Introduction
The extracellular matrix (ECM) not only provides
structural supports and tissue organization but is also important
for regulation of vital processes, including cell proliferation,
migration, differentiation and apoptosis, through specific
receptor-mediated interactions (1). The constitution and property of the
ECM constantly change under normal conditions such as development
and aging and also under pathological conditions such as cancer,
wound healing, and fibrosis (2).
Tenascins are a family of glycoproteins that act as
modifiers of cell adhesiveness, and the family of tenascins
comprises four members, tenascin-C (TNC), tenascin-R (TNR),
tenascin-X (TNX) and tenascin-W (TNW), in vertebrates (3). Tenascins have a characteristic
domain organization with heptad repeats, epidermal growth factor
(EGF)-like repeats, fibronectin type III (FNIII)-like repeats, and
a fibrinogen (FG)-related domain from the N-terminal to C-terminal
regions.
TNX is the largest member of the tenascin family and
is ubiquitously expressed, but it is expressed prominently in
muscle and loose connective tissue (4). Intriguingly, TNX is mainly
downregulated during cancer progression, and a high TNX expression
level is associated with a good prognosis (5). As a physiological function of TNX,
TNX regulates the fibril spacing between collagen fibrils through
direct binding to collagen or through indirect binding to
collagen-associated proteins including type XII collagen and
decorin (6). Functions of TNX in
collagen fibrillogenesis (7,8),
stiffness of collagen (9), and
elastic fiber remodeling (10)
have also been suggested. Taken together, results of previous
studies indicate that TNX contributes to collagen deposition,
collagen stability, and the mechanical property of collagen. The
absence of TNX causes a form of Ehlers-Danlos Syndrome (EDS) termed
classical-like EDS (clEDS) (11–13). clEDS is inherited in an autosomal
recessive pattern and is characterized by hypermobile joints,
hyperextensible skin, and easy bruising without atrophic scarring
(12).
Additional roles of TNX in pain (14), behavior (15), blood vessel formation and
neovascularization (16–18), triglyceride synthesis (19), bone homeostasis (20), and tumor suppression (21,22) have also been proposed (23).
Importantly, our group has shown that TNX plays a
role in hepatic fibrosis that develops in mice fed a high-fat and
high-cholesterol diet with high levels of phosphorus and calcium
(HFCD) (24). In that study,
livers in HFCD-fed wild-type (WT) mice showed greater dysfunction,
more type I collagen deposition, and greater inflammatory response
than those in TNX-deficient mice (24).
Several signaling pathways including transforming
growth factor-β (TGF-β) and Wingless/Int (WNT) have been identified
as key mediators that are linked to fibrosis progression (25). Alcaraz et al (26) demonstrated that the FG-related
domain of TNX activates latent TGF-β1 via integrin α11β1 as a novel
receptor of TNX, thereafter leading to activation of the TGF-β/Smad
signaling pathway followed by epithelial-mesenchymal transition
(EMT) in epithelial cells (6).
Therefore, it is reasonable to assume that the TGF-β signaling
pathway is also involved in TNX-elicited hepatic fibrosis in
HFCD-fed WT mice. Recently, the Yes-associated protein 1
(YAP)/transcriptional coactivator with PDZ-binding motif (TAZ)
signaling pathway has been revealed to be another important
signaling cascade during the development of liver fibrosis
(27). YAP/TAZ acts as a
downstream effector of the Hippo pathway (28). The YAP/TAZ signaling pathway is
activated during the progression of matrix stiffness and then
transformation of hepatic stellate cells (HSCs) into myofibroblasts
occurs with augmentation of collagen synthesis and deposition
(29). Furthermore, integrin
α11β1 preferentially binds to type I collagen (30) and mediates pro-fibrotic signals
from the collagen matrix via YAP1 and P21-activated kinase (PAK) in
liver fibrosis (31). Notably,
Romaine et al (32)
reported that overexpression of integrin α11 in mice resulted in
left ventricular hypertrophy and cardiac fibrosis.
In the present study, we investigated whether the
TNX-FG domain can induce type I collagen α1 chain (COL1A1)
expression in LX-2 human hepatic stellate cells. It was revealed
that overexpression of both a 15-amino acid (aa) peptide derived
from the TNX-FG domain (hTNX-FGFFFF) and integrin α11 (ITGA11)
induces the expression of COL1A1 through the YAP signaling
pathway.
Materials and methods
Cell culture
The human hepatic stellate cell line LX-2 (Merck)
was grown at 37°C in a 5% CO2 humidified atmosphere in
Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with
4.5 g/l D-glucose, L-glutamine, 2% fetal bovine serum (Gibco), 50
U/ml penicillin, and 50 µg/ml streptomycin.
Plasmid construction
A full-length human integrin α 11 cDNA (pBJ1-ITGA11)
(Fig. S1) cloned into the pBJ-1
vector provided by Dr Ning Lu (University of Bergen, Norway) was
used to express integrin α11 in LX-2 cells (33). An empty vector, pBJΔ plasmid, was
constructed by complete removal of ITGA11 cDNA region from
pBJ1-ITGA11 plasmid (Fig. S1).
Overexpression of ITGA11 was confirmed by western blot analysis
(Fig. S2).
Expression plasmids encoding various regions derived
from the fibrinogen domain of human TNX (TNX-FG) were constructed
as follows. The coding regions of the clones are shown in Fig. 1. The 5′-upstream FLAG-tagged pSecF
vector (16) derived from the
pSecTag2/Hygro B vector (Thermo Fisher Scientific, Inc.) served as
the vector backbone for the expression constructs. A PCR-generated
fragment with EcoRI and XhoI sites at each end was
inserted into the EcoRI-XhoI sites of the pSecF
vector. The DNA templates and primer pairs for PCR used for the
construction of expression plasmids, pSecF-hTNX-FG2,
pSecF-hTNX-FGF-2, pSec-hTNX-FGL-2, pSecF-hTNX-FGFF-3,
pSecF-hTNX-FGFL-1, pSecF-hTNX-FGFFF-2, pSecF-hTNX-FGFFL-3,
pSecF-hTNX-FGFFFF-8, pSecF-hTNX-FGFFFM-1 and pSecF-hTNX-FGFFFL-5,
are shown in Table I.
pSecF-hTNX-FGpeptide2-5 was constructed by the primer annealing
method. The forward fhTNX-FGpeptide2 primer and the reverse
rhTNX-FGpeptide2 primer (Table I)
were phosphorylated at the 5′ terminal region and annealed. Then
the annealed sample was cloned into the pSecF vector. Each primer
was synthesized by Hokkaido System Science. PCR was done using 100
ng of a DNA template and 0.2 mM of each primer with TaKaRa Ex
Taq® according to the instruction manual (Takara). The
following thermocycling conditions were used for PCR: initial
denaturation at 94°C for 1 min and then 30 cycles of 98°C for 20
sec and 68°C for 5 min, and 72°C for 10 min.
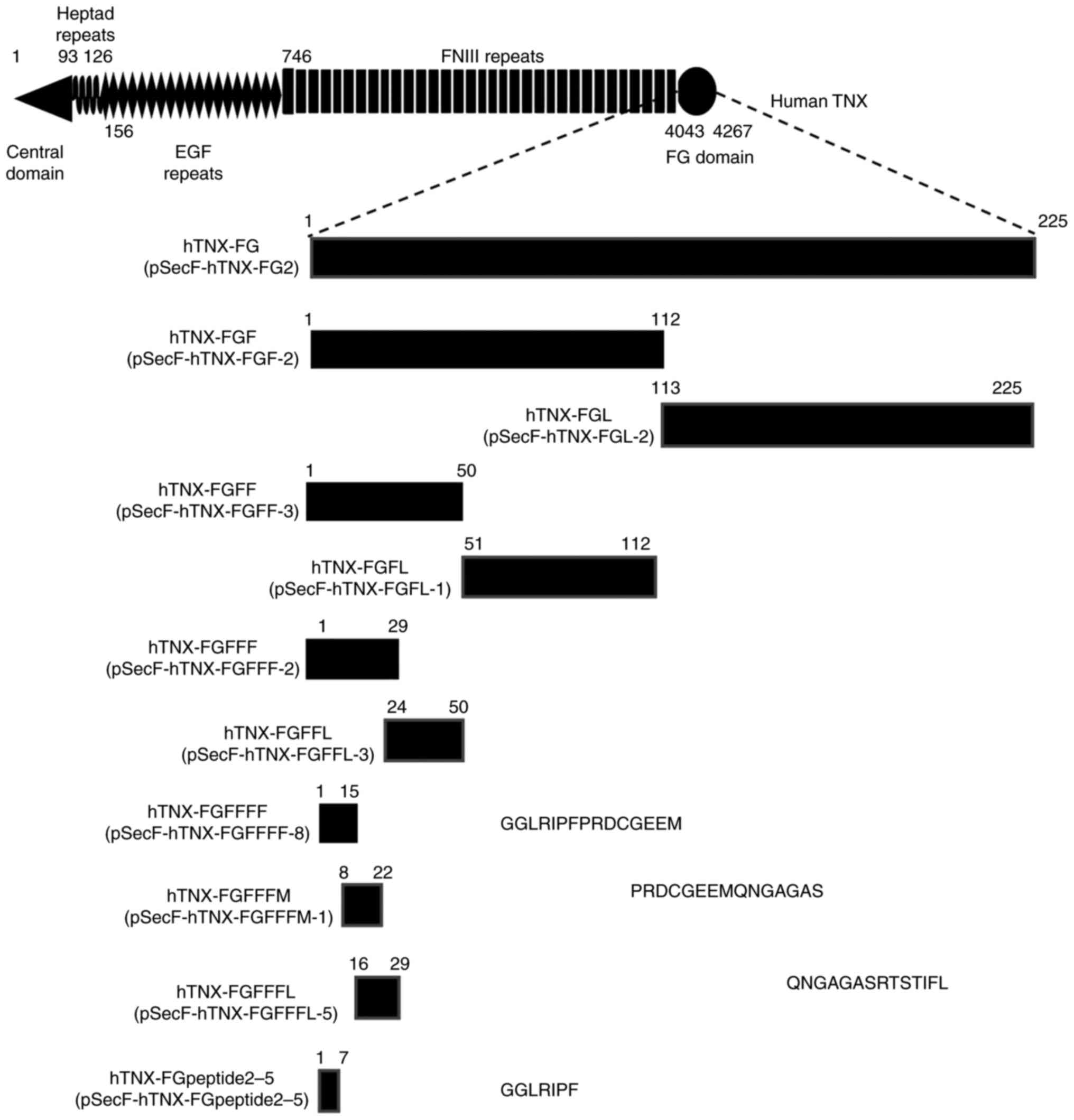 | Figure 1.Encompassed TNX-FG region of
constructed expression plasmid clones. A schematic diagram of the
domain structure of human TNX based on our previous study (50) from the N-terminus to C-terminus
showing the central domain (aa no. 1-92), heptad repeats (aa no.
93-126), 18.5 EGF-like repeats (aa no. 156-745), 33 FNIII-like
repeats (aa no. 746-4,042) and an FG-related domain (aa no.
4,043-4,267). The horizontal black boxes under the diagram indicate
the encompassed regions of the expression plasmids. Their plasmid
names (e.g., pSecF-hTNX-FG2) are shown in parentheses. The names of
expressed proteins or peptides (e.g., hTNX-FG) are also shown. The
number above each black box sets aa number 4,043 (starting position
of the FG-related domain) and aa number 4,267 (last position of the
FG-related domain) in the full-length human TNX diagram to aa
numbers 1 and 225, respectively, and then it indicates the aa
numbers of the start position and last position in the TNX-FG
domain. The aa sequences of hTNX-FGFFFF, hTNX-FGFFFM, hTNX-FGFFFL
and hTNX-FGpeptide2-5 are also shown. EGF, epidermal growth factor;
FNIII, fibronectin type III repeat; FG, fibrinogen; TNX,
tenascin-X; hTNX-FG, fibrinogen-related domain of human tenascin-X;
hTNX-FGF, first half of hTNX-FG; hTNX-FGL, latter half of hTNX-FG;
hTNX-FGFF, first half of hTNX-FGF; hTNX-FGFL, latter half of
hTNX-FGF; hTNX-FGFFF, first half of hTNX-FGFF; hTNX-FGFFL, latter
half of hTNX-FGFF; hTNX-FGFFFF, GGLRIPFPRDCGEEM peptide from
hTNX-FG; hTNX-FGFFFM, PRDCGEEMQNGAGAS peptide from hTNX-FG;
hTNX-FGFFFL, QNGAGASRTSTIFL peptide from hTNX-FG;
hTNX-FGpeptide2-5, GGLRIPF peptide from hTNX-FG. |
 | Table I.Constructed expression plasmids with
DNA templates and primer pairs for PCR. |
Table I.
Constructed expression plasmids with
DNA templates and primer pairs for PCR.
| Protein or peptide
(expression plasmid) | DNA template
(Refs.) | Primer name | Primer sequence
(5′-3′) | Plasmid backbone
(Refs.) |
|---|
| hTNX-FG
(pSecF-hTNX-FG2) | pcDNA3-F-XB-S
(51) | fEcohTNXFG-1 | F:
GGGGAATTCGGTGGGCTGCGGATC | pSecF (16) |
|
|
| rXhohTNXFG-1 | R:
GGGCTCGAGTCAGCCTCCCCCCGC |
|
| hTNX-FGF
(pSecF-hTNX-FGF-2) | pcDNA3-F-XB-S
(51) | fEcohTNXFG-1 | F:
GGGGAATTCGGTGGGCTGCGGATC | pSecF (16) |
|
|
| rXhohTNXFG-2 | R:
GGGCTCGAGTCAGGCGAACACAGCCTC |
|
| hTNX-FGL
(pSec-hTNX-FGL-2) | pcDNA3-F-XB-S
(51) | fEcohTNXFG-2 | F:
GGGGAATTCCAGTACGACTCCTTC | pSecF (16) |
|
|
| rXhohTNXFG-1 | R:
GGGCTCGAGTCAGCCTCCCCCCGC |
|
| hTNX-FGFF
(pSecF-hTNX-FGFF-3) | pcDNA3-F-XB-S
(51) | fEcohTNXFG-1 | F:
GGGGAATTCGGTGGGCTGCGGATC | pSecF (16) |
|
|
| rXhohTNXFGFF-1 | R:
GGGCTCGAGTCACCAGCCGCCCCCATC |
|
| hTNX-FGFL
(pSecF-hTNX-FGFL-1) | pcDNA3-F-XB-S
(51) | fEcohTNXFGFL-1 | F:
GGGGAATTCCTGGTGTTCCAGCGC | pSecF (16) |
|
|
| rXhohTNXFG-2 | R:
GGGCTCGAGTCAGGCGAACACAGCCTC |
|
| hTNX-FGFFF
(pSecF-hTNX-FGFFF-2) | pcDNA3-F-XB-S
(51) | fEcohTNXFG-1 | F:
GGGGAATTCGGTGGGCTGCGGATC | pSecF (16) |
|
|
|
rXhohTNXFGFFF-1 | R:
GGGCTCGAGTCAGAGGAAGATGGTGC |
|
| hTNX-FGFFL
(pSecF-hTNX-FGFFL-3) | pcDNA3-F-XB-S
(51) |
fEcohTNXFGFFL-1 | F:
GGGGAATTCACCAGCACCATCTTC | pSecF (16) |
|
|
| rXhohTNXFGFF-1 | R:
GGGCTCGAGTCACCAGCCGCCCCCATC |
|
| hTNX-FGFFFF
(pSecF-hTNX-FGFFFF-8) |
pSecF-hTNX-FGFFF-2 | fEcohTNXFG-1 | F:
GGGGAATTCGGTGGGCTGCGGATC | pSecF (16) |
|
| (prepared in the
present study) |
rXhohTNXFGFFFF-1 | R:
GGGCTCGAGTCACATCTCCTCCCCGC |
|
| hTNX-FGFFFM
(pSecF-hTNX-FGFFFM-1) |
pSecF-hTNX-FGFFF-2 |
fEcohTNXFGFFFM-1 | F:
GGGGAATTCCCCAGGGACTGCGGG | pSecF (16) |
|
| (prepared in the
present study) |
rXhohTNXFGFFFM-1 | R:
GGGCTCGAGTCAGGAGGCACCGGCTCC |
|
| hTNX-FGFFFL
(pSecF-hTNX-FGFFFL-5) |
pSecF-hTNX-FGFFF-2 |
fEcohTNXFGFFFL-1 | F:
GGGGAATTCCAGAACGGAGCCGGT | pSecF (16) |
|
| (prepared in the
present study) |
rXhohTNXFGFFF-1 | R:
GGGCTCGAGTCAGAGGAAGATGGTGC |
|
| hTNX-FGpeptide2-5
(pSecF-hTNX-FGpeptide2-5) | Primer
annealing |
fhTNX-FGpeptide2 | F:
AATTCGGTGGGCTGCGGATCCCCTTCTGAC | pSecF (16) |
|
| (prepared in the
present study) |
rhTNX-FGpeptide2 | R:
TCGAGTCAGAAGGGGATCCGCAGCCCACCG |
|
Transfection with expression plasmid
DNA and/or small interfering RNA (siRNA)
Five ×105 LX-2 cells were seeded on
6-well plates in DMEM with 2% FBS, followed by culturing overnight.
Then the medium was changed to DMEM with 0.5% FBS. One µg of each
expression plasmid DNA was transfected using
Lipofectamine® LTX with Plus reagent according to the
instruction manual (Thermo Fisher Scientific, Inc.). The
transfected cells were cultured for 48 h and then subjected to RNA
extraction.
On the other hand, for the co-transfection of siRNA
and expression plasmid DNA, 5×105 LX-2 cells were
cultured overnight on 6-well plates in DMEM with 2% FBS. Then the
medium was changed to DMEM with 0.5% FBS. Fifty nM YAP1 siRNA
(sc-38637; Santa Cruz Biotechnology, Inc.), which is a pool of 3
target-specific siRNAs (sc-38637A, sc-38637B, and sc-38637C), or
control siRNA (firefly luciferase GL3, Nippon Gene, Tokyo, Japan)
was transfected using Lipofectamine® RNAiMAX reagent
according to the instruction manual (Thermo Fisher Scientific,
Inc.). The siRNA-transfected cells were cultured for 24 h. Then 1
µg of expression plasmid DNA was further transfected using
Lipofectamine® LTX with Plus reagent. The co-transfected
cells were further cultured for 48 h and then subjected to RNA
extraction. The sequences of siRNAs used in this study are shown in
Table II.
| Table II.siRNAs used in the present study. |
Table II.
siRNAs used in the present study.
| Gene | Cat. no.
(manufacturer) | siRNA sequence
(5′-3′) |
|---|
| Human
YAP1 | sc-38637A (Santa
Cruz Biotechnology, Inc.) | Sense
CCACCAAGCUAGAUAAAGAdTdT |
|
|
| Antisense
UCUUUAUCUAGCUUGGUGGdTdT |
| Human
YAP1 | sc-38637B (Santa
Cruz Biotechnology, Inc.) | Sense
GCAUGAGACAAUUUCCAUAdTdT |
|
|
| Antisense
UAUGGAAAUUGUCUCAUGCdTdT |
| Human
YAP1 | sc-38637C (Santa
Cruz Biotechnology, Inc.) | Sense
GGGUGUGCCUAUCAUAACAdTdT |
|
|
| Antisense
UGUUAUGAUAGGCACACCCdTdT |
| Firefly Luciferase
GL3 | Control siRNA
duplex, 318-05931 (Nippon Gene Co., Ltd.) | Sense
CUUACGCUGAGUACUUCGAdTdT |
|
|
| Antisense
UCGAAGUACUCAGCGUAAGdTdT |
RNA extraction and RT-qPCR
Total RNA was extracted from cells using Isogen
(Nippon Gene) and RNA was treated with the Turbo
DNA-free™ kit according to the manufacturer's
instructions (Thermo Fisher Scientific, Inc.). Thereafter, 1 µg of
RNA was used for the synthesis of cDNA with the
PrimeScript™ RT reagent kit (Perfect Real Time) (Takara)
by iCycler 170-8720JA (Bio-Rad Laboratories). The following
temperature protocol was used for RT: 37°C for 15 min and 85°C for
5 sec. Subsequently, qPCR analysis was performed with Thermal
Cycler Dicer Real Time System TP860 (Takara) and TB Green Premix Ex
Taq™ II (Tli RNaseH Plus) kit (Takara). The reaction and
operation of the apparatus were performed according to the
instruction manuals (Takara). The following thermocycling
conditions were used for qPCR: initial denaturation at 95°C for 30
sec, then 40 cycles of 95°C for 5 sec and 60°C for 30 sec, and 95°C
for 15 sec, 60°C for 30 sec, and 95°C for 15 sec. Gene expression
levels were normalized by the expression level of
glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The
sequences of primers to amplify the target genes, human α-smooth
muscle actin (ACTA2), human type I collagen α1 chain
(COL1A1), human tenascin-X (TNXB), human integrin β1
(ITGB1), human integrin α11 (ITGA11), human TGF-β1
(TGFB1), human YAP1 (YAP1) and human GAPDH
(GAPDH), are shown in Table
III. Relative expression was calculated using the
2−ΔΔCq method with normalization to GAPDH
(34).
| Table III.Primers used for quantitative PCR
analyses. |
Table III.
Primers used for quantitative PCR
analyses.
| Gene | Primer sequence
(5′-3′) | DDBJ/Genbank
accession number |
|---|
| ACTA2 | F:
ACTGCCGCATCCTCATCC | X13839 |
|
| R:
ATGCTGTTGTAGGTGGTTTCAT |
|
| COL1A1 | F:
CCCCTGGAAAGAATGGAGAT | Z74615 |
|
| R:
AATCCTCGAGCACCCTGA |
|
| TNXB | F:
GTGGTCCAGTATGAGGACACG | BC130037 |
|
| R:
CTGGTGGTCACGTACGTCAC |
|
| ITGB1 | F:
GAAAACAGCGCATATCTGGAAAT | X07979 |
|
| R:
CAGCCAATCAGTGATCCACAA |
|
| ITGA11 | F:
GACGGGAGACGTGTACAAGTGTC | AF137378 |
|
| R:
CCGAGGCGCATGTTGTC |
|
| TGFB1 | F:
ACATTGACTTCCGCAAGGAC | X02812 |
|
| R:
GTCCAGGCTCCAAATGTAGG |
|
| YAP1 | F:
GAACTCGGCTTCAGGTCCTC | NM_001195045 |
|
| R:
AGGGTCAAGCCTTGGGTCTA |
|
| GAPDH | F:
ACAACTTTGGTATCGTGGAAGG | X01677 |
|
| R:
GCCATCACGCCACAGTTTC |
|
Furthermore, we confirmed the successful
overexpression of both hTNX-FGFFFF and ITGA11 in LX-2 cells in
co-transfection with siRNA by RT-qPCR analyses. qPCR for
hTNX-FGFFFF was done with specific primers, forward fhTNX-FGFFF
primer (5′-GGTGGGCTGCGGATCCCCTTC-3′) and reverse rhTNX-FGFFFF
primer (5′-CATCTCCTCCCCGC-3′).
Confirmation of successful
overexpression of constructed expression plasmids in LX-2
cells
As for FLAG-tagged pSecF-hTNX-FG2, pSecF-hTNX-FGF-2,
pSec-hTNX-FGL-2, pSecF-hTNX-FGFF-3 and pSecF-hTNX-FGFL-1, after
transfection in LX-2 cells followed by culture as described above,
protein extracts from cells were prepared with Pierce™
RIPA buffer (Thermo Fisher Scientific, Inc.) with protease
inhibitor Complete (Roche Diagnostics). The samples were sonicated
and then incubated at 4°C for 2 h and centrifuged, and the
supernatant was collected in new tubes. The protein concentration
was measured with the Pierce™ BCA protein assay kit
(Thermo Fisher Scientific, Inc.). Then the protein extract after
transfection of pSecF-hTNX-FG2, pSecF-hTNX-FGF-2, pSec-hTNX-FGL-2,
pSecF-hTNX-FGFF-3 and pSecF-hTNX-FGFL-1 was subjected to 15%
SDS-PAGE under a reducing. After electrophoresis, the proteins
after transfection of pSecF-hTNX-FG2, pSecF-hTNX-FGF-2,
pSec-hTNX-FGL-2, pSecF-hTNX-FGFF-3 and pSecF-hTNX-FGFL-1 were
transferred to Immobilon-PSQ membrane (Merck Millipore).
The membrane was blocked with 5% skim milk (Snow Brand Milk
Products) and incubated with primary antibody overnight at 4°C
followed by incubation with a secondary antibody. The primary
antibody used was anti-FLAG M2 mouse monoclonal antibody (1:1,500,
#F3165, Sigma-Aldrich). Horseradish peroxidase (HRP)-conjugated
anti-mouse goat IgG (H+L chain) (1:25,000, #330, Medical and
Biological Laboratories) were used as a secondary antibody. The
blot was washed in TBST [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.1%
Tween-20]. HRP reaction was performed with ECL Prime Western
Blotting Detection Reagents (Cytiva, Marlborough, MA, USA), and the
developed chemiluminescent intensity was detected by
Amersham™ ImageQuant™ 800 (Cytiva).
Successful overexpression of FLAG-tagged proteins including
hTNX-FG, hTNX-FGF, hTNX-FGL, hTNX-FGFF and hTNX-FGFL was confirmed
by western blot analyses (data not shown).
As for pSecF-hTNX-FGFFF-2, pSecF-hTNX-FGFFL-3,
pSec-hTNX-FGFFFF-8, pSecF-hTNX-FGFFFM-1 and pSecF-hTNX-FGFFFL-5 and
pSecF-hTNX-FGpeptide2-5, we verified their successful
overexpression by RT-qPCR analyses with suitable primer pairs for
each transcript, since we failed to detect their proteins by
Western blot analyses due to the products with small molecular
weights of less than 10 kDa (data not shown).
Confirmation of successful
overexpression of pBJ1-ITGA11 plasmid in LX-2 cells
The protein extracts from non-transfected LX-2
cells, pBJΔ (empty vector)-transfected LX-2 cells and
pBJ1-ITGA11-transfected LX-2 cells were prepared with
Pierce™ RIPA buffer with protease inhibitor complete.
Subsequently, 60 µg of the protein extracts was subjected to 10%
SDS-PAGE under non-reducing conditions. After electrophoresis, the
proteins were transferred to Amersham™
Protran™ nitrocellulose membrane (Cytiva). The membrane
was blocked with 5% skim milk and incubated with primary antibody
for 2 h followed by incubation with a secondary antibody. The
primary antibody used was anti-integrin α11 rat monoclonal antibody
(1:1,000; #396214; R&D Systems, Inc.). HRP-conjugated anti-rat
goat IgG (H+L chain) (1:25,000; #31470; Thermo Fisher Scientific,
Inc.) was used as a secondary antibody. The blot was washed in
TBST. The HRP reaction was performed with ECL Prime Western
Blotting Detection Reagents, and the developed chemiluminescent
intensity was detected using the Amersham™
ImageQuant™ 800.
Experiments with inhibitors
To investigate the possible signaling pathway
involved in the induction of COL1A1 expression by
overexpression of both hTNX-FGFFFF and integrin α11 in LX-2 cells,
a TGF-β receptor type 1 (TGFBRI) inhibitor (SB525334)
(MedChemExpress) and a YAP inhibitor (verteporfin) (Cayman
Chemical) were used. Each of the inhibitors was dissolved in
dimethyl sulfoxide (DMSO). After transfection as described above,
LX-2 cells were cultured in DMEM with 0.5% FBS for 24 h, and then
the culture was further continued with 2 µM verteporfin for 19 h or
with 10 µM SB525334 for 42 h. The inhibitor-treated LX-2 cells were
collected and then RNA was extracted from the cells.
Enzyme-linked immunosorbent assay
(ELISA)
ELISA was carried out to determine the concentration
of TGF-β1 in the conditioned medium of LX-2 cells transfected with
the expression plasmids by using the Quantikine ELISA human TGF-β1
immunoassay (R&D Systems) according to the manufacturer's
manual. To activate latent TGF-β1 to the immunoreactive form,
samples were treated with HCl for 10 min prior to the assay.
Statistical analysis and software
Data are expressed as means ± standard deviation
(SD). Statistical analysis was performed using one-way ANOVA with
the Tukey-Kramer, Dunnett or Bonferroni post hoc test for the
comparison of multiple groups in Mac ToukeiKaiseki Ver. 3.0 (ESUMI
Co., Ltd., Nakano, Tokyo, Japan) to define statistical differences
for the groups. P<0.05 was considered to indicate a
statistically significant difference. The maps of pBJ1-ITGA11 and
pBJΔ plasmids were prepared using the SnapGene Viewer software
(version 5.1.3) (https://www.snapgene.com/snapgene-viewer).
Results
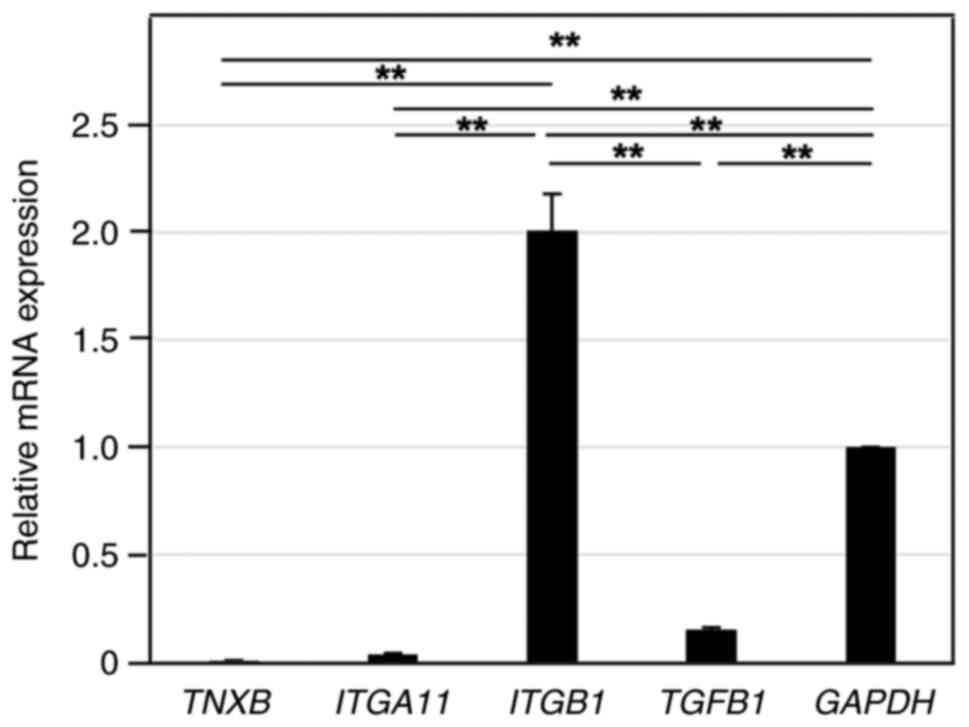
Endogenous expression levels of TNXB
and ITGA11 are very low compared with that of ITGB in LX-2
cells
First, we examined the endogenous expression levels
of genes relevant to this study, including TNXB, ITGA11,
ITGB1 and TGFB1, in LX-2 cells by RT-qPCR. As shown in
Fig. 2, ITGB1 and
TGFB1 expression levels were 2-times and 0.15-times higher,
respectively, than the expression level of control GAPDH.
However, the expression levels of TNXB and ITGA11
were very low, and they were only 0.002-times and 0.017-times
higher, respectively, than the expression level of ITGB1.
Generally, integrin α11 forms a heterodimer with integrin β1, which
is endogenously abundant in cells (35). It has been reported that
overexpressed integrin α11 in cells functionally interacts with
endogenous integrin β1, forming integrin α11β1 (36). Therefore, we attempted to
overexpress both integrin α11 and the fibrinogen domain of TNX in
LX-2 cells for the sake of further analysis. The overexpression of
an expression plasmid, pBJ1-ITGA11 (Fig. S1), for ITGA11 in LX-2 cells was
confirmed (Fig. S2). On the
other hand, we could not observe any specific expression of ITGA11
in non-transfected cells and an empty vector, pBJΔ
plasmid-transfected cells (Fig.
S2). In this study, we used a conditioned medium with 0.5% FBS
that contained TGF-β1, eventually resulting in the concentration of
TGF-β1 in the cell condition medium being 502.4 nM as determined by
ELISA for TGF-β1 (data not shown). Therefore, since we considered
that the amount of TGF-β1 in the cell conditioned medium is
sufficient for this study, TGF-β1 was not overexpressed in LX-2
cells anymore.
Identification of the region in TNX-FG
involved in the induction of COL1A1 expression in LX-2 cells
In order to determine whether overexpression of both
the TNX-FG domain and integrin α11β1 is able to provoke induction
of COL1A1 expression in LX-2 cells, both hTNX-FG (Fig. 1) and integrin α11 were
overexpressed and then the expression levels of fibrosis marker
genes including ACTA2, COL1A1 and TGFB1 were
investigated by RT-qPCR (Fig.
3A). Contrary to our expectation, induction of the expression
of these genes in LX-2 cells was not observed by overexpression of
both hTNX-FG and integrin α11 compared with that in LX-2 cells
alone (Fig. 3A). Next,
considering the possibility that TNX-FG coding a 225-aa protein
contains not only a positive region(s) involved in induced
expression but also a negative region(s) involved in decreased
expression of fibrosis maker genes, TNX-FG was divided into the
first half (referred to as hTNX-FGF) and the latter half (hTNX-FGL)
(Fig. 1), and then these regions
were assessed for their activity to induce expression of fibrosis
marker genes (Fig. 3B). As
expected, significant induction of COL1A1 expression was
detected when both hTNX-FGF and integrin α11 were overexpressed in
LX-2 cells compared with that in control cells without transfection
(1.37-fold, P<0.05 vs. control), but induction of ACTA2
and TGFB1 expression was not observed (Fig. 3B). On the other hand, in the case
of overexpression of both hTNX-FGL and integrin α11, induction of
COL1A1 expression as well as ACTA2 and TGFB1
expression was not observed.
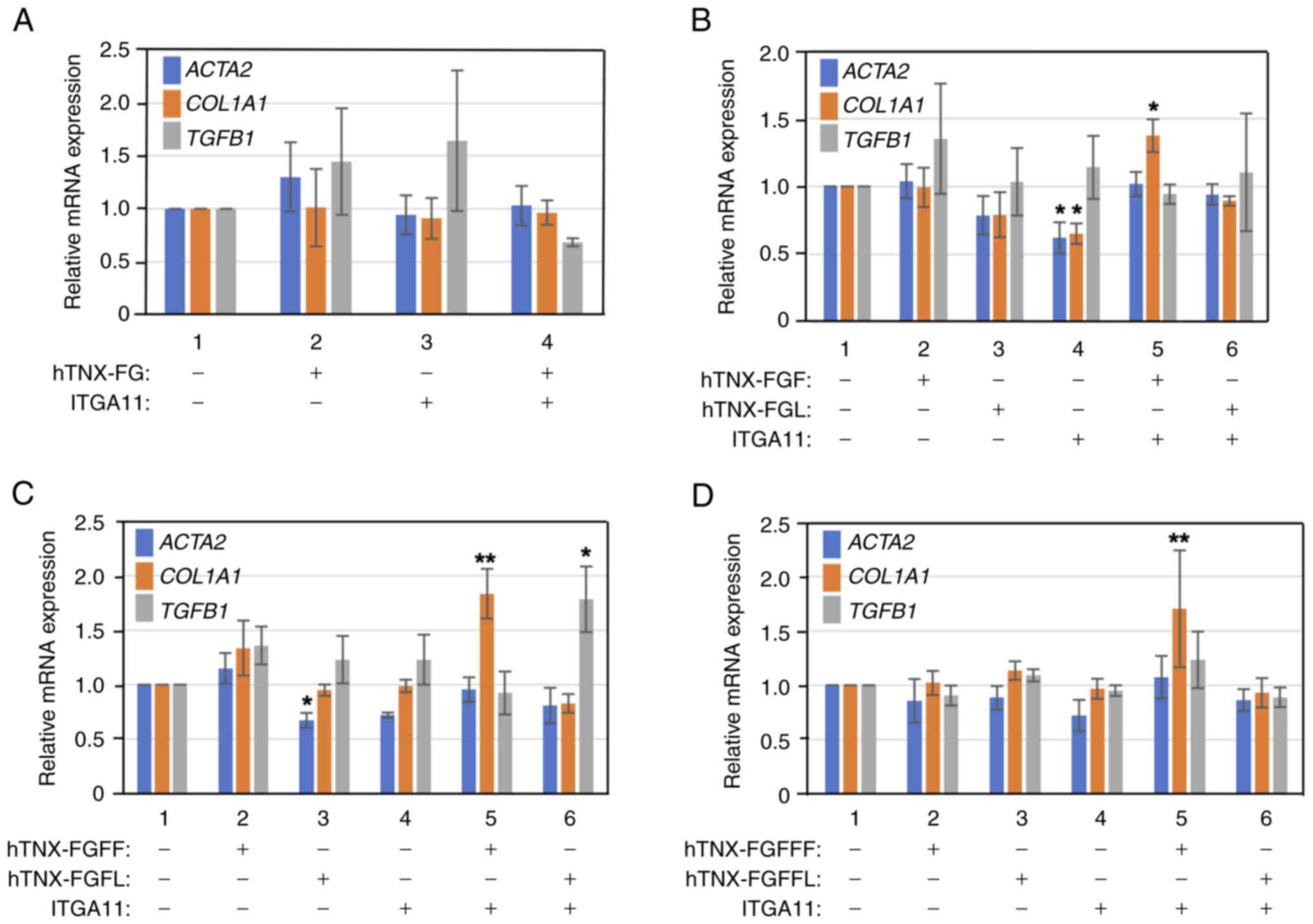 | Figure 3.Narrowing down of the domain involved
in the induction of COL1A1 expression in LX-2 cells. (A)
Overexpression of both full-length fibrinogen domain of TNX
[TNX-FG] and ITGA11 failed to induce the expression of fibrosis
marker genes, including ACTA2, COL1A1 and TGFB1. LX-2
cells were transfected with expression vectors for hTNX-FG (lane
2), ITGA11 (lane 3) and hTNX-FG and ITGA11 (lane 4) in DMEM/0.5%
FBS. (B) Induction of COL1A1 expression by overexpression of
both hTNX-FGF and ITGA11. LX-2 cells were transfected with
expression vectors for hTNX-FGF (lane 2), hTNX-FGL (lane 3), ITGA11
(lane 4), hTNX-FGF and ITGA11 (lane 5) and hTNX-FGL and ITGA11
(lane 6) in DMEM/0.5% FBS. (C) Induction of COL1A1
expression by overexpression of both hTNX-FGFF and ITGA11. LX-2
cells were transfected with expression vectors for hTNX-FGFF (lane
2), hTNX-FGFL (lane 3), ITGA11 (lane 4), hTNX-FGFF and ITGA11 (lane
5) and hTNX-FGFL and ITGA11 (lane 6) in DMEM/0.5% FBS. (D)
Induction of COL1A1 expression by overexpression of both
hTNX-FGFFF and ITGA11. LX-2 cells were transfected with expression
vectors for hTNX-FGFFF (lane 2), hTNX-FGFFL (lane 3), ITGA11 (lane
4), hTNX-FGFFF and ITGA11 (lane 5) and hTNX-FGFFL and ITGA11 (lane
6) in DMEM/0.5% FBS. (A-D) As a control, RNA from the cells without
transfection (lane 1) was used. The cell lysate was prepared 48 h
after transfection and then RNA was purified. Subsequently, the
expression levels of ACTA2, COL1A1 and TGFB1 were
examined by reverse transcription-quantitative PCR. The expression
level of each gene in the control was set to 1.0, and the relative
expression level of each gene compared with that of the control is
shown (n=3). Data are presented as the mean ± SD. *P<0.05,
**P<0.01 vs. control (lane 1), one-way ANOVA with Dunnett's post
hoc test. COL1A1, type I collagen α1 chain; TNX, tenascin-X;
ITGA11, integrin α11; ACTA2, α-smooth muscle actin; hTNX-FG,
fibrinogen-related domain of human tenascin-X; hTNX-FGF, first half
of hTNX-FG; hTNX-FGL, latter half of hTNX-FG; hTNX-FGFF, first half
of hTNX-FGF; hTNX-FGFL, latter half of hTNX-FGF; hTNX-FGFFF, first
half of hTNX-FGFF; hTNX-FGFFL, latter half of hTNX-FGFF. |
In order to narrow down the region in hTNX-FGF that
is responsible for the induction of expression of COL1A1, we
constructed expression plasmids for hTNX-FGFF (first half of
hTNX-FGF) and hTNX-FGFL (latter half of hTNX-FGF) (Fig. 1), and then we examined the
activity of hTNX-FGFF and hTNX-FGFL for induction of the expression
of fibrosis marker genes (Fig.
3C). As shown in Fig. 3C,
overexpression of both hTNX-FGFL and integrin α11 was not effective
for induction of ACTA2 and COL1A1 expression other
than TGFB1 expression (1.79-fold, P<0.05 vs. control),
but overexpression of both hTNX-FGFF and integrin α11 triggered the
induction of COL1A1 expression (1.84-fold, P<0.01 vs.
control). Subsequently, the hTNX-FGFF region was divided into the
first half (hTNX-FGFFF) and latter half (hTNX-FGFFL) (Fig. 1). When analyzed in the same way,
it was found out that overexpression of both hTNX-FGFFF and
integrin α11 evoked the induction of COL1A1 expression
(1.67-fold, P<0.01 vs. control) (Fig. 3D).
Ultimately, the hTNX-FGFFF region was divided into
the first half (hTNX-FGFFFF) and latter half (hTNX-FGFFFL)
(Fig. 1). After similar analysis,
it was revealed that overexpression of both hTNX-FGFFFF and
integrin α11 significantly evoked the induction of COL1A1
expression (1.79-fold, P<0.01 vs. control) (Fig. 4A). hTNX-FGFFFF codes
GGLRIPFPRDCGEEM from the N-terminal to C-terminal regions with a
length of 15-aa. In order to determine further narrowed sequences
involved in the induction of COL1A1 expression, we prepared
an expression plasmid, pSecF-hTNX-FGpeptide2-5 (Fig. 1), coding GGLRIPF within
hTNX-FGFFFF and an expression plasmid, pSecF-hTNX-FGFFFM-1
(Fig. 1), coding the 15-aa
sequence partially overlapped with the hTNX-FGFFFF sequence.
However, neither of the expression plasmids could significantly
induce expression of the three fibrosis marker genes (Fig. 4B and C). These results indicated
that the minimal sequence in the TNX-FG domain expressed with
integrin α11 for induction of COL1A1 expression in LX-2
cells is GGLRIPFPRDCGEEM.
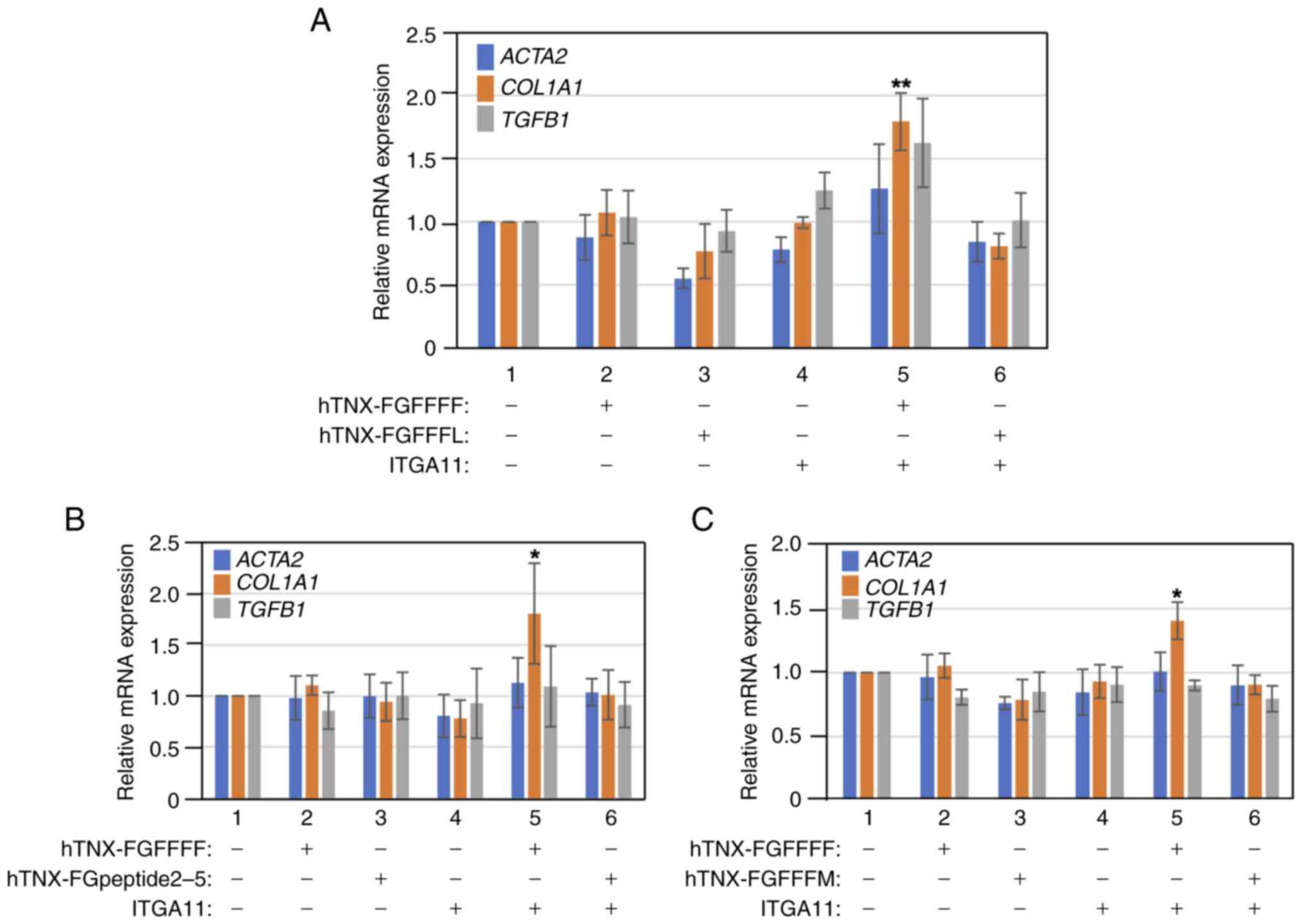 | Figure 4.Identification of the minimal
sequence responsible for induction of COL1A1 expression in
LX-2 cells. (A) Induction of COL1A1 expression by
overexpression of both hTNX-FGFFFF and ITGA11. LX-2 cells were
transfected with expression vectors for hTNX-FGFFFF (lane 2),
hTNX-FGFFFL (lane 3), ITGA11 (lane 4), hTNX-FGFFFF and ITGA11 (lane
5) and hTNX-FGFFFL and ITGA11 (lane 6) in DMEM/0.5% FBS. (B)
Overexpression of both hTNX-FGpeptide2-5 and ITGA11 did not cause
induction of COL1A1 expression. LX-2 cells were transfected
with expression vectors for hTNX-FGFFFF (lane 2), hTNX-FGpeptide2-5
(lane 3), ITGA11 (lane 4), hTNX-FGFFFF and ITGA11 (lane 5) and
hTNX-FGpeptide2-5 and ITGA11 (lane 6) in DMEM/0.5% FBS. (C)
Overexpression of both hTNX-FGFFFM and ITGA11 did not cause
induction of COL1A1 expression. LX-2 cells were transfected
with expression vectors for hTNX-FGFFFF (lane 2), hTNX-FGFFFM (lane
3), ITGA11 (lane 4), hTNX-FGFFFF and ITGA11 (lane 5) and
hTNX-FGFFFM and ITGA11 (lane 6) in DMEM/0.5% FBS. After
transfection followed by cell culture, cell lysate extraction and
RNA purification, the expression levels of ACTA2, COL1A1 and
TGFB1 were examined by reverse transcription-quantitative
PCR. (A-C) The expression level of each gene in the control (lane
1, RNA from cells without transfection) was set to 1.0, and the
relative expression level of each gene compared with that of the
control is shown (n=3). Data are presented as the mean ± SD.
*P<0.05, **P<0.01 vs. control (lane 1), one-way ANOVA with
Dunnett's post hoc test. COL1A1, type I collagen α1 chain;
TNX, tenascin-X; ITGA11, integrin α11; ACTA2, α-smooth
muscle actin; hTNX-FGFFFF, GGLRIPFPRDCGEEM peptide from
fibrinogen-related domain of human tenascin-X (hTNX-FG);
hTNX-FGFFFM, PRDCGEEMQNGAGAS peptide from hTNX-FG; hTNX-FGFFFL,
QNGAGASRTSTIFL peptide from hTNX-FG; hTNX-FGpeptide2-5, GGLRIPF
peptide from hTNX-FG. |
Furthermore, we synthesized the 15-aa peptide
(hTNX-FGFFFF peptide) and then examined whether the addition of
hTNX-FGFFFF peptide in the medium with co-transfection of integrin
α11 can induce COL1A1 expression in LX-2 cells. However, we
failed to obtain conclusive results showing that COL1A1
expression is induced by the addition of the hTNX-FGFFFF peptide
(data not shown). Our results indicate that addition of synthetic
15-amino acid peptide in the medium is not sufficient for induction
of COL1A1 expression and that expression of the 15-amino
acid peptide in cells is necessary for its induction.
YAP inhibitor verteporfin strongly
suppresses COL1A1 expression triggered by overexpression of both
hTNX-FGFFFF and integrin α11
First, to reveal whether the TGF-β signaling pathway
is involved in the induction of COL1A1 expression when both
hTNX-FGFFFF and integrin α11 were overexpressed, a TGFBRI inhibitor
(SB525334) was added to the culture medium after the transfection
of expression plasmids for both hTNX-FGFFFF and integrin α11 in
LX-2 cells. After incubation, the cells were collected and then the
expression levels of ACTA2, COL1A1 and TGFB1 were
analyzed by RT-qPCR. As shown in Fig.
5A the expression levels of COL1A1 and TGFB1 were
significantly suppressed to 52.9 and 54.1%, respectively, by the
addition of DMSO and SB525334 (lane 3) compared with that of DMSO
alone (lane 2) (P<0.01). Next, we investigated the involvement
of the YAP signaling pathway by the addition of a YAP inhibitor
(verteporfin). As shown in Fig.
5B, verteporfin (lane 3) significantly suppressed YAP1
expression to 41.6% (P<0.01) and ACTA2 expression to
66.7% (P<0.01) and strongly suppressed COL1A1 expression
to 11.8% (P<0.01) compared with that of DMSO (lane 2). These
results indicated that the TGF-β1 signaling pathway is partly
involved in the induction of COL1A1 expression but that the
YAP signaling pathway is greatly involved in the induction of
COL1A1 expression by overexpression of both hTNX-FGFFFF and
integrin α11.
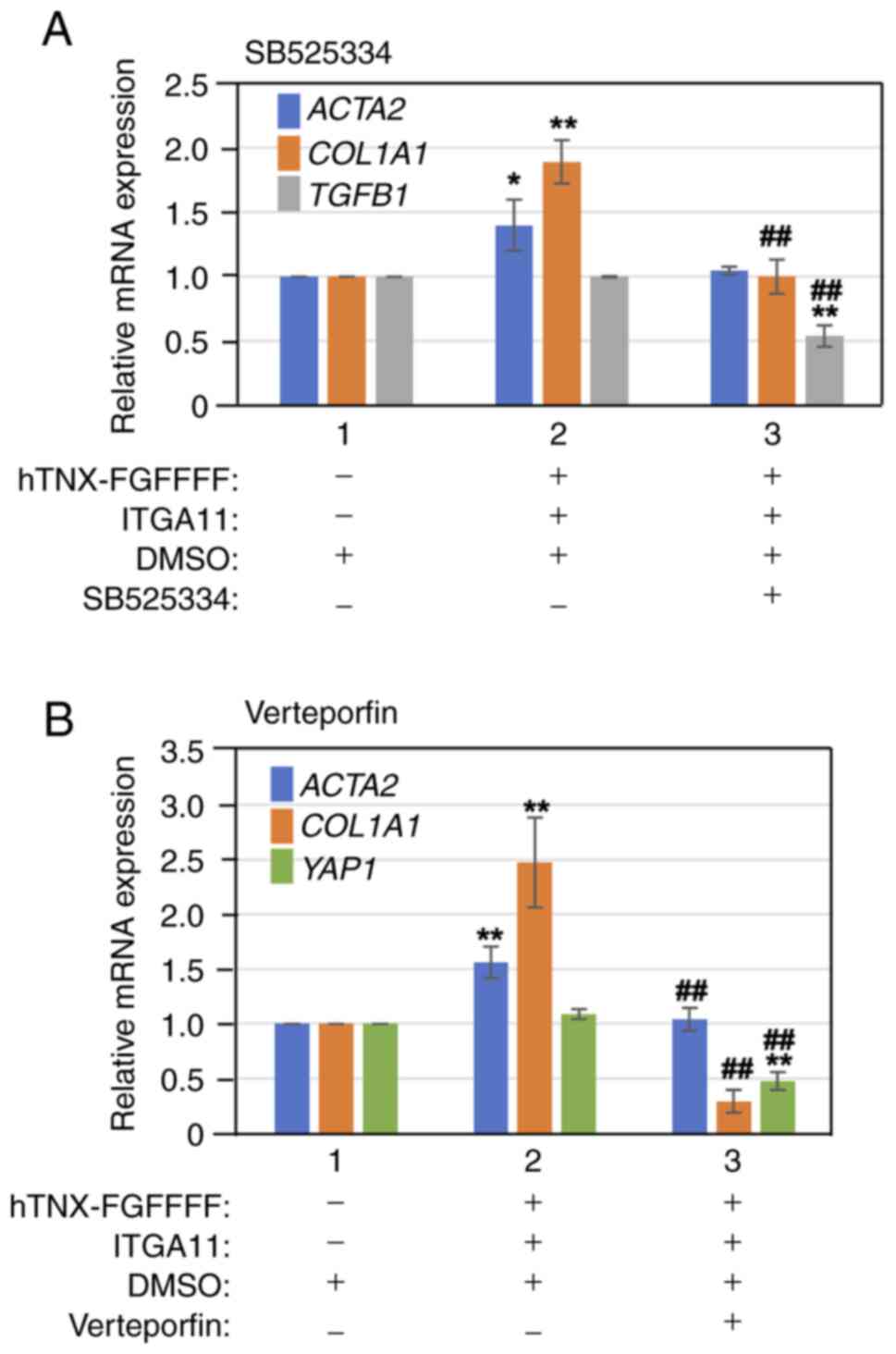 | Figure 5.Induction of COL1A1 expression
by overexpression of both hTNX-FGFFFF and ITGA11 in addition to
inhibitors. (A) Induction of COL1A1 expression by
overexpression of both hTNX-FGFFFF and ITGA11 with a TGFBRI
inhibitor (SB525334). DMSO (lanes 1, 2 and 3) and SB525334 (lane 3)
were added to the culture medium (DMEM/0.5% FBS) after the
transfection of expression plasmids for both hTNX-FGFFFF and ITGA11
in LX-2 cells (lanes 2 and 3). (B) Induction of COL1A1
expression by overexpression of both hTNX-FGFFFF and ITGA11 with a
YAP inhibitor (verteporfin). DMSO (lanes 1, 2 and 3) and vertepofin
(lane 3) were added to the culture medium (DMEM/0.5% FBS) after the
transfection of expression plasmids for both hTNX-FGFFFF and ITGA11
in LX-2 cells (lanes 2 and 3). Subsequently, the cells were
cultured followed by cell lysate extraction, RNA purification and
reverse transcription-quantitative PCR. (A and B) The expression
level of each gene [ACTA2, COL1A1 and TGFB1 for (A)
and ACTA2, COL1A1 and YAP1 for (B)] in the control
(lane 1) was set to 1.0, and the relative expression level of each
gene compared with that of the control (lane 1) is shown (n=3).
Data are presented as the mean ± SD. *P<0.05, **P<0.01 vs.
control (lane 1); ##P<0.01 vs. lane 2, one-way ANOVA
with the Bonferroni post hoc test. COL1A1, type I collagen
α1 chain; TNX, tenascin-X; ITGA11, integrin α11; ACTA2,
α-smooth muscle actin; YAP, Yes-associated protein; hTNX-FGFFFF,
GGLRIPFPRDCGEEM peptide from fibrinogen-related domain of human
tenascin-X (hTNX-FG). |
YAP1 knockdown with YAP1 siRNA
suppresses the induction of COL1A1expression by overexpression of
both hTNX-FGFFFF and integrin α11
Next, YAP1 was knocked down with YAP1 siRNA prior to
overexpression of both hTNX-FGFFFF and integrin α11, and then the
expression levels of ACTA2, COL1A1 and YAP1 were
analyzed by RT-qPCR in LX-2 cells. As shown in Fig. 6, knockdown of YAP1 with siRNA
(lane 3) significantly suppressed YAP1 expression to 45.4%
(P<0.01) and COL1A1 expression to 72.4% (P<0.05)
compared to that with control siRNA treatment (lane 2). The results
of experiments with the YAP inhibitor verteporfin as well as with
YAP1 siRNA indicated that the YAP signaling pathway is mainly
involved in the induction of COL1A1 expression by
overexpression of both hTNX-FGFFFF and integrin α11.
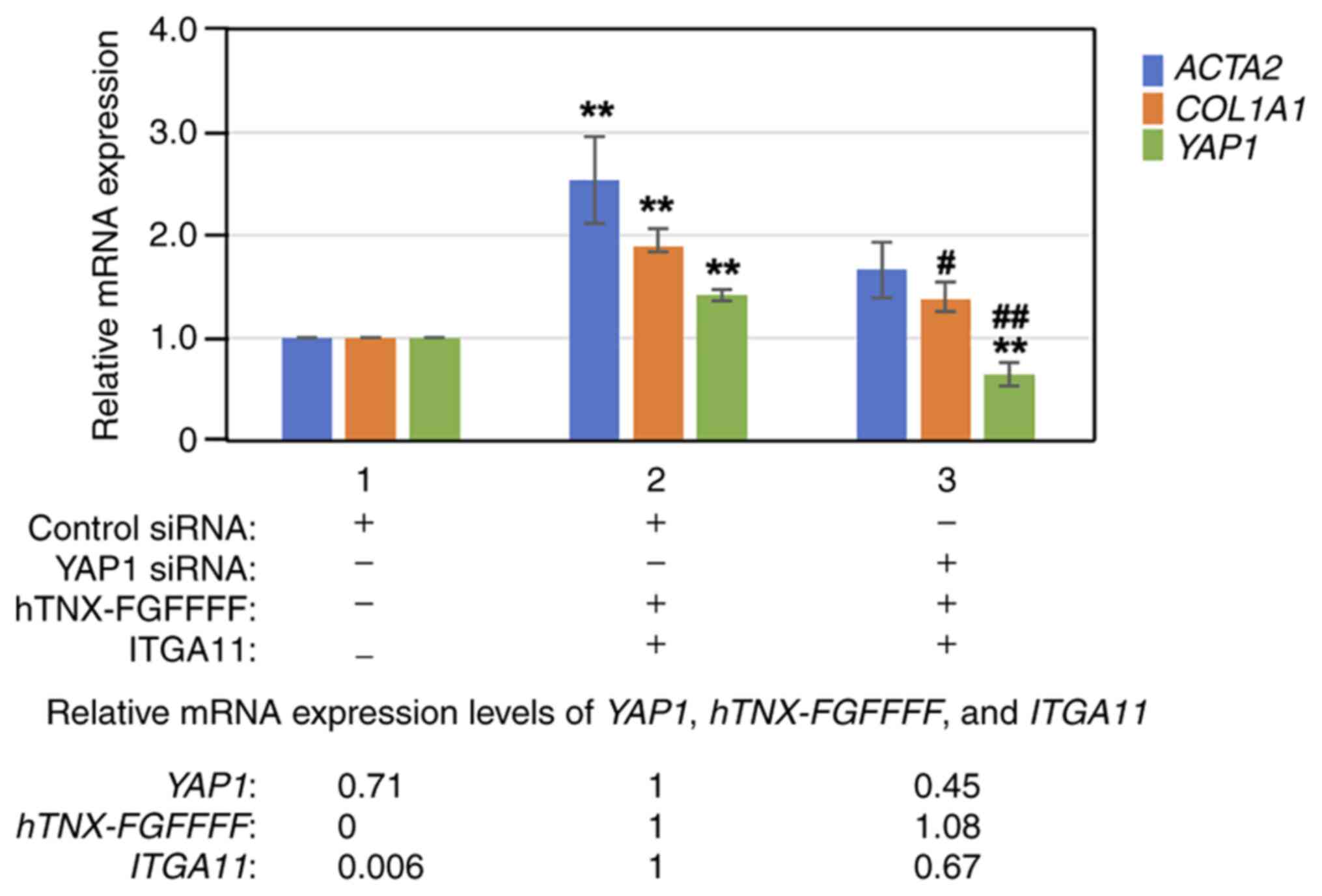 | Figure 6.Effect of YAP1 knockdown on induction
of COL1A1 expression. YAP1 was knocked down with YAP1 siRNA
prior to overexpression of both hTNX-FGFFFF and ITGA11, and then
the expression levels of ACTA2 and COL1A1 were
analyzed by reverse transcription-quantitative PCR in LX-2 cells.
RNA from cells treated with transfection of control siRNA only
(lane 1), transfection of control siRNA followed by co-transfection
with both hTNX-FGFFFF and ITGA11 expression plasmids (lane 2) and
transfection of YAP1 siRNA followed by co-transfection with both
hTNX-FGFFFF and ITGA11 expression plasmids (lane 3) was used. The
expression level of each gene (ACTA2, COL1A1 and
YAP1) in the control (control siRNA only) (lane 1) was set
to 1.0, and the relative expression level of each gene compared
with that of the control (control siRNA) is shown (n=3). Data are
presented as the mean ± SD. **P<0.01 vs. control (lane 1);
#P<0.05, ##P<0.01 vs. lane 2, one-way
ANOVA with the Bonferroni post hoc test. At the bottom of the
figure, the relative expression levels of YAP1, hTNX-FGFFFF
and ITGA11 are also shown, setting lane 2 to 1.0, since
hTNX-FGFFFF expression was not detected in lane 1.
COL1A1, type I collagen α1 chain; TNX, tenascin-X; ITGA11,
integrin α11; ACTA2, α-smooth muscle actin; YAP1,
Yes-associated protein 1; siRNA, small interfering RNA;
hTNX-FGFFFF, GGLRIPFPRDCGEEM peptide from fibrinogen-related domain
of human tenascin-X (hTNX-FG). |
Discussion
In this study, we revealed that overexpression of
both a 15-aa peptide (hTNX-FGFFFF) derived from the TNX-FG domain
and integrin α11 induces the expression of type I collagen α1 chain
(COL1A1) via mainly the YAP signaling pathway and partly by
the TGF-β1 signaling pathway in LX-2 cells. It is known that
overexpressed integrin α11 functionally interacts with endogenous
integrin β1, resulting in the formation of integrin α11β1 (36). Therefore, the expression of
COL1A1 induced by hTNX-FGFFFF would be caused via the YAP
signaling pathway through integrin α11β1.
It has been shown that TNC, the most
well-characterized member of the tenascin family, can promote
fibrosis (37) in not only the
liver (38) but also the heart
(39) and lung (40). In mice with immune-mediated
chronic hepatitis, El-Karef et al (38) showed that TNC accelerates liver
fibrosis by an increased inflammatory response through recruitment
of activated hepatic stellate cells (HSCs) and upregulation of
TGF-β expression. However, the region in TNC that is responsible
for the pathogenesis of fibrosis remains to be determined.
Eponymous fibrinogen is involved in the pathogenesis of fibrosis in
addition to the formation of a provisional fibrin matrix for
creating a hemostatic blood clot (41). Craciun et al (42) showed that the expression of
fibrinogen in the kidney is induced after the appearance of a renal
lesion such as unilateral ureteral obstruction due to the induction
of fibroblast proliferation and activation of the TGF-β canonical
signaling pathway, leading to renal fibrosis. However, also in
fibrinogen, the region involved in the pathogenesis of fibrosis has
not yet been identified.
On the other hand, Aubert et al (43) recently showed that the FG domain
of each tenascin family member, which is highly conserved among the
four members, is associated with latent TGF-β1 and that the latent
TGF-β1 is activated to its mature form to transmit TGF-β1 signals
into cells. Molecular modeling and dynamic approaches have revealed
that some residues mainly located in loop 9 of each FG domain, a
region located at the C-terminal part of the domain, interact with
some residues located in helix α1 of latency-associated peptide
(LAP) and some residues in the mature TGF-β1 moiety (43). In the present study, we identified
a 15-aa peptide (referred to as hTNX-FGFFFF with an aa sequence of
GGLRIPFPRDCGEEM) located at the N-terminal part of the TNX-FG
domain as the minimum region responsible for the induction of
COL1A1 expression. Interestingly, this minimum region in the
TNX-FG domain was not overlapped with some residues in loop 9 in
the TNX-FG domain reported by Aubert et al (43), indicating that the 15-aa peptide
would fail to interact directly with LAP and mature TGF-β1.
However, the fact that SB525334 weakly suppresses the induction of
COL1A1 expression with overexpression of hTNX-FGFFFF and
integrin α11β1 as shown in Fig.
5A indicates that the TGF-β1 signaling pathway has only a small
contribution to the induction of COL1A1 expression. The
reason why the TGF-β1 signaling pathway is weakly activated by
overexpression of hTNX-FGFFFF and integrin α11β1 remains unclear,
but we speculate that overexpression of hTNX-FGFFFF and integrin
α11β1 weakly leads to the activation of TGF-β1 signaling following
an unidentified interaction of hTNX-FGFFFF and integrin α11β1 with,
for example, latent TGF-β binding protein (LTBP), a reservoir of
latent TGF-β into the matrix (44). It is important to explore the
possibility of tripartite interactions among hTNX-FGFFFF, integrin
α11β1, and LTBP.
It has not been determined whether hTNX-FGFFFF
(GGLRIPFPRDCGEEM sequence) interacts with integrin α11β1 directly
or indirectly. Integrin α11β1 recognizes the GFOGER sequence (where
O represents hydroxyproline) in fibrillar collagens such as type I
collagen (COL1) and type II collagen (COL2) via its I domain
(45). A comparison of the
hTNX-FGFFFF peptide sequence and the GFOGER sequence shows that
there is almost no homology between the two sequences. Therefore,
we speculate that hTNX-FGFFFF peptide fails to interact with
integrin α11 directly. In contrast, it is possible that hTNX-FGFFFF
peptide indirectly interacts with integrin α11 via COL1A1. Human
COL1A1 (UniProtKB: P02452) has a GFOGER sequence, and integrin α11
can thus interact with COL1A1. In addition, hTNX-FGFFFF peptide
might interact with COL1A1 since the TNX-FG domain is known to be
involved in the interaction of TNX with COL1 (46). With the interactions of
hTNX-FGFFFF peptide-COL1A1-integrin α11, the stimuli from
hTNX-FGFFFF peptide might be transmitted to YAP via integrin α11β1,
leading to the induction of COL1A1 expression, because it
has been reported that YAP signaling downstream of integrin α11β1
promotes the fibrotic phenotype in myofibroblasts (31). As a future plan, we are
considering to examine the interaction of hTNX-FGFFFF and COL1A1 by
biochemical analyses.
Both COL1A1 and ACTA2 are well known
as fibrosis marker genes in myofibroblasts. However, it is thought
that stimulation of YAP signaling sometimes leads to induction of
ACTA2 expression (47) and
sometimes does not (48) in
myofibroblasts. It has also been reported that only some
collagen-producing fibroblasts co-express ACTA2 in the
fibrotic lung and kidney (49).
Therefore, the results of the present study showing that
overexpression of both hTNX-FGFFFF and integrin α11 causes
induction of COL1A1 expression but not induction of
ACTA2 expression also indicate distinct regulation of the
gene expression of COL1A1 and ACTA2 in LX-2
cells.
Previously, our group showed that HFCD-fed wild-type
(WT) mice display liver dysfunction including type I collagen
deposition and inflammatory response compared with those in
TNX-deficient mice, indicating that TNX promotes liver fibrosis
in vivo (24). Therefore,
the results obtained in the present in vitro study showing
induction of COL1A1 expression by a 15-aa peptide located in
the TNX-FG domain in LX-2 cells may be related to the promotion of
liver fibrosis observed in HFCD-fed wild-type (WT) mice in
vivo. As described in the Results section, addition of the
synthetic 15-amino acid peptide in the conditioned medium with
co-transfection of integrin α11 was not sufficient for induction of
COL1A1 expression in LX-2 cells. We speculated that
expression of the 15-amino acid peptide in cells is necessary for
its induction. Thereby, we speculate that mere administration of
the synthetic 15-amino acid peptide to mice would not induce
COL1A1 expression in vivo as well. At present, this
causes the lack of animal experiments as a limitation of this
study.
In conclusion, the present in vitro study
showed that a 15-aa peptide located in the N-terminal part of the
TNX-FG domain induces the expression of COL1A1 via YAP
signaling downstream of integrin α11β1. Therefore, the suppression
of TNX expression may be a novel therapeutic target for improving
fibrosis in the liver.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Ning Lu
(Department of Biomedicine, Centre for Cancer Biomarkers, Norwegian
Centre of Excellence, University of Bergen, Bergen, Norway) for
providing the cDNA (pBJ1-ITGA11) encoding full-length human
integrin α11 protein and its gene map.
Funding
This work was supported by the Japan Society for the Promotion
of Science KAKENHI (grant no. 19K08470) from the Ministry of
Education, Culture, Sports, Science and Technology of Japan.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KIM designed the experiments, performed the
experiments and wrote the manuscript. KIM, KK and KY analyzed the
data. HT contributed to the conception and experimental design on
the construction of expression plasmids, and criti-cally revised
the manuscript for intellectual content. KIM and KY confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Adams JC and Watt FM: Regulation of
development and differentiation by the extracellular matrix.
Development. 117:1183–1198. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mavrogonatou E, Pratsinis H, Papadopoulou
A, Karamanos NK and Kletsas D: Extracellular matrix alterations in
senescent cells and their significance in tissue homeostasis.
Matrix Biol. 75–76. 27–42. 2019.PubMed/NCBI
|
|
3
|
Chiquet-Ehrismann R and Tucker RP:
Tenascins and the importance of adhesion modulation. Cold Spring
Harb Perspect Biol. 3:a0049602011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Matsumoto K, Saga Y, Ikemura T, Sakakura T
and Chiquet-Ehrismann R: The distribution of tenascin-X is distinct
and often reciprocal to that of tenascin-C. J Cell Biol.
125:483–493. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liot S, Aubert A, Hervieu V, Kholti NE,
Schalkwijk J, Verrier B, Valcourt U and Lambert E: Loss of
tenascin-X expression during tumor progression: A new pan-cancer
marker. Matrix Biol Plus. 6–7. 1000212020.PubMed/NCBI
|
|
6
|
Valcourt U, Alcaraz LB, Exposito JY,
Lethias C and Bartholin L: Tenascin-X: Beyond the architectural
function. Cell Adh Migr. 9:154–165. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Minamitani T, Ikuta T, Saito Y, Takebe G,
Sato M, Sawa H, Nishimura T, Nakamura F, Takahashi K, Ariga H and
Matsumoto K: Modulation of collagen fibrillogenesis by tenascin-X
and type VI collagen. Exp Cell Res. 298:305–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Egging D, van den Berkmortel F, Taylor G,
Bristow J and Schalkwijk J: Interactions of human tenascin-X
domains with dermal extracellular matrix molecules. Arch Dermatol
Res. 298:389–396. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Margaron Y, Bostan L, Exposito JY,
Malbouyres M, Trunfio-Sfarghiu AM, Berthier Y and Lethias C:
Tenascin-X increases the stiffness of collagen gels without
affecting fibrillogenesis. Biophys Chem. 147:87–91. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zweers MC, van Vlijmen-Willems IM, van
Kuppevelt TH, Mecham RP, Steijlen PM, Bristow J and Schalkwijk J:
Deficiency of tenascin-X causes abnormalities in dermal elastic
fiber morphology. J Invest Dermatol. 122:885–891. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Burch GH, Gong Y, Liu W, Dettman RW, Curry
CJ, Smith L, Miller WL and Bristow J: Tenascin-X deficiency is
associated with Ehlers-Danlos syndrome. Nat Genet. 17:104–108.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schalkwijk J, Zweers MC, Steijlen PM, Dean
WB, Taylor G, van Vlijmen IM, van Haren B, Miller WL and Bristow J:
A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X
deficiency. N Engl J Med. 345:1167–1175. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Malfait F, Francomano C, Byers P, Belmont
J, Berglund B, Black J, Bloom L, Bowen JM, Brady AF, Burrows NP, et
al: The 2017 international classification of the Ehlers-Danlos
syndromes. Am J Med Genet C Semin Med Genet. 175:8–26. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Okuda-Ashitaka E, Kakuchi Y, Kakumoto H,
Yamanishi S, Kamada H, Yoshidu T, Matsukawa S, Ogura N, Uto S,
Minami T, et al: Mechanical allodynia in mice with tenascin-X
deficiency associated with Ehlers-Danlos syndrome. Sci Rep.
10:65692020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kawakami K and Matsumoto K: Behavioral
alterations in mice lacking the gene for tenascin-X. Biol Pharm
Bull. 34:590–593. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ikuta T, Ariga H and Matsumoto K:
Extracellular matrix tenascin-X in combination with vascular
endothelial growth factor B enhances endothelial cell
proliferation. Genes Cells. 5:913–927. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sakai H, Yokota S, Kajitani N, Yoneyama T,
Kawakami K, Yasui Y and Matsumoto KI: A potential contribution of
tenascin-X to blood vessel formation in peripheral nerves. Neurosci
Res. 124:1–7. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sumioka T, Iwanishi H, Okada Y, Nidegawa
Y, Miyajima M, Matsumoto KI and Saika S: Loss of tenascin X gene
function impairs injury-induced stromal angiogenesis in mouse
corneas. J Cell Mol Med. 22:948–956. 2018.PubMed/NCBI
|
|
19
|
Matsumoto K, Sato T, Oka S, Orba Y, Sawa
H, Kabayama K, Inokuchi J and Ariga H: Triglyceride accumulation
and altered composition of triglyceride-associated fatty acids in
the skin of tenascin-X-deficient mice. Genes Cells. 9:737–748.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kajitani N, Yamada T, Kawakami K and
Matsumoto KI: TNX deficiency results in bone loss due to an
increase in multinucleated osteoclasts. Biochem Biophys Res Commun.
512:659–664. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Matsumoto K, Takayama N, Ohnishi J,
Ohnishi E, Shirayoshi Y, Nakatsuji N and Ariga H: Tumour invasion
and metastasis are promoted in mice deficient in tenascin-X. Genes
Cells. 6:1101–1111. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Matsumoto K, Minamitani T, Orba Y, Sato M,
Sawa H and Ariga H: Induction of matrix metalloproteinase-2 by
tenascin-X deficiency is mediated through the c-Jun N-terminal
kinase and protein tyrosine kinase phosphorylation pathway. Exp
Cell Res. 297:404–414. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Matsumoto KI and Aoki H: The roles of
tenascins in cardiovascular, inflammatory, and heritable connective
tissue diseases. Front Immunol. 11:6097522020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yamaguchi S, Kawakami K, Satoh K, Fukunaga
N, Akama K and Matsumoto KI: Suppression of hepatic dysfunction in
tenascin-X-deficient mice fed a high-fat diet. Mol Med Rep.
16:4061–4067. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Piersma B, Bank RA and Boersema M:
Signaling in fibrosis: TGF-β, WNT, and YAP/TAZ converge. Front Med
(Lausanne). 2:592015.PubMed/NCBI
|
|
26
|
Alcaraz LB, Exposito JY, Chuvin N, Pommier
RM, Cluzel C, Martel S, Sentis S, Bartholin L, Lethias C and
Valcourt U: Tenascin-X promotes epithelial-to-mesenchymal
transition by activating latent TGF-β. J Cell Biol. 205:409–428.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mannaerts I, Leite SB, Verhulst S,
Claerhout S, Eysackers N, Thoen LF, Hoorens A, Reynaert H, Halder G
and van Grunsven LA: The hippo pathway effector YAP controls mouse
hepatic stellate cell activation. J Hepatol. 63:679–688. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nguyen-Lefebvre AT, Selzner N, Wrana JL
and Bhat M: The hippo pathway: A master regulator of liver
metabolism, regeneration, and disease. FASEB J. 35:e215702021.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen G, Xia B, Fu Q, Huang X, Wang F, Chen
Z and Lv Y: Matrix mechanics as regulatory factors and therapeutic
targets in hepatic fibrosis. Int J Biol Sci. 15:2509–2521. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schulz JN, Plomann M, Sengle G, Gullberg
D, Krieg T and Eckes B: New developments on skin fibrosis-essential
signals emanating from the extracellular matrix for the control of
myofibroblasts. Matrix Biol. 68–69. 522–532. 2018.PubMed/NCBI
|
|
31
|
Martin K, Pritchett J, Llewellyn J, Mullan
AF, Athwal VS, Dobie R, Harvey E, Zeef L, Farrow S, Streuli C, et
al: PAK proteins and YAP-1 signalling downstream of integrin beta-1
in myofibroblasts promote liver fibrosis. Nat Commun. 7:125022016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Romaine A, Sørensen IW, Zeltz C, Lu N,
Erusappan PM, Melleby AO, Zhang L, Bendiksen B, Robinson EL,
Aronsen JM, et al: Overexpression of integrin α11 induces cardiac
fibrosis in mice. Acta Physiol (Oxf). 222:2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu CQ, Popova SN, Brown ER,
Barsyte-Lovejoy D, Navab R, Shih W, Li M, Lu M, Jurisica I, Penn
LZ, et al: Integrin alpha 11 regulates IGF2 expression in
fibroblasts to enhance tumorigenicity of human non-small-cell lung
cancer cells. Proc Natl Acad Sci USA. 104:11754–11759. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Barczyk M, Carracedo S and Gullberg D:
Integrins. Cell Tissue Res. 339:269–280. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tiger CF, Fougerousse F, Grundström G,
Velling T and Gullberg D: Alpha11beta1 integrin is a receptor for
interstitial collagens involved in cell migration and collagen
reorganization on mesenchymal nonmuscle cells. Dev Biol.
237:116–129. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kasprzycka M, Hammarström C and Haraldsen
G: Tenascins in fibrotic disorders-from bench to bedside. Cell Adh
Migr. 9:83–89. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
El-Karef A, Yoshida T, Gabazza EC,
Nishioka T, Inada H, Sakakura T and Imanaka-Yoshida K: Deficiency
of tenascin-C attenuates liver fibrosis in immune-mediated chronic
hepatitis in mice. J Pathol. 211:86–94. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nishioka T, Onishi K, Shimojo N, Nagano Y,
Matsusaka H, Ikeuchi M, Ide T, Tsutsui H, Hiroe M, Yoshida T and
Imanaka-Yoshida K: Tenascin-C may aggravate left ventricular
remodeling and function after myocardial infarction in mice. Am J
Physiol Heart Circ Physiol. 298:H1072–H1078. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Carey WA, Taylor GD, Dean WB and Bristow
JD: Tenascin-C deficiency attenuates TGF-ß-mediated fibrosis
following murine lung injury. Am J Physiol Lung Cell Mol Physiol.
299:L785–L793. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Masamune A, Kikuta K, Watanabe T, Satoh K,
Hirota M, Hamada S and Shimosegawa T: Fibrinogen induces cytokine
and collagen production in pancreatic stellate cells. Gut.
58:550–559. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Craciun FL, Ajay AK, Hoffmann D, Saikumar
J, Fabian SL, Bijol V, Humphreys BD and Vaidya VS: Pharmacological
and genetic depletion of fibrinogen protects from kidney fibrosis.
Am J Physiol Renal Physiol. 307:F471–F484. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Aubert A, Mercier-Gouy P, Aguero S,
Berthier L, Liot S, Prigent L, Alcaraz LB, Verrier B, Terreux R,
Moali C, et al: Latent TGF-β activation is a hallmark of the
tenascin family. Front Immunol. 12:6134382021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Todorovic V and Rifkin DB: LTBPs, more
than just an escort service. J Cell Biochem. 113:410–418. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang WM, Kapyla J, Puranen JS, Knight CG,
Tiger CF, Pentikainen OT, Johnson MS, Farndale RW, Heino J and
Gullberg D: Alpha 11beta 1 integrin recognizes the GFOGER sequence
in interstitial collagens. J Biol Chem. 278:7270–7277. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lethias C, Carisey A, Comte J, Cluzel C
and Exposito JY: A model of tenascin-X integration within the
collagenous network. FEBS Lett. 580:6281–6285. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu X, Long X, Liu W, Zhao Y, Hayashi T,
Yamato M, Mizuno K, Fujisaki H, Hattori S, Tashiro SI, et al: Type
I collagen induces mesenchymal cell differentiation into
myofibroblasts through YAP-induced TGF-β1 activation. Biochimie.
150:110–130. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Muppala S, Raghunathan VK, Jalilian I,
Thomasy S and Murphy CJ: YAP and TAZ are distinct effectors of
corneal myofibroblast transformation. Exp Eye Res. 180:102–109.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sun KH, Chang Y, Reed NI and Sheppard D:
α-Smooth muscle actin is an inconsistent marker of fibroblasts
responsible for force-dependent TGFβ activation or collagen
production across multiple models of organ fibrosis. Am J Physiol
Lung Cell Mol Physiol. 310:L824–L836. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ikuta T, Sogawa N, Ariga H, Ikemura T and
Matsumoto K: Structural analysis of mouse tenascin-X: Evolutionary
aspects of reduplication of FNIII repeats in the tenascin gene
family. Gene. 217:1–13. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Endo T, Ariga H and Matsumoto K: Truncated
form of tenascin-X, XB-S, interacts with mitotic motor kinesin Eg5.
Mol Cell Biochem. 320:53–66. 2009. View Article : Google Scholar : PubMed/NCBI
|