 and KLF15 was verified in the present study, whether
other factors are related to the pathological mechanism in
myocardial I/R injury is unclear. Moreover, whether the
KLF15/CTRP12 signaling pathway affects other apoptotic proteins by
endoplasmic reticulum stress-related or mitochondria-related
apoptotic signaling is unknown. To the best of our knowledge, there
are no studies that have reported the association between other
CTRPs and KLFs, especially in myocardial I/R injury. Therefore, it
is necessary to further study the associations of other CTRPs and
KLFs in myocardial I/R injury and identify the downstream genes of
the KLF15/CTRP12 signaling pathway in myocardial I/R injury.
Second, even though the expression changes of CTRP12 and KLF15 in
I/R tissues were explored, localization studies of CTRP and KLF15
proteins were not performed and the expression level of KLF15 in
CTRP12-overexpressed H/R cells was not investigated. In the present
study KLF15 overexpression and its effects were investigated;
however, the role of downregulated KLF15 was not and therefore the
expression of the KLF15 inhibitor microRNA-223-3p will be
investigated to assess the effects of KLF15 silencing both in
vivo and in vitro. Future work will also explore more
signaling pathways that are potentially controlled by KLF15 in
myocardial I/R injury. Third, in the present study, it was
demonstrated that KLF15 and KLF15-mediated CRTP12 significantly
regulated the production of inflammatory cytokines, but the
mechanism which decreases inflammatory cytokine levels by CTRP12
was not explored. Fourth the interaction between the CTRP12
promotor and KLF15 was preliminarily verified using the
dual-luciferase reporter assay but needs to be further confirmed.
Finally, both KLF15 and CTRP12 expression should be considered
following hypoxia or reperfusion alone, in order to evaluate the
effect of reperfusion-induced ROS. Furthermore, the effects of
CTRP12 and KLF15 on different parts of the heart, such as the
infarct, the border zone and on the volume of infarcted heart, were
not explored, as well as the effects of the time and degree of I/R
and H/R treatment on the expression and role of CTRP12 and KLF15.
These issues are important and will be investigated in future
studies.
In conclusion, the present study demonstrated that
CTRP12 may potentially exert a protective effect against
H/R-induced H9c2 cell injury via attenuating inflammation and
apoptosis. These effects were regulated by KLF15. Overall, the
aforementioned findings suggested that CTRP12 may serve as a novel
target for treating ischemic heart diseases.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BL and XT designed the study, performed the
experiments, wrote and revised the manuscript. BL analyzed the
datasets. XT searched the literature. Both authors read and
approved the final manuscript. BL and XT confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
This study was approved by the Animal Care and Use
Committee of Shenzhen Peking University, The Hong Kong University
of Science and Technology Medical Center (Shenzhen, China; approval
no. 2020-630).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Reed GW, Rossi JE and Cannon CP: Acute
myocardial infarction. Lancet. 389:197–210. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
White HD and Chew DP: Acute myocardial
infarction. Lancet. 372:570–584. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang Z, Yu J, Wu J, Qi F, Wang H, Wang Z
and Xu Z: Scutellarin protects cardiomyocyte ischemia-reperfusion
injury by reducing apoptosis and oxidative stress. Life Sci.
157:200–207. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ibanez B, Fuster V, Jiménez-Borreguero J
and Badimon JJ: Lethal myocardial reperfusion injury: A necessary
evil? Int J Cardiol. 151:3–11. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Prech M, Marszałek A, Schröder J, Filas V,
Lesiak M, Jemielity M, Araszkiewicz A and Grajek S: Apoptosis as a
mechanism for the elimination of cardiomyocytes after acute
myocardial infarction. Am J Cardiol. 105:1240–1245. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Scarabelli TM, Stephanou A, Pasini E,
Comini L, Raddino R, Knight RA and Latchman DS: Different signaling
pathways induce apoptosis in endothelial cells and cardiac myocytes
during ischemia/reperfusion injury. Circ Res. 90:745–748. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Anselmi A, Abbate A, Girola F, Nasso G,
Biondi-Zoccai GG, Possati G and Gaudino M: Myocardial ischemia,
stunning, inflammation, and apoptosis during cardiac surgery: A
review of evidence. Eur J Cardiothorac Surg. 25:304–311. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hoffman JW Jr, Gilbert TB, Poston RS and
Silldorff EP: Myocardial reperfusion injury: Etiology, mechanisms,
and therapies. J Extra Corpor Technol. 36:391–411. 2004.PubMed/NCBI
|
|
10
|
Al-Salam S and Hashmi S: Myocardial
ischemia reperfusion injury: Apoptotic, inflammatory and oxidative
stress role of galectin-3. Cell Physiol Biochem. 50:1123–1139.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kawano J and Arora R: The role of
adiponectin in obesity, diabetes, and cardiovascular disease. J
Cardiometab Syndr. 4:44–49. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Omidifar A, Toolabi K, Rahimipour A,
Emamgholipour S and Shanaki M: The gene expression of CTRP12 but
not CTRP13 is upregulated in both visceral and subcutaneous adipose
tissue of obese subjects. Diabetes Metab Syndr. 13:2593–2599. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Du J, Xu J, Wang X, Liu Y, Zhao X and
Zhang H: Reduced serum CTRP12 levels in type 2 diabetes are
associated with renal dysfunction. Int Urol Nephrol. 52:2321–2327.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang G, Chen JJ, Deng WY, Ren K, Yin SH
and Yu XH: CTRP12 ameliorates atherosclerosis by promoting
cholesterol efflux and inhibiting inflammatory response via the
miR-155-5p/LXRα pathway. Cell Death Dis. 12:2542021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Enomoto T, Ohashi K, Shibata R, Higuchi A,
Maruyama S, Izumiya Y, Walsh K, Murohara T and Ouchi N:
Adipolin/C1qdc2/CTRP12 protein functions as an adipokine that
improves glucose metabolism. J Biol Chem. 286:34552–34558. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fadaei R, Moradi N, Kazemi T, Chamani E,
Azdaki N, Moezibady SA, Shahmohamadnejad S and Fallah S: Decreased
serum levels of CTRP12/adipolin in patients with coronary artery
disease in relation to inflammatory cytokines and insulin
resistance. Cytokine. 113:326–331. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou MQ, Jin E, Wu J, Ren F, Yang YZ and
Duan DD: CTRP12 ameliorated lipopolysaccharide-induced
cardiomyocyte injury. Chem Pharm Bull (Tokyo). 68:133–139. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McConnell BB and Yang VW: Mammalian
Krüppel-like factors in health and diseases. Physiol Rev.
90:1337–1381. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Enomoto T, Ohashi K, Shibata R, Kambara T,
Uemura Y, Yuasa D, Kataoka Y, Miyabe M, Matsuo K, Joki Y, et al:
Transcriptional regulation of an insulin-sensitizing adipokine
adipolin/CTRP12 in adipocytes by Krüppel-like factor 15. PLoS One.
8:e831832013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang Q, Li MY, Su YF, Fu J, Zou ZY, Wang Y
and Li SN: Absence of miR-223-3p ameliorates hypoxia-induced injury
through repressing cardiomyocyte apoptosis and oxidative stress by
targeting KLF15. Eur J Pharmacol. 841:67–74. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bayne K, Ramachandra GS, Rivera EA and
Wang JF: The evolution of animal welfare and the 3Rs in Brazil,
China, and India. J Am Assoc Lab Anim Sci. 54:181–191.
2015.PubMed/NCBI
|
|
22
|
Cao X, Li B, Han X, Zhang X, Dang M, Wang
H, Du F, Zeng X and Guo C: Soluble receptor for advanced glycation
end-products promotes angiogenesis through activation of STAT3 in
myocardial ischemia/reperfusion injury. Apoptosis. 25:341–353.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Roughan JV and Flecknell PA:
Buprenorphine: A reappraisal of its antinociceptive effects and
therapeutic use in alleviating post-operative pain in animals. Lab
Anim. 36:322–343. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Haubner BJ, Adamowicz-Brice M, Khadayate
S, Tiefenthaler V, Metzler B, Aitman T and Penninger JM: Complete
cardiac regeneration in a mouse model of myocardial infarction.
Aging (Albany NY). 4:966–977. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Haubner BJ, Schuetz T and Penninger JM: A
reproducible protocol for neonatal ischemic injury and cardiac
regeneration in neonatal mice. Basic Res Cardiol. 111:642016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ding M, Chen Y, Luan H, Zhang X, Zhao Z
and Wu Y: Dexmedetomidine reduces inflammation in traumatic brain
injury by regulating the inflammatory responses of macrophages and
splenocytes. Exp Ther Med. 18:2323–2331. 2019.PubMed/NCBI
|
|
27
|
Zhang C, Liang R, Gan X, Yang X, Chen L
and Jian J: MicroRNA-384-5p/beclin-1 as potential indicators for
epigallocatechin gallate against cardiomyocytes ischemia
reperfusion injury by inhibiting autophagy via PI3K/Akt pathway.
Drug Des Devel Ther. 13:3607–3623. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shen S, He F, Cheng C, Xu B and Sheng J:
Uric acid aggravates myocardial ischemia-reperfusion injury via
ROS/NLRP3 pyroptosis pathway. Biomed Pharmacother. 133:1109902021.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shi Y, Liu M, Huang Y, Zhang J and Yin L:
Promotion of cell autophagy and apoptosis in cervical cancer by
inhibition of long noncoding RNA LINC00511 via transcription factor
RXRA-regulated PLD1. J Cell Physiol. 235:6592–6604. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cadenas S: ROS and redox signaling in
myocardial ischemia-reperfusion injury and cardioprotection. Free
Radic Biol Med. 117:76–89. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gao X, Zhang H, Zhuang W, Yuan G, Sun T,
Jiang X, Zhou Z, Yuan H, Zhang Z and Dong H: PEDF and PEDF-derived
peptide 44mer protect cardiomyocytes against hypoxia-induced
apoptosis and necroptosis via anti-oxidative effect. Sci Rep.
4:56372014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jing L, Li Q, He L, Sun W, Jia Z and Ma H:
Protective effect of tempol against hypoxia-induced oxidative
stress and apoptosis in H9c2 cells. Med Sci Monit Basic Res.
23:159–165. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang ZQ, Xu W, Wu JL, Lu X and Chen XM:
MicroRNA-374a protects against myocardial ischemia-reperfusion
injury in mice by targeting the MAPK6 pathway. Life Sci.
232:1166192019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Maisch B, Seferović PM, Ristić AD, Erbel
R, Rienmüller R, Adler Y, Tomkowski WZ, Thiene G and Yacoub MH;
Task Force on the Diagnosis and Management of Pricardial Diseases
of the European Society of Cardiology, : Guidelines on the
diagnosis and management of pericardial diseases executive summary;
The Task force on the diagnosis and management of pericardial
diseases of the European society of cardiology. Eur Heart J.
25:587–610. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jin AP, Zhang QR, Yang CL, Ye S, Cheng HJ
and Zheng YY: Up-regulation of CTRP12 ameliorates
hypoxia/re-oxygenation-induced cardiomyocyte injury by inhibiting
apoptosis, oxidative stress, and inflammation via the enhancement
of Nrf2 signaling. Hum Exp Toxicol. 40:2087–2098. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ogawa H, Ohashi K, Ito M, Shibata R,
Kanemura N, Yuasa D, Kambara T, Matsuo K, Hayakawa S, Hiramatsu-Ito
M, et al: Adipolin/CTRP12 protects against pathological vascular
remodelling through suppression of smooth muscle cell growth and
macrophage inflammatory response. Cardiovasc Res. 116:237–249.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang X, Huang T and Xie H: CTRP12
alleviates isoproterenol induced cardiac fibrosis via inhibiting
the activation of P38 pathway. Chem Pharm Bull (Tokyo). 69:178–184.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kovac S, Angelova PR, Holmström KM, Zhang
Y, Dinkova-Kostova AT and Abramov AY: Nrf2 regulates ROS production
by mitochondria and NADPH oxidase. Biochim Biophys Acta.
1850:794–801. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Schmidlin CJ, Dodson MB, Madhavan L and
Zhang DD: Redox regulation by NRF2 in aging and disease. Free Radic
Biol Med. 134:702–707. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Abrescia P, Treppiccione L, Rossi M and
Bergamo P: Modulatory role of dietary polyunsaturated fatty acids
in Nrf2-mediated redox homeostasis. Prog Lipid Res. 80:1010662020.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Krijnen PA, Nijmeijer R, Meijer CJ, Visser
CA, Hack CE and Niessen HW: Apoptosis in myocardial ischaemia and
infarction. J Clin Pathol. 55:801–811. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhou Q, Peng X, Liu X, Chen L, Xiong Q,
Shen Y, Xie J, Xu Z, Huang L, Hu J, et al: FAT10 attenuates
hypoxia-induced cardiomyocyte apoptosis by stabilizing caveolin-3.
J Mol Cell Cardiol. 116:115–124. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Leenders JJ, Wijnen WJ, Hiller M, van der
Made I, Lentink V, van Leeuwen REW, Herias V, Pokharel S, Heymans
S, de Windt LJ, et al: Regulation of cardiac gene expression by
KLF15, a repressor of myocardin activity. J Biol Chem.
285:27449–27456. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Haldar SM, Lu Y, Jeyaraj D, Kawanami D,
Cui Y, Eapen SJ, Hao C, Li Y, Doughman YQ, Watanabe M, et al: Klf15
deficiency is a molecular link between heart failure and aortic
aneurysm formation. Sci Transl Med. 2:26ra262010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Noack C, Haupt LP, Zimmermann WH,
Streckfuss-Bömeke K and Zelarayán LC: Generation of a KLF15
homozygous knockout human embryonic stem cell line using paired
CRISPR/Cas9n, and human cardiomyocytes derivation. Stem Cell Res.
23:127–131. 2017. View Article : Google Scholar : PubMed/NCBI
|