Introduction
Periodontitis is a chronic oral inflammatory
response of the periodontium that affects almost 50% of US adults
≥30 years of age (1,2). This chronic, multifactorial disease
is characterized by gingival inflammation and alveolar bone loss
(3). Periodontitis can trigger an
immune-inflammatory response that ultimately leads to a
non-reversible change of bone supporting tissues, resulting in
progressive destruction of alveolar bone and finally tooth loss
(4). Periodontitis has also been
reported to be associated with the development of several systemic
diseases, such as rheumatoid arthritis, psoriasis, systemic
sclerosis, Alzheimer's disease and cardiovascular diseases (CVD)
(5). Among the systemic diseases
associated with periodontitis, CVD has received the most attention,
as a number of epidemiological and clinical studies have indicated
a strong association between periodontitis and CVD (6,7).
Periodontitis clearly imparts adverse risks for CVD in clinical
evidence in addition to animal models (8,9).
Atherosclerosis is a degenerative vascular disease
accompanying chronic and progressive vascular inflammatory
conditions, which results in fatal consequences such as myocardial
infarction and stroke due to the development of atherosclerotic
plaque (10). Although
hyperlipidemia is known as a major risk factor for the development
of atherosclerosis, previous studies have indicated that systemic
inflammation plays a notable role in the initiation and progression
of atherosclerosis and is also one of the precipitating factors for
atherogenesis (11,12). Systemic inflammation (such as
increased serum proinflammatory cytokine levels) is known to induce
vascular inflammation, resulting in vascular endothelial cell
dysfunction, an early important step of atherogenesis. Also,
vascular inflammation causes atheroma progression and complications
by inducing the proliferation and migration of vascular smooth
muscle cells, resulting in thrombosis (13).
There are several systemic inflammatory diseases
that are known to increase the risk of atherosclerosis development,
such as rheumatoid arthritis, psoriasis, systemic lupus
erythematosus (SLE) and systemic sclerosis (14). Periodontitis, a local and systemic
condition, has also been suggested to be one of the putative risk
factors for atherogenesis (15–18). We previously reported that
ligature-induced periodontitis induces severe systemic inflammation
and exacerbates atherosclerosis in hyperlipidemic Apolipoprotein
E-deficient (ApoE−/−) mice (19); however, to the best of our
knowledge, the effect of severe periodontitis on atherogenesis in
normolipidemic condition remains unknown. To understand whether
hyperlipidemic condition is necessary for the development and
exacerbation of atherosclerosis associated with periodontitis, the
present study investigated the effect of ligature-induced
periodontitis on atherogenesis in normolipidemic wild-type (WT) and
hyperlipidemic ApoE−/− mice.
Materials and methods
Animals
A total of 20 male WT and ApoE−/− C57BL/6
mice (Jackson Laboratory) that were 7 weeks old (mean weight, 19
g), were housed in the vivarium at The University of California
(Division of Laboratory Animal Medicine, Los Angeles, USA). All
mice were housed in a pathogen-free animal experimental facility of
University of California, Los Angeles, under a 12 h light/dark
cycle at 20°C, with 40% humidity, and proper ventilation and air
circulation. The mice were fed with a high-fat diet (HFD) for 14
weeks (cat. no. D12079B; Research Diets, Inc.) to facilitate the
development of atherosclerosis (19). All mice had free access to the
food and drinking water. Mice health and behavior were monitored
every other day throughout the whole duration of the experiment (14
weeks). No mice were found dead during the experimental period.
Isoflurane (99.9%) and a mixture of ketamine (100 mg/kg) and
xylazine (5 mg/kg) were used as anesthetics during ligature
placement. Carprofen (3 mg/kg), a pain relief drug, was also used
once a day for 3 days after ligature placement to minimize pain of
the mice. All mice were euthanized under general anesthesia using
isoflurane (99.9%) to minimize suffering. Euthanasia was performed
via cardiac perfusion, and the heartbeats of the mice were assessed
for 5 min to verify death. All experiments were approved by the
Chancellor's Animal Research Committee of the University of
California, Los Angeles (approval no. ARC # 2019-057-01A).
Induction of periodontitis in
mice
At 1 week after starting the HFD, the mice were
divided into four groups (five mice per group) as follows: i) WT
control mice (WT/no lig.); ii) ligature-placed WT mice (WT/with
lig.); iii) ApoE−/− mice (ApoE−/−/no lig.);
and iv) ligature-placed ApoE−/− mice
(ApoE−/−/with lig.). The 6–0 silk ligature
(MilliporeSigma) was placed around the second molars under general
anesthesia using Ketamine/Xylazine (100 and 5 mg per kg,
respectively) via intraperitoneal injection as described previously
(20).
Tissue collection and histological
analysis
Whole blood was collected from mice by cardiac
puncture under general anesthesia with isoflurane (Abbott
Laboratories). The mice were then perfused and fixed with 4%
paraformaldehyde at room temperature in phosphate-buffered saline
(PBS) via the left ventricle for 5 min. After the perfusion, the
heart, the carotid artery and the full-length of the aorta-to-iliac
bifurcation were exposed and dissected carefully from any
surrounding tissues. The heart and artery samples were embedded in
Scigen Tissue-Plus O.C.T. Compound (Thermo Fisher Scientific,
Inc.), and sectioned at 7-µm thickness. A total of 12 sections at
100 µm intervals were collected from each mouse and stained with
Oil red O (Sigma-Aldrich; Merck KGaA) at room temperature for 15
min to quantify atherosclerotic burden at the sinus lesion. The
maxillae were excised and half of the palatal tissues were
harvested using a blade for gene expression analysis. For
micro-computed tomography (µCT) analysis, the maxillae were fixed
with 4% paraformaldehyde in PBS, pH 7.4, at 4°C overnight and
stored in 70% ethanol solution at 4°C.
After µCT scanning, the maxillae were decalcified
using 5% EDTA and 4% sucrose in PBS (pH 7.4) for 3 weeks at 4°C.
The decalcification solution was changed daily. Decalcified
maxillae were processed for paraffin embedding blocks at the UCLA
Translational Procurement Core Laboratory. Blocks were sectioned at
5-µm intervals using a microtome (Thermo Fisher Scientific, Inc.).
The sections were dewaxed at 60°C for 30 min, and were then
rehydrated at room temperature by incubating the sections twice in
xylene for 5 min, twice in 100% ethanol for 2 min, twice in 95%
ethanol for 2 min and in 70% ethanol for 2 min. After the sections
were rinsed with running tap water for 1 min at room temperature,
the sections were stained with hematoxylin and eosin for 30 min at
room temperature (Sigma-Aldrich; Merck KGaA). For
tartrate-resistant acid phosphatase staining, the sections were
stained using an acid phosphatase kit (cat. no. 378A;
Sigma-Aldrich; Merck KGaA), according to the manufacturer's
protocol, and then counterstained using hematoxylin for 20 min at
room temperature. The digital images of the stained sections were
obtained using the DP72 light microscope (Olympus Corporation). The
clinical attachment loss was observed under the microscope by
measuring cement-enamel junction (CEJ) to the base of the pocket
depth by an investigator (JS). The reading was confirmed in a
double blind manner by another individual (N-HP).
µCT and histological analysis of
maxillae
The fixed maxillae were subjected to µCT scanning
(Skyscan1275; Bruker Corporation) using a voxel size of 20
µm3 and a 0.5 mm aluminum filter at 55 kVp and 145 µA.
Two-dimensional slices from each maxilla were combined using NRecon
and CTAn/CTVol programs (Bruker Corporation) to form a
three-dimensional reconstruction. Alveolar bone loss was quantified
by measuring the distance from the palatal and mesiobuccal CEJ to
the alveolar bone crest (ABC) of the second molars by an
investigator (JS). The reading was confirmed in a double blind
manner by another individual (N-HP).
En face analysis
The full-length of the aorta-to-iliac bifurcation
was opened along the ventral midline and dissected free of the
animal under a stereomicroscope (Stemi 305; Zeiss AG). For en face
analysis, the aorta was stained with Sudan IV (Sigma-Aldrich; Merck
KGaA) as previously described (9)
and pinned out flat, intimal side up, between cover slides. Aortic
images were captured using a Nikon digital camera (Nikon
Corporation) and the intensity of lipid staining was analyzed using
ImageJ software version 1.48 (National Institutes of Health).
Serum lipid and cytokine
measurements
Levels of total cholesterol (TC), triglycerides
(TG), high density lipoprotein (HDL) and low-density lipoprotein
(LDL) were measured using a Cholesterol Assay kit (Abcam) in the
UCLA Cardiovascular Core Facility (21). The serum levels of TNF-α, IL-1β
and IL-6 were measured by ELISA using Ready-SET-go kits [TNF-α
Mouse Uncoated ELISA kit with Plates (cat. no. 88732422); IL-6
Mouse Uncoated ELISA Kit with Plates (cat. no. 88706422); IL-1β
Mouse Uncoated ELISA kit with Plates (cat. no. 88701322); Thermo
Fisher Scientific, Inc.] according to the manufacturer's protocol.
The color reaction was stopped with the addition of Stop solution
(BioLegend, Inc.), and absorbance was read immediately using a
plate reader at 450 nm (Bio-Rad Laboratories, Inc.). The standard
curve was calculated by plotting the standards against the
absorbance values, and the cytokine levels were measured in
pg/ml.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA from mouse tissues was extracted using
RNeasy micro kit (cat. no. 74106; Qiagen GmbH) and
reverse-transcribed with the following cycle: 5 min at 65°C, 2 min
at 25°C and 50 min at 45°C using SuperScript® III
Reverse Transcriptase Synthesis kit (cat. no. 18064014; Thermo
Fisher Scientific, Inc.). Subsequently, qPCR was performed under
the following conditions: Pre-incubation at 95°C for 2 min;
amplification at 95°C for 15 sec, 57°C for 15 sec and 72°C for 1
min, for 55 cycles; melting curve at 95°C for 15 sec, 60°C for 1
min and 95°C for 15 sec using PowerUp™ SYBR Green Master
Mix (cat. no. A25741; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. The following primers were used for
qPCR: TNF-α forward, 5′-TCAGGTTGCCTCTGTCTCAG-3′, and reverse,
5′-GCTCTGTGAGGAAGGCTGTG-3′; IL-1β forward,
5′-CACAGCAGCACATCAACAAG-3′, and reverse,
5′-GTGCTCATGTCCTCATCCTG-3′; IL-6 forward,
5′-TGGGACTGATGCTGGTGACA-3′, and reverse
5′-GCCTCCGACTTGTGAAGTGGT-3′; and β-actin forward,
5′-CATTGCTGACAGGATGCAGAAGG-3′, and reverse
5′-TGCTGGAAGGTGGACAGTGAGG-3′. β-actin served as control and the
fold induction was calculated using the comparative ΔCq method
(22). Data were presented as
relative transcript levels (2−ΔΔCq).
Immunohistochemical (IHC)
staining
The slides containing the maxilla sections were
submerged in a citric acid-based antigen unmasking solution (Vector
Laboratories) at 65°C overnight for antigen retrieval and then
incubated with anti-CD68 antibody (cat. no. ab125212; Abcam) or p65
(cat. no. ab32536; Abcam) as primary antibodies. The tissue
sections were visualized using 3-amino-9-ethylcarbazole (AEC; cat.
no. SK-4205; Vector Laboratories, Inc.) at room temperature for 1
min, followed by counterstaining with hematoxylin for 1 min at room
temperature, then sealed with ImmunoHistoMount™ solution
(Agilent Technologies, Inc.). The digital images of the stained
sections were obtained using the DP72 light microscope (Olympus
Corporation).
Statistical analysis
All graphs were created and statistical analyses
were calculated using GraphPad Prism 9.3.1 (GraphPad Software,
Inc.), and for multiple comparisons, one-way ANOVA with Tukey's
post hoc test was used. P<0.05 was considered to indicate a
statistically significant difference. All in vitro results were
confirmed by at least three independent experiments. Error bars
represent mean ± SEM.
Results
Development of periodontitis is
induced by the ligature placement in WT and ApoE−/−
mice
To investigate the effect of periodontitis on
atherogenesis in both normolipidemic and hyperlipidemic conditions,
periodontitis was induced in WT and ApoE−/− mice by
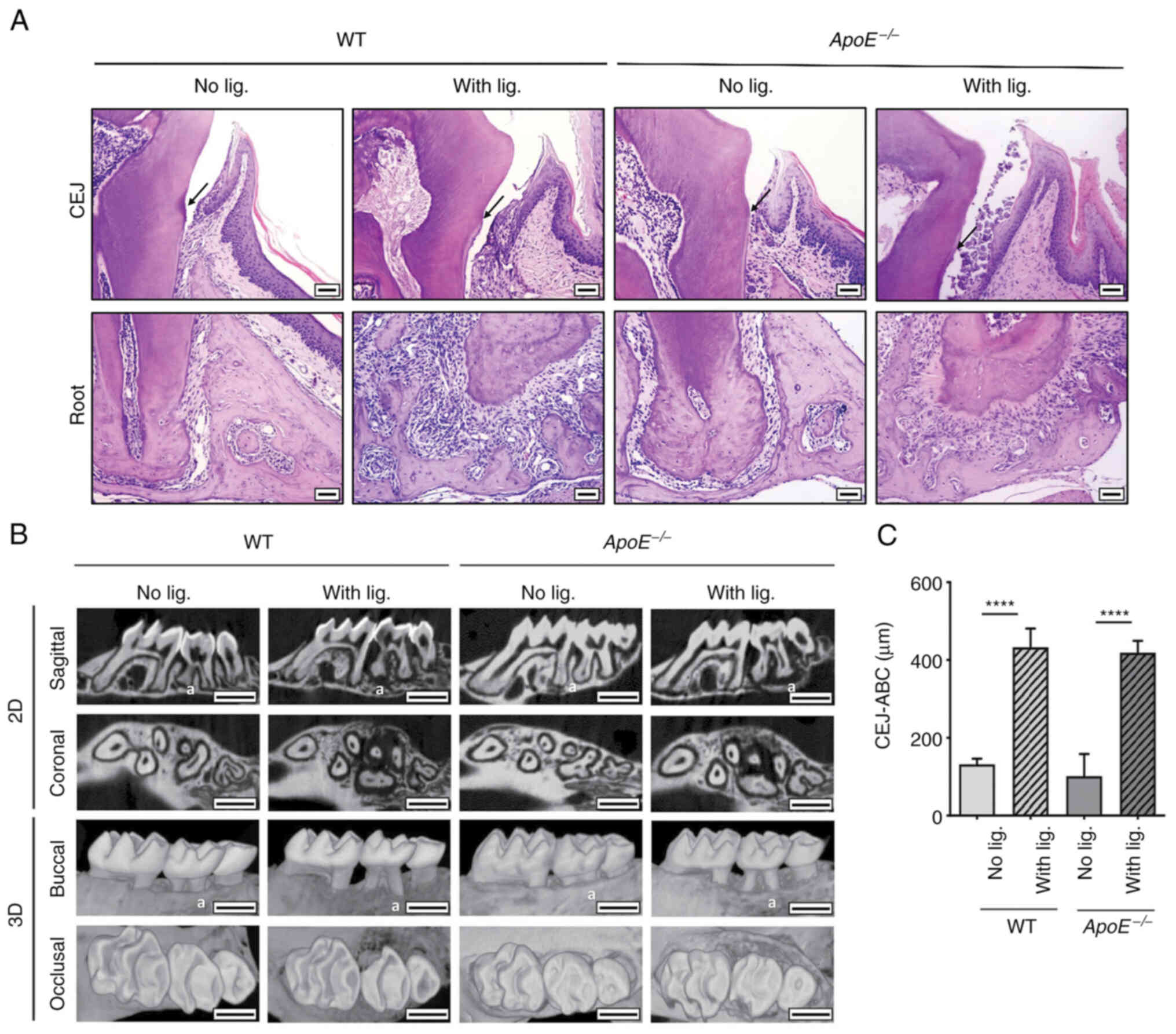
placing ligature for 14 weeks as described previously (19). Histological examination revealed
that the ligature placement induced alterations in the organization
of the junctional epithelium and deep subgingival pockets, such as
clinical epithelial attachment loss (Fig. 1A) and alveolar bone loss (Fig. 1B) in both WT and
ApoE−/− mice compared with the control mice without
ligature placement. The µCT analysis revealed a similar alveolar
bone loss in both WT and ApoE−/− mice with ligature
compared with the control mice, as measured by the distance between
the CEJ and the ABC (Fig. 1B and
C). These data indicate that long-term ligature placement
induced a similar degree of periodontitis in both groups of
mice.
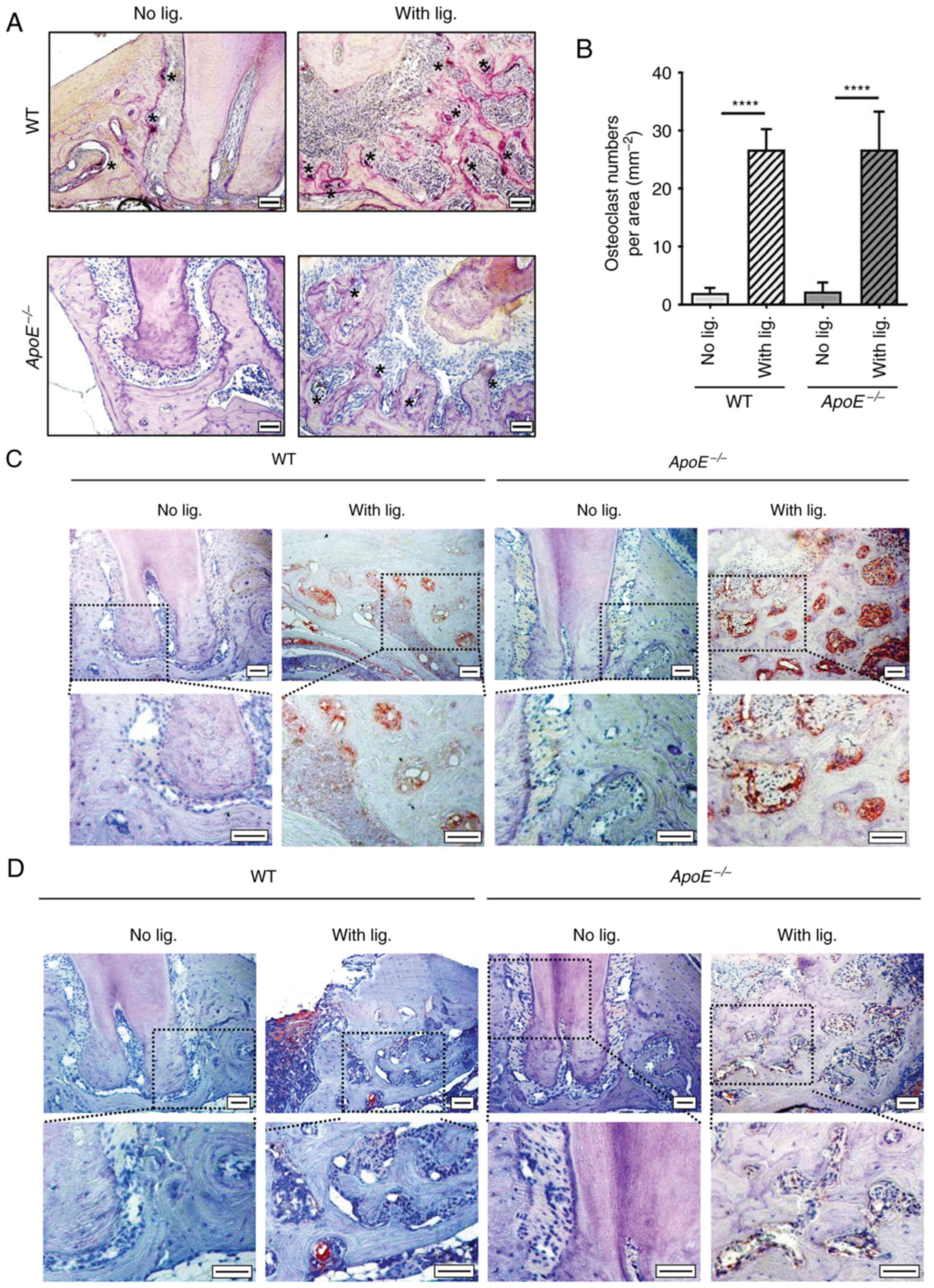
As local inflammation of periodontium induces
alveolar bone loss (23), the
presence of osteoclasts, macrophages and inflammation-specific
markers, such as p65, were evaluated to examine the status of local
inflammation in both WT and ApoE−/− mice. Ligature
placement significantly increased the numbers of osteoclasts around
the ligated tooth in both WT and ApoE−/− mice
experimental groups, but not in the non-ligated groups (Fig. 2A and B). Furthermore, IHC staining
of CD68, a macrophage surface marker, revealed that ligature
placement similarly increased the recruitment of macrophages in
both WT and ApoE−/− mice (Fig. 2C). Ligature placement also
upregulated the expression of p65, a subunit of the major
inflammatory transcriptional factor, NF-κB (24) in the alveolar bone of both WT and
ApoE−/− mice (Fig.
2D). These data indicated that ligature placement induced
similar degree of periodontitis and local inflammation in both WT
and ApoE−/− mice.
Differential systemic status of lipid
and systemic inflammation in WT and ApoE−/− mice by
ligature placement
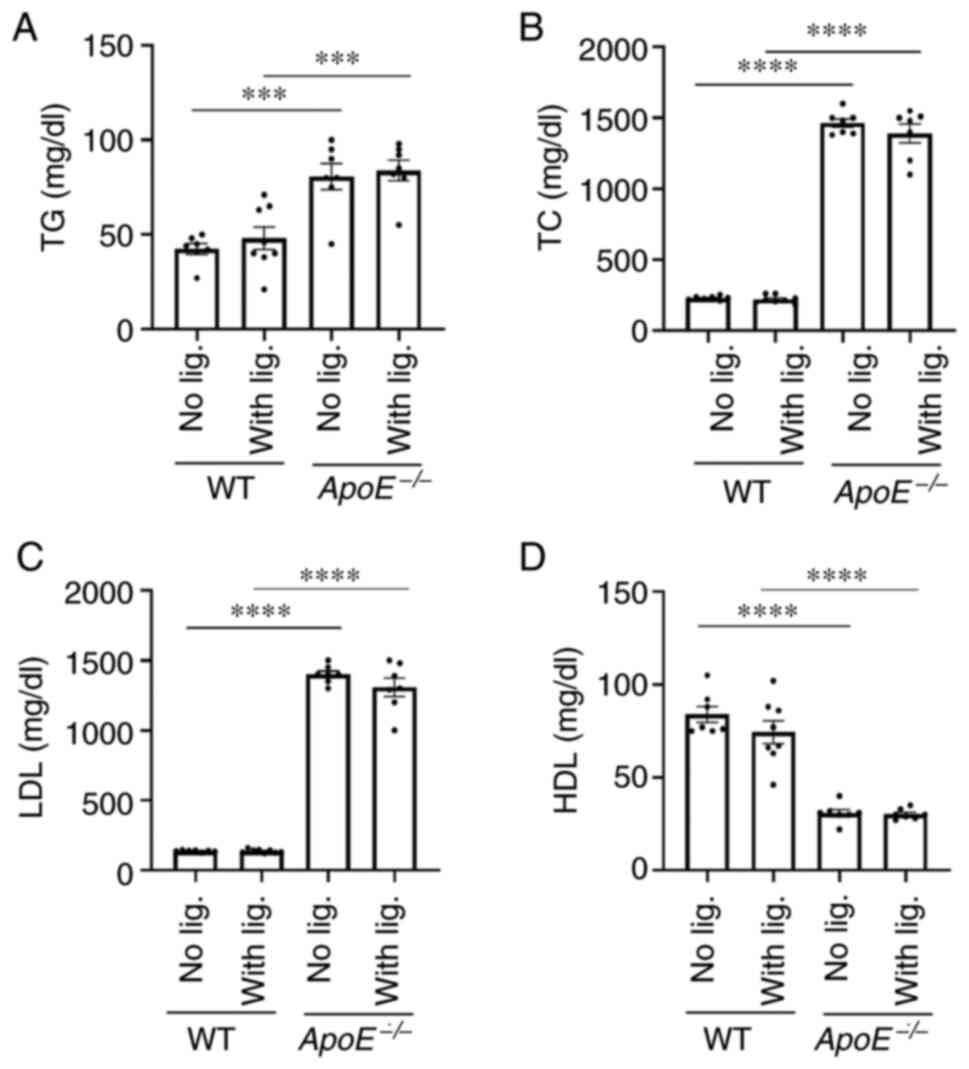
To determine whether the ligature placement altered
lipid content in the blood stream, enzymatic assays were performed
to detect the levels of several types of lipids. As expected, the
control ApoE−/− mice without ligature placement had
significantly higher levels of TG, TC and LDL levels in the serum
compared with WT control mice, although WT control mice revealed
significantly higher HDL levels compared with the
ApoE−/− control mice (Fig.
3A-D). Notably, the long-term ligature placement did not alter
the lipid profiles in both experimental WT and ApoE−/−
mice when compared with those of control mice without ligature
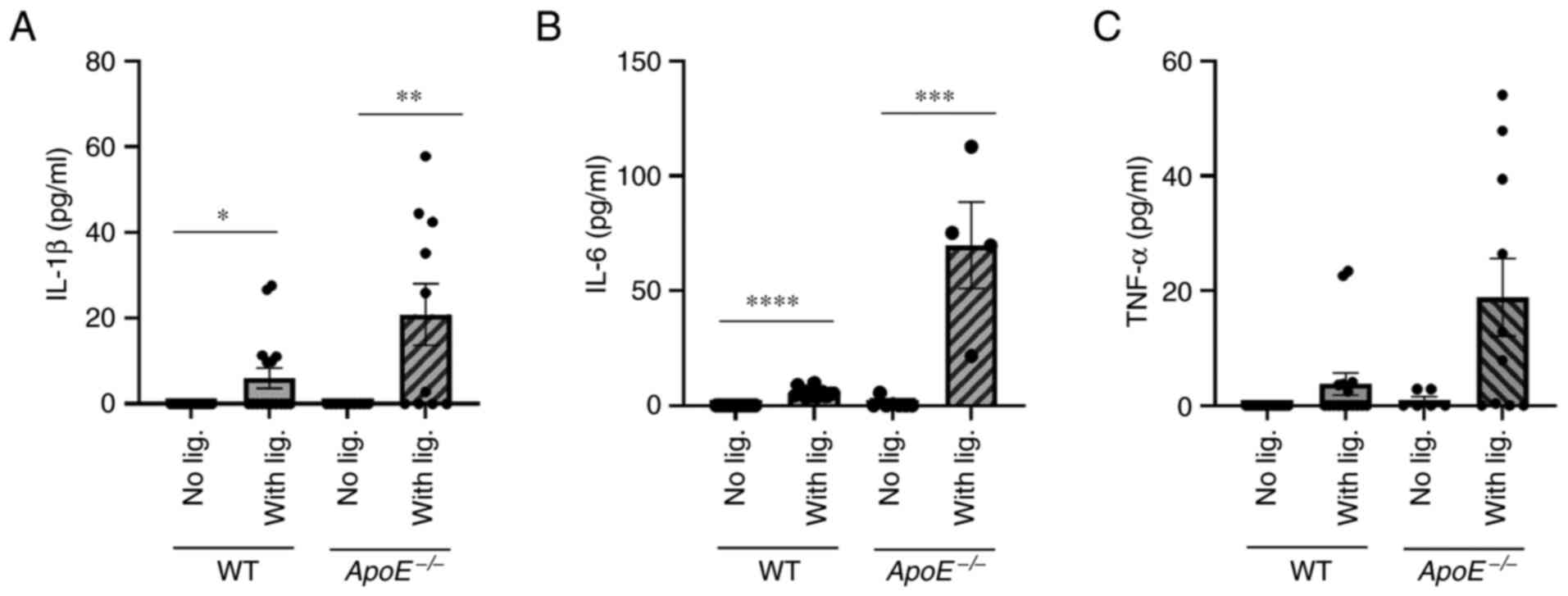
placement (Fig. 3). By contrast,
ligature placement significantly increased the serum levels of
proinflammatory cytokines, such as, IL-1β, IL-6 and TNFα in the
serum of both WT and ApoE−/− mice compared in the mice
that did not receive ligature placement, although the cytokine
levels were markedly higher in ApoE−/− mice compared
with WT mice (Fig. 4). These data
indicated that ligature-induced periodontitis induced systemic
inflammation in both WT and ApoE−/− mice without
altering lipid profiles.
Ligature induced periodontitis
enhances arterial lipid deposition and vascular inflammation in
ApoE−/− mice, not in WT mice
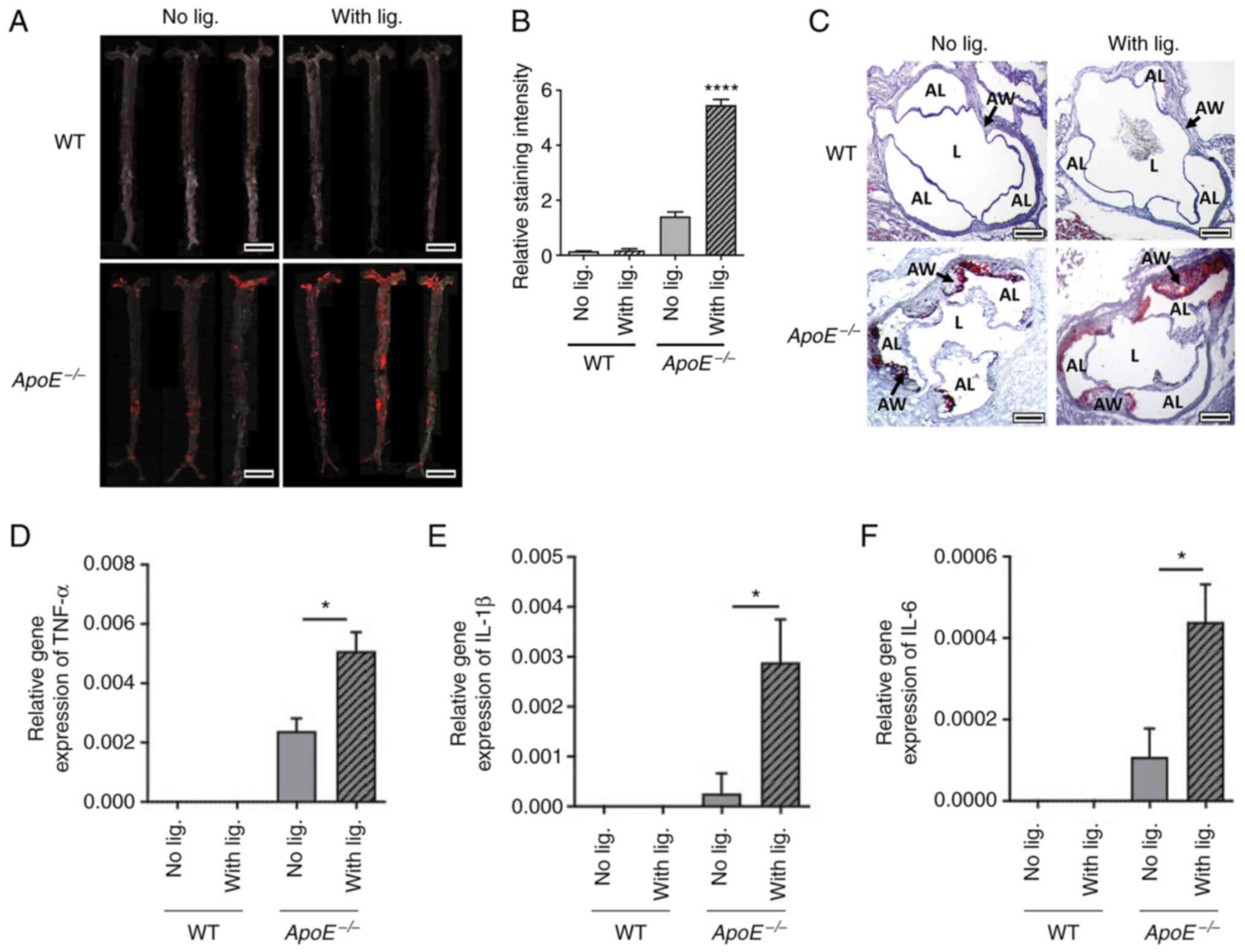
To examine the atherosclerosis development, en face
analysis was performed to quantify the lipid deposition on the
arterial wall. WT mice did not develop notable lipid depositions in
both with or without ligature placement groups. By contrast,
ApoE−/− mice revealed significantly increased lipid
deposition in ligatured mice compared with the non-ligatured group
(Fig. 5A and B). Similarly, the
Oil Red-O staining of aortic roots also demonstrated increased
deposition of lipid in the vessel walls of ApoE−/− mice
receiving ligature placement compared with those in
ApoE−/− mice without ligature placement, but no notable
lipid deposition was observed in the WT mice regardless of ligature
placement (Fig. 5C).
When gene expression levels of the pro-inflammatory
cytokines were measured from the aorta using RT-qPCR, the present
study revealed negligible levels in WT mice whether the ligature
was placed or not (Fig. 5D-F). By
contrast, there was a significant increase in the expression levels
of TNF-α, IL-1β and IL-6 in the arterial wall in ApoE−/−
mice with ligature placement compared with ApoE−/− mice
without ligature placement (Fig.
5D-F). These data indicated that ligature-induced periodontitis
increased vascular inflammation and aortic lipid deposition in
ApoE−/− mice, but not in WT mice.
Discussion
In the present study, severe periodontitis was
introduced in both WT and ApoE−/− mice by a long-term
ligature placement around the second molars. WT mice did not
develop atherosclerosis even under extreme conditions such as
severe periodontitis and HFD Meanwhile, HFD alone moderately
developed atherosclerosis in ApoE−/− mice, but the
ligature-induced severe periodontal disease significantly
exacerbated the development of atherosclerosis.
It is worth noting that WT mice seldomly develop
atherosclerosis, which is attributed to their innate capacity to
lowering lipid contents (25,26). In the present study, serum lipids
in WT mice were significantly decreased compared with those in
ApoE−/− mice when both groups were exposed to HFD. One
of the major functions of ApoE protein is to serve as a ligand for
the LDL receptor and to facilitate uptake and clearance of various
lipoprotein complexes (27).
Subsequently, the current study clearly demonstrated and verified
that elevated lipid levels are a bona fide pre-requisite for
atherosclerosis development.
Notably, severe periodontitis induced by the
ligature placement did not alter the levels of lipids in WT mice
when compared with WT mice without periodontitis, despite the
significant increase of systemic inflammation by periodontitis.
Previous clinical studies demonstrate rather conflicting results on
the association between periodontitis and lipid levels; some
studies have revealed a positive correlation while others
demonstrated it to be insignificant (28–31). Based on the present data, it was
hypothesized that ligature-induced periodontitis did not directly
affect the lipid levels in serum and that ligature-induced
periodontitis and lipid levels may be independent risk factors for
the atherosclerosis development in mice.
The present study indicated that the levels of
pro-inflammatory cytokines such as IL-1β, IL-6 and TNF-α were all
significantly elevated in ApoE−/− mice in the presence
of severe periodontitis, which suggested that these cytokines were
putative therapeutic targets for the atherosclerosis. Indeed, the
utilization of anti-IL-6 or anti-TNF-α therapy such as infliximab
or tocilizumab have been suggested in clinics; however, however,
their negative side effects decrease their likelihood of being used
(32,33). By contrast, a previous clinical
trial, Canakinumab Anti-inflammatory Thrombosis Outcomes Study,
demonstrated that Canakinumab, an anti-IL-1β antibody, effectively
reduces recurrent major adverse cardiovascular events (34). Overall, these clinical studies
underscore the importance of systemic inflammation in mediating the
development of atherosclerosis.
Although the present study demonstrated an
association between severe periodontitis and the development of
atherosclerosis in ApoE−/− mice, the molecular mechanism
of this link remains unknown. However, there are several
explanations for this association. First, severe periodontitis
induces chronic inflammation, which results in persistent
pro-inflammatory cytokines at the local level and releases them
systemically throughout the body, such that it ultimately affects
atherosclerosis in hyperlipidemic condition. Second, the oral
bacteria responsible for periodontal disease might induce the
development of atherosclerosis. Indeed, Actinobacillus
actinomycetemcomitans and Porphyromonas gingivalis have been
revealed in atherosclerotic lesions in humans (35). In addition, oral inoculation of
Porphyromonas gingivalis induces atherosclerosis in experimental
hyperlipidemic mouse models (36,37). However, given that other ‘sterile’
inflammatory conditions, such as SLE, are also a risk factor for
atherosclerosis (38), the sole
role of bacteria requires further clarification. Lastly, chronic
and persistent periodontal disease may trigger alterations in local
immunity (via epigenetics) such that it permanently changes the
behaviors of the key players in atherosclerosis (39,40). The fundamental mechanisms of
periodontitis-induced atherosclerosis development need to be
further investigated.
There are several limitations to the present study.
First, although ApoE−/− mice are well-established to
represent studies on atherosclerosis, global knockout of ApoE in
other cells make it challenging to rule out involvement of
confounding cells (41,42). As such, it may require additional
validation of the present findings with other mouse models such as
LDLR−/− mice. Second, 14 weeks of ligature placement
were used to mimic chronic periodontal disease conditions. However,
it remains to be determined whether 14 weeks of ligature in mice
truly represents chronic periodontal disease in humans. Third,
involvement of the oral microbiome cannot be ruled out because, as
aforementioned, several known oral bacterial species are found in
the atherosclerotic lesions (35–37) and play a direct pathologic role in
atherosclerosis development.
In summary, the present study suggested that severe
periodontal diseases were a significant risk factor for
exacerbating atherosclerosis under specific conditions, such as
hyperlipidemia. In addition, periodontal disease and high
cholesterol were seemingly independent risk factors for
atherosclerosis development, and the coupling of systemic
inflammation and hyperlipidemia may be necessary for the
development and exacerbation of atherosclerosis induced by
periodontitis. Alleviating chronic periodontal disease could
potentially be a novel therapeutic method to intervene in
atherosclerosis development in high-risk groups.
Acknowledgements
Not applicable.
Funding
This work was supported in part by the research funds awarded
from the UCLA Chancellor's Office and National Institutes of
Health/National Institute of Dental and Craniofacial Research
(grant no. R01DE 023348).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
NHP and RK were involved in the conceptualization of
the study. JS, SK, and SHL performed the experiments and
participated in data analysis. JS, SK and SHL confirmed the
authenticity of all the raw data. JS, SK, SHL, RK and NHP were
involved in the discussion and interpretation of the results. JS,
SK, RK, and NHP drafted the manuscript. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
All procedures were performed in compliance with the
institution's policy and applicable provisions of the United States
Department of Agriculture (USDA) Animal Welfare Act Regulations and
the Public Health Service (PHS) Policy. The experimental protocols
were approved by the Animal Research Committee of the University of
California, Los Angeles (approval no. ARC# 2019-057).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Eke PI, Dye BA, Wei L, Thornton-Evans GO
and Genco RJ; CDC Periodontal Disease Surveillance workgroup, :
James Beck (University of North Carolina, Chapel Hill, USA), Gordon
Douglass (Past President, American Academy of Periodontology), Roy
Page (University of Washin: Prevalence of periodontitis in adults
in the United States: 2009 and 2010. J Dent Res. 91:914–920. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Eke PI, Dye BA, Wei L, Slade GD,
Thornton-Evans GO, Borgnakke WS, Taylor GW, Page RC, Beck JD and
Genco RJ: Update on prevalence of periodontitis in adults in the
United States: NHANES 2009 to 2012. J Periodontol. 86:611–622.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dzink JL, Tanner AC, Haffajee AD and
Socransky SS: Gram negative species associated with active
destructive periodontal lesions. J Clin Periodontol. 12:648–659.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kinane DF, Zhang P, Benakanakere M,
Singleton J, Biesbrock A, Nonnenmacher C and He T: Experimental
gingivitis, bacteremia and systemic biomarkers: A randomized
clinical trial. J Periodontal Res. 50:864–869. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Slots J: Periodontitis: Facts, fallacies
and the future. Periodontol. 75:7–23. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rydén L, Buhlin K, Ekstrand E, de Faire U,
Gustafsson A, Holmer J, Kjellström B, Lindahl B, Norhammar A,
Nygren Å, et al: Periodontitis increases the risk of a first
myocardial infarction: A report from the PAROKRANK study.
Circulation. 133:576–583. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Slocum C, Kramer C and Genco CA: Immune
dysregulation mediated by the oral microbiome: Potential link to
chronic inflammation and atherosclerosis. J Intern Med.
280:114–128. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jain A, Batista EL Jr, Serhan C, Stahl GL
and Van Dyke TE: Role for periodontitis in the progression of lipid
deposition in an animal model. Infect Immun. 71:6012–6018. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hasturk H, Abdallah R, Kantarci A, Nguyen
D, Giordano N, Hamilton J and Van Dyke TE: Resolvin E1 (RvE1)
attenuates atherosclerotic plaque formation in diet and
inflammation-induced atherogenesis. Arterioscler Thromb Vasc Biol.
35:1123–1133. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Joshi NV, Toor I, Shah AS, Carruthers K,
Vesey AT, Alam SR, Sills A, Hoo TY, Melville AJ, Langlands SP, et
al: Systemic atherosclerotic inflammation following acute
myocardial infarction: Myocardial infarction begets myocardial
infarction. J Am Heart Assoc. 4:e0019562015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Soehnlein O and Libby P: Targeting
inflammation in atherosclerosis-from experimental insights to the
clinic. Nat Rev Drug Discov. 20:589–610. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hansson BG, Rosenquist K, Antonsson A,
Wennerberg J, Schildt EB, Bladström A and Andersson G: Strong
association between infection with human papillomavirus and oral
and oropharyngeal squamous cell carcinoma: A population-based
case-control study in southern Sweden. Acta Otolaryngol.
125:1337–1344. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Libby P, Ridker PM and Hansson GK:
Progress and challenges in translating the biology of
atherosclerosis. Nature. 473:317–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gargiulo P, Marsico F, Parente A, Paolillo
S, Cecere M, Casaretti L, Pellegrino AM, Formisano T, Fabiani I,
Soricelli A, et al: Ischemic heart disease in systemic inflammatory
diseases. An appraisal. Int J Cardiol. 170:286–290. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Offenbacher S, Beck JD, Moss K, Mendoza L,
Paquette DW, Barrow DA, Couper DJ, Stewart DD, Falkner KL, Graham
SP, et al: Results from the Periodontitis and Vascular Events
(PAVE) Study: A pilot multicentered, randomized, controlled trial
to study effects of periodontal therapy in a secondary prevention
model of cardiovascular disease. J Periodontol. 80:190–201. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Beck J, Garcia R, Heiss G, Vokonas PS and
Offenbacher S: Periodontal disease and cardiovascular disease. J
Periodontol. 67 (Suppl 10):S1123–S1137. 1996. View Article : Google Scholar
|
|
17
|
Suh JS, Kim S, Boström KI, Wang CY, Kim RH
and Park NH: Periodontitis-induced systemic inflammation
exacerbates atherosclerosis partly via endothelial-mesenchymal
transition in mice. Int J Oral Sci. 11:212019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oberoi R, Vlacil AK, Schuett J, Schösser
F, Schuett H, Tietge UJF, Schieffer B and Grote K: Anti-tumor
necrosis factor-α therapy increases plaque burden in a mouse model
of experimental atherosclerosis. Atherosclerosis. 277:80–89. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Suh JS, Lee SH, Fouladian Z, Lee JY, Kim
T, Kang MK, Lusis AJ, Boström KI, Kim RH and Park NH: Rosuvastatin
prevents the exacerbation of atherosclerosis in ligature-induced
periodontal disease mouse model. Sci Rep. 10:63832020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim T, Kim S, Song M, Lee C, Yagita H,
Williams DW, Sung EC, Hong C, Shin KH, Kang MK, et al: Removal of
pre-existing periodontal inflammatory condition before tooth
extraction ameliorates medication-related osteonecrosis of the
jaw-like lesion in mice. Am J Pathol. 188:2318–2327. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huang Q, Qin L, Dai S, Zhang H, Pasula S,
Zhou H, Chen H and Min W: AIP1 suppresses atherosclerosis by
limiting hyperlipidemia-induced inflammation and vascular
endothelial dysfunction. Arterioscler Thromb Vasc Biol. 33:795–804.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C (T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lamont RJ, Koo H and Hajishengallis G: The
oral microbiota: Dynamic communities and host interactions. Nat Rev
Microbiol. 16:745–759. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mussbacher M, Salzmann M, Brostjan C,
Hoesel B, Schoergenhofer C, Datler H, Hohensinner P, Basílio J,
Petzelbauer P, Assinger A and Schmid JA: Cell type-specific roles
of NF-κB linking inflammation and thrombosis. Front Immunol.
10:852019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Getz GS and Reardon CA: Animal models of
atherosclerosis. Arterioscler Thromb Vasc Biol. 32:1104–1115. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pendse AA, Arbones-Mainar JM, Johnson LA,
Altenburg MK and Maeda N: Apolipoprotein E knock-out and knock-in
mice: Atherosclerosis, metabolic syndrome, and beyond. J Lipid Res.
50 (Suppl):S178–S182. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mahley RW: Apolipoprotein E: Cholesterol
transport protein with expanding role in cell biology. Science.
240:622–630. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Machado AC, Quirino MR and Nascimento LF:
Relation between chronic periodontal disease and plasmatic levels
of triglycerides, total cholesterol and fractions. Braz Oral Res.
19:284–289. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Valentaviciene G, Paipaliene P,
Nedzelskiene I, Zilinskas J and Anuseviciene OV: The relationship
between blood serum lipids and periodontal condition.
Stomatologija. 8:96–100. 2006.PubMed/NCBI
|
|
30
|
Thapa S and Wei F: Association between
high serum total cholesterol and periodontitis: National Health and
nutrition examination survey 2011 to 2012 study of American adults.
J Periodontol. 87:1286–1294. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee S, Im A, Burm E and Ha M: Association
between periodontitis and blood lipid levels in a Korean
population. J Periodontol. 89:28–35. 2018.PubMed/NCBI
|
|
32
|
IL6R Genetics Consortium Emerging Risk
Factors Collaboration, . Sarwar N, Butterworth AS, Freitag DF,
Gregson J, Willeit P, Gorman DN, Gao P, Saleheen D, Rendon A, et
al: Interleukin-6 receptor pathways in coronary heart disease: A
collaborative meta-analysis of 82 studies. Lancet. 379:1205–1213.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mann DL: Innate immunity and the failing
heart: The cytokine hypothesis revisited. Circ Res. 116:1254–1268.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ridker PM, Everett BM, Thuren T, MacFadyen
JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker
SD, et al: Antiinflammatory therapy with canakinumab for
atherosclerotic disease. N Engl J Med. 377:1119–1131. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kozarov EV, Dorn BR, Shelburne CE, Dunn WA
Jr and Progulske-Fox A: Human atherosclerotic plaque contains
viable invasive Actinobacillus actinomycetemcomitans and
Porphyromonas gingivalis. Arterioscler Thromb Vasc Biol.
25:e17–e18. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gibson FC III, Hong C, Chou HH, Yumoto H,
Chen J, Lien E, Wong J and Genco CA: Innate immune recognition of
invasive bacteria accelerates atherosclerosis in apolipoprotein
E-deficient mice. Circulation. 109:2801–2806. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lalla E, Lamster IB, Hofmann MA,
Bucciarelli L, Jerud AP, Tucker S, Lu Y, Papapanou PN and Schmidt
AM: Oral infection with a periodontal pathogen accelerates early
atherosclerosis in apolipoprotein E-null mice. Arterioscler Thromb
Vasc Biol. 23:1405–1411. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wigren M, Nilsson J and Kaplan MJ:
Pathogenic immunity in systemic lupus erythematosus and
atherosclerosis: Common mechanisms and possible targets for
intervention. J Intern Med. 278:494–506. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xu S, Pelisek J and Jin ZG:
Atherosclerosis is an epigenetic disease. Trends Endocrinol Metab.
29:739–742. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Koelwyn GJ, Corr EM, Erbay E and Moore KJ:
Regulation of macrophage immunometabolism in atherosclerosis. Nat
Immunol. 19:526–537. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Daugherty A, Tall AR, Daemen MJAP, Falk E,
Fisher EA, García-Cardeña G, Lusis AJ, Owens AP III, Rosenfeld ME,
Virmani R, et al: Recommendation on design, execution, and
reporting of animal atherosclerosis studies: A scientific statement
from the american heart association. Circ Res. 121:e53–e79. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hinder LM, Vincent AM, Hayes JM, McLean LL
and Feldman EL: Apolipoprotein E knockout as the basis for mouse
models of dyslipidemia-induced neuropathy. Exp Neurol. 239:102–110.
2013. View Article : Google Scholar : PubMed/NCBI
|