Introduction
The 46, XY disorder of sex development (DSD) is
defined as the abnormal sexual differentiation process of 46, XY
males, with patients presenting with gonads and/or phenotypic sex
that is inconsistent with a male genotype (1). Patients with 46, XY DSD are usually
born with birth defects and are at a high risk of gonadal tumors,
which not only increases physical and mental harm to patients, but
also subsequently burdens the family. Notably, an early diagnosis
is important for a positive outcome for patients with 46, XY DSD
(2). However, the clinical
phenotype of 46, XY DSD is highly heterogeneous. The abnormal
external genitalia of patients with 46, XY DSD can be manifested as
completely male or female; however, more commonly, a phenotype
between male and female is presented, with characteristics
including hypospadias, cryptorchidism, clitoral enlargement and
labia majora fusion (3). Clinical
diagnosis of 46, XY DSD is difficult and >50% of cases are not
accurately diagnosed (4).
The most frequent causes of 46, XY DSD are androgen
insensitivity and gonadal dysgenesis (5), which may involve mutations in
numerous genes, including sex-determining region Y (SRY),
androgen receptor (AR), SRY-related HMG-box gene 9
(SOX9), steroid 5 α-reductase 2, nuclear receptor subfamily
5 group A member 1 and mitogen-activated protein kinase kinase
kinase 1 (MAP3K1) (6).
Among them, MAP3K1 (NM_005921.1) variants are a common cause
and account for 15–20% of 46, XY DSD [Online Mendelian Inheritance
in Man (OMIM) 400044] cases (7).
These variants may lead to protein function acquisition that
changes co-factor binding and increases phosphorylation of targets
in the downstream MAPK pathway, which can lead to an imbalance
between SOX9/FGF9 and WNT/β-catenin signaling and abnormal human
sex determination (8). Although
these findings strongly suggest that MAP3K1 variants are
involved in the etiology of testicular dysgenesis, it is still
necessary to better understand the mechanism underlying the effects
of the MAPK pathway on the gene regulatory network of human
testicular development (6,9).
Therefore, identification of more cases will help to better
understand the relationship between genotype and phenotype in DSD
caused by MAP3K1 mutations.
The present study detected a novel pathogenic
MAP3K1 variant (c.3020A>G) in a Chinese patient with 46,
XY DSD and partial growth hormone (GH) deficiency. The variant was
revealed to be pathogenic and p.Gln1007 in MAP3K1 was shown to be
conserved across various species in bioinformatics analysis. The
present research expands the mutation spectrum of MAP3K1 and
may be conducive to improved genetic diagnosis and counseling in
the future.
Subjects and methods
Patients
The proband was a premature infant who was delivered
by caesarean section at 29 weeks' gestation with no testicles
detected at birth. At the age of 1 year, the parents of the proband
came to Shandong Provincial Hospital (Shandong, China) to receive a
diagnosis. The non-consanguineous parents of the patient had a
normal phenotype and denied a family history of genetic diseases.
All family members received careful clinical examinations and
peripheral blood samples were obtained for genetic analyses. The
hormone levels of the patient were detected; ultrasonography and
pituitary magnetic resonance (MR), histopathological examination
and laparoscopy were applied to evaluate the patient's genital
development and pituitary development, respectively. The Ethics
Committee of Shandong Provincial Hospital Affiliated to Shandong
University approved the present study (approval no. 2019-147) and
written informed consent was obtained from the parents of the
patient. The GH stimulation was conducted as follows: The patient
fasted for 8 h; on the first day, the patient received an injection
of arginine hydrochloride (0.5 g/kg); before arginine injection,
and 30, 60 and 90 min after injection, blood was collected to
measure GH and blood pressure was monitored. On the third day, oral
levodopa (10 mg/kg) was administered, and the same method of blood
collection as mentioned for the first day was performed, and blood
pressure was monitored.
Clinical follow-up
The patient was followed up by regular visits to
determine their specific characteristics and prognosis. In
particular, their intellectual and urogenital development,
treatment options and potential complications, such as gender
anxiety, psychological and cognitive impairment, and the presence
of gonadal tumors, were followed up on over 4 years.
Whole-exome sequencing (WES) and
Sanger sequencing
Using QIAamp DNA Mini kit (Qiagen GmbH), genomic DNA
was isolated from peripheral blood leukocytes for genetic analysis
of the pedigree (II-1, II-2 and III-1). Subsequently, DNA from
peripheral blood was used for WES; the genes associated with GH
deficiency were also studied by WES. Genomic DNA fragmentation,
paired-end adaptor ligation, amplification and purification were
performed, and human exons were then captured using SeqCap EZ Med
Exome Enrichment Kit (Roche Diagnostics). Following post-capture
amplification and purification, a DNA library was generated and
sequenced using the Illumina HiSeq sequencing platform (Illumina,
Inc.). To obtain the coverage and mean read depth of target
regions, sequence data alignment to the human reference genome
(hg19) and variant calling were performed using NextGene V2.3.4
software (SofGenetics). The average coverage of the exome was
>100×, which allowed a deep enough examination of the target
region to accurately match >99% of the target exons. Mutations
with low coverage in the target area were screened and filtered to
ensure the accuracy of data analysis. The quality/integrity of the
processed samples were verified by QuBit4 Fluorometer (Thermo
Fisher Scientific, Inc.) and Nanodrop one (Thermo Fisher
Scientific, Inc.); the type of sequencing nucleotide length was 300
bp (PE150) and the direction of sequencing was paired end; the
catalogue number of the sequencing kit was NovaSeq 6000 S4 Reagent
kit v1.5 (300 cycles; cat. no. 20028312; Illumina Inc.). The
loading concentration of the final library were ~10–20 pM for DNA
sequencing.
In addition, the frequency of normal populations
(whether there are variants included in the normal population
database) were obtained by NextGene V2.3.4 and our in-house scripts
from data from Genome Aggregation Database (GnomAD, http://gnomad.broadinstitute.org/), Exome
Aggregation Consortium (ExAC, http://exac.broadinstitute.org), Trans-Omics for
Precision Medicine (TOPMED, http://topmed.nhlbi.nih.gov/), Human Gene Mutation
Database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php), Clinvar and
OMIM databases (http://www.omim.org).
A variant was identified as a mutation when it was
not found in 500 Chinese controls with data from sequencing
companies in dbSNP (https://www.ncbi.nlm.nih.gov/snp/) and in the exome
variant server (https://evs.gs.washington.edu/EVS/), or when the
allele frequency was <0.001 in the aforementioned databases.
According to Standards and Guidelines for the Interpretation of
Sequence Variants published by the American College of Medical
Genetics and Genomics (ACMG) in 2015 (10), pathogenic variants were determined
with Human Genome Variation Society nomenclature.
Candidate variants were detected by WES.
Subsequently, detected pathogenic or suspected pathogenic variants
were verified by Sanger sequencing. The following tagged sequencing
primers were designed for MAP3K1: Forward primer,
5′-CAACAACAACAACAACAGAG-3′; reverse primer,
5′-TGAGGAGATGCAGAAGGT-3. Polymerase chain reaction (PCR) was
carried out in a 50-µl reaction system including 5 µl 10X PCR
buffer, 4 µl genomic DNA obtained as aforementioned, 4 µl dNTPs, 1
µl forward and reverse primers, and 0.3 µl Taq Hot Start (all from
Takara Bio, Inc.). The PCR conditions consisted of an initial
denaturation step (95°C for 5 min), followed by 40 cycles of
denaturation (95°C, 30 sec), annealing (65°C, 30 sec) and
elongation (72°C, 30 sec). Using an ABI 3730 system (Applied
Biosystems; Thermo Fisher Scientific, Inc.), amplicons were
sequenced. Sequence analysis was performed using the autoassembler
software Chromas 2.6.6 (Technelysium Pty, Ltd.).
Bioinformatics analysis
To confirm the conservation of amino acids at
mutated positions, Clustal W (UCD) software (https://www.ebi.ac.uk/Tools/msa/clustalo/) was applied
to compare 20 species. In addition, online software, such as
Mutation Taster (https://www.mutationtaster.org/), PolyPhen v.2
(http://genetics.bwh.harvard.edu/pph2/) and PROVEAN
(http://provean.jcvi.org/index.php),
was used to predict the potential pathogenic effects of the
variants. The Humvar model in PolyPhen v.2 is adapted to assess
variations associated with Mendelian genetic diseases and can
distinguish severely harmful variations from other human variants.
The HumDiv model is suitable for evaluating rare variants that may
be involved in complex diseases or in high-density regions of GWAS
studies. Therefore, this study is more suitable for Humvar
model.
Using I-TASSER software (https://zhanglab.ccmb.med.umich.edu/I-TASSER/),
modeling of wild type and mutant proteins was performed. PyMOL
Viewer 2.0 (http://pymol.sourceforge.net). was employed to observe
the effect of the variants on the protein.
Results
Clinical manifestation
Due to reduced fetal movement during pregnancy, the
proband of the present study was delivered by cesarean section at
29 weeks' gestation with a birth weight of 1.26 kg. The newborn
classification was low birth weight and small for gestational age.
After birth, the infant was found to have a short penis, low muscle
tension and no testicles. Growth and development lagged behind
those of their peers; however, the proband's intellectual
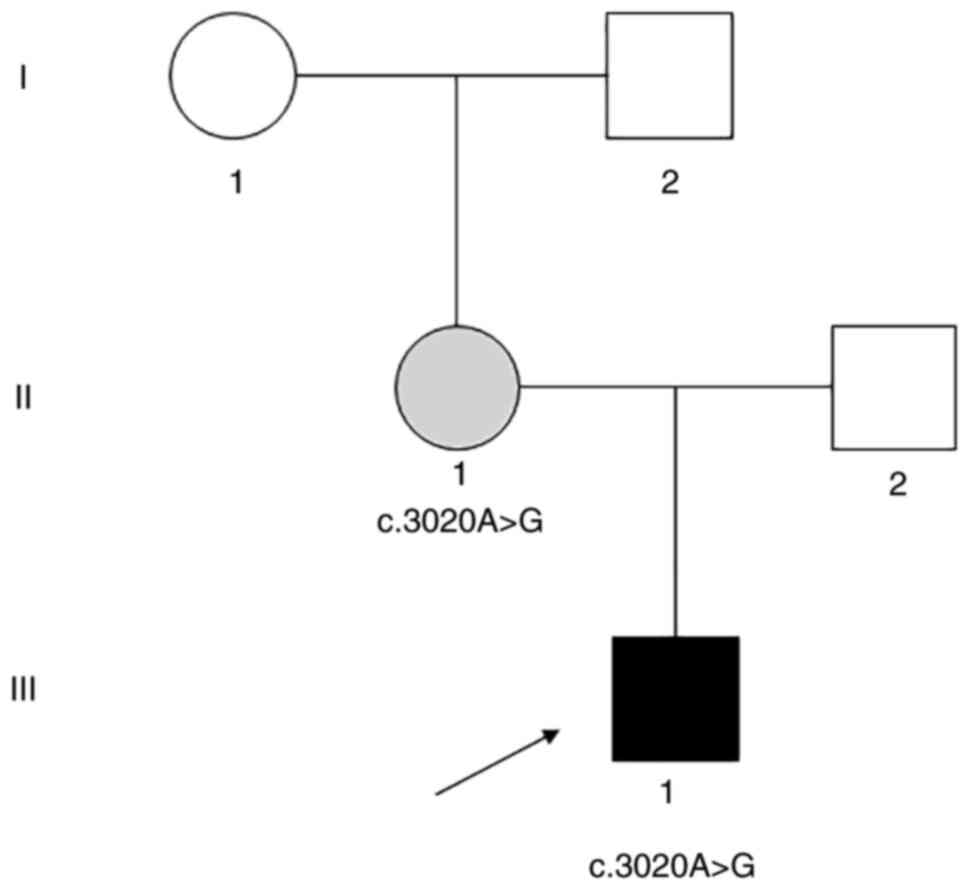
development was basically normal. The lineage of the patient is
shown in Fig. 1.
The patient was assessed at our hospital at the age
of 1 year. Physical examination showed a small penis, lack of
bilateral testicles, slightly reduced muscle tension in the limbs
and short stature. Their height was 65.7 cm (target height,
76.5±2.6 cm) and body weight was 8.9 kg (target weight, 10.05±1.05
kg); in addition, their parents' height and body weight were
relatively low (father: height, 176 cm and weight, 65 kg; mother:
height, 159 cm and weight, 54 kg). Bone age test showed an age of 6
months. According to an abdominal ultrasound, the patient presented
with bilateral scrotum emptiness, and no testicular tissue was
detected in groin and pelvic regions.
The karyotype analysis revealed 46, XY in 50
metaphases. The estradiol, inhibin B and insulin-like growth factor
1 levels were low for a child, whereas the luteinizing hormone
(LH), follicle-stimulating hormone (FSH) and cortisol levels were
high. Furthermore, testosterone, prolactin and adrenocorticotropic
hormone levels were normal for a child (Table I).
 | Table I.Hormone measurements of the
patient. |
Table I.
Hormone measurements of the
patient.
| Patient age | LH, mIU/l | FSH, mIU/l | T, ng/ml | E, pg/ml | AMH, ng/ml | ACTH, pg/ml | COR, nmol/l | PRL, ng/ml | IGF-1, ng/ml | Inhibin B,
pg/ml | TSH, mIU/ml | FT3, pmol/l | FT4, pmol/l |
|---|
| 1.0 year | 20.76 | 160.51 | 0.78 | 1.54 | 0.01 | 33.09 | 503.90 | 11.21 | <25.0 | <10.0 | 2.981 | 6.1 | 14.21 |
| 2.0 year | 35.68 | 141.59 | 0.11 | 1.68 | 1.20 | - | - | - | - | 10.0 | - | - | - |
GH stimulation test indicated that in response to
stimuli, a low level of GH was detected, and the presence of
partial GH deficiency was suggested. The gonadotropin-releasing
hormone excitation test showed that the patient had a normal
pituitary response (Table II).
Pituitary MR examination revealed that the upper edge of the
pituitary gland was raised.
| Table II.Results of GH stimulation test and
gonadotropin-releasing hormone excitation test of the patient. |
Table II.
Results of GH stimulation test and
gonadotropin-releasing hormone excitation test of the patient.
| Hormone | 0 min | 30 min | 60 min | 90 min |
|---|
| GH, ng/ml | 1.70 | 2.04 | 5.01 | 3.82 |
| FSH, mIU/l | 160.51 | >205.0 | >205.0 | >205.0 |
| LH, mIU/l | 20.76 | 105.52 | 137.03 | 140.46 |
Follow up
The patient underwent laparoscopy at the age of 2.0
years and no testes were detected in the abdominal cavity and
inguinal region. Their height was 80.2 cm (target height, 88.5±3.4
cm) and body weight was 11.40 kg (target weight, 12.54±1.23 kg).
Histopathological examination revealed there were oviduct,
epididymis and fibrous vascular tissue on both sides of the
abdomen, but no obvious convoluted tubule and follicular tissue.
Hormone measurements at 2.0 years indicated low estradiol AMH and
inhibin B-levels for a child, whereas the levels of LH and FSH were
high. Testosterone levels was decreased from year 1-year 2
(Table I).
The patient identified as male at the last follow-up
when they were 5 years of age. Their height was 110.2 cm (target
height, 111.3±4.4 cm) and body weight was 16.5 kg (target weight,
18.98±1.97 kg). Gynecological examination confirmed male external
sexual organs with a lack of testicles. The length of the penis was
1.5 cm.
The mental development of the patient was normal.
During the follow-up period, no medication was used, and
psychological or cognitive impairment, gender anxiety disorder and
gonadal tumors were not observed.
Mutation detection
The WES technique was applied to the genetic
analysis of the pedigree to further search for the disease-causing
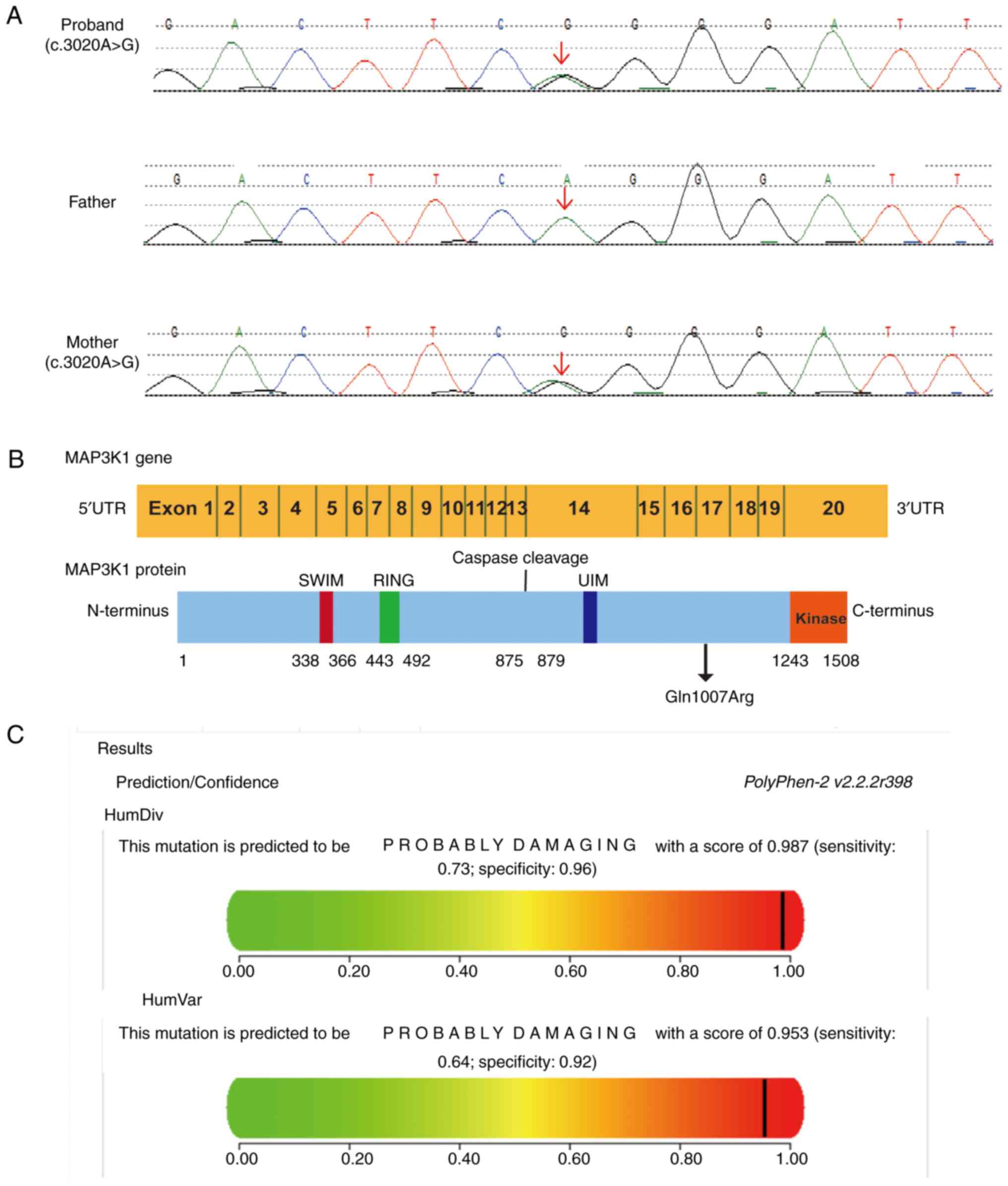
genes. Notably, a novel heterozygous MAP3K1 variant
(c.3020A>G) was identified in the patient and their mother,
which resulted in a change in the 1,007th amino acid of the encoded
protein from glutamine to arginase (Gln1007Arg; Fig. 2A). Since no substitution was
detected in TOPMED, GnomAD and ExAC, this variant was not
considered polymorphic. The C-terminus of MAP3K1 has a
serine/threonine kinase domain; upstream of this domain is a
conserved caspase-3 cleavage site and a ubiquitin interaction
motif. Two zinc finger structures, including one RING structure and
one SWIM region, are located at the N-terminus of MAP3K1 (11). The heterozygous variant was
located at position 1,007 (Gln1007Arg), upstream of the
serine/threonine kinase domain (Fig.
2B). PolyPhen v.2 predicted the hemizygous MAP3K1
variant to be potentially damaging with a score of 0.953 (Fig. 2C), and this variant was predicted
to be neutral by PROVEAN with a score of −0.557. Mutation Taster
showed that the pathogenic variant could influence protein
properties. According to ACMG criteria, the MAP3K1 variant
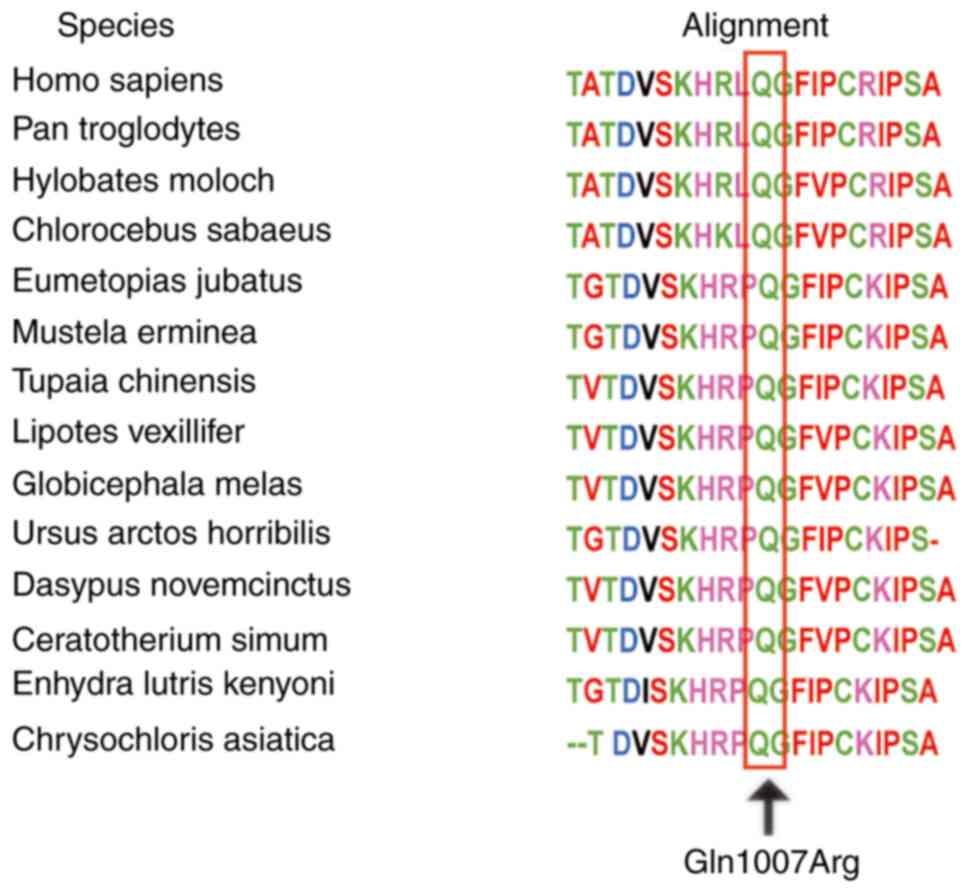
had ‘unknown significance’. Using the Clustal W tool, multiple
amino acid sequence alignments suggested that p.Gln1007 is
conserved across various species (Fig. 3). Currently, the 3D MAP3K1 protein
cannot be crystalized; therefore, structural analysis could not be
performed. In addition, there were no other mutations associated
with hypogonadism in the exome of the patient. Therefore, it was
hypothesized that this rare MAP3K1 mutation (c.3020A>G)
may be associated with the occurrence of 46, XY DSD. The presence
of allelic variants in the main genes associated with GH deficiency
were excluded.
Discussion
The present study identified a novel heterozygous
MAP3K1 variant (c.3020A>G, p.Gln1007Arg) in a patient
with 46, XY DSD. The GH levels of the patient were evaluated and a
partial GH deficiency was detected. Follow-up for 4 years indicated
that the patient had a good prognosis except for developmental
delay. The present findings enrich the MAP3K1 variant
spectrum and contribute to the study of genotype-phenotype
relationships in patients with 46, XY DSD.
The expression of the SRY gene (MIM 480000)
on the Y chromosome controls male sex determination, leading to the
development of undifferentiated gonads into testes (12,13). Sertoli cells in the testis produce
anti-Müllerian hormone, which causes the degradation of Müllerian
ducts, whereas Leydig cells secrete testosterone to promote
Wolffian duct differentiation into seminal vesicles, vas deferens
and epididymis. In the absence of the Y chromosome and SRY
expression, undifferentiated gonads develop into ovaries, Müllerian
ducts form the uterus, fallopian tubes and the upper third of the
vagina, and Wolffian ducts degenerate. This process occurs
approximately from the 4th week to the 12th week of pregnancy, and
is performed in a strictly timed and gene dose-dependent manner
(12). If there is a problem with
gonad development, it can lead to DSDs (14,15). Notably, 46, XY DSD may be caused
by functional mutations in genes (16,17) and a missense mutation of the
MAP3K1 gene is a common cause of 46, XY DSD (18). In the present study, a missense
mutation, c.3020A>G, was found in exon 14, which changed the
1,007th amino acid of the encoded protein from glutamine to
arginase; glutamine is an uncharged neutral amino acid in wild type
MAP3K1, which in this mutation is replaced by the positively
charged basic amino acid arginine. The secondary structure of the
protein may be affected by this change in charge. The conservation
of the p.Gln1007 residue and a high pathogenic probability of this
MAP3K1 mutation indicates the importance of the residue in
the structure and function of the protein. Similarly, Das et
al (19) identified a
missense mutation in exon 14 of the MAP3K1 gene
(c.2416G>A, p.Asp806Asn) in four patients with hypospadias with
gonadal abnormalities, which resulted in aspartic acid at position
806 of the amino acid sequence being replaced by asparagine;
computer analysis indicated this missense mutation was a pathogenic
mutation.
To date, numerous mutations have been identified in
the open reading frame or non-coding region of the MAP3K1
gene from the HGMD database; however, only 15 missense mutations of
the MAP3K1 gene have been reported to cause 46, XY DSD
(Table III). Pearlman et
al (20) identified three
missense mutations of MAP3K1 in a family from New Zealand,
which included five women with complete gonadal dysfunction
(c.1846G>A, p.Gly616Arg) and two patients with sporadic 46, XY
gonadal dysgenesis (c.566T>C, p.Leu189Pro and c.566T>G,
p.Leu189Arg). PolyPhen and SIFT revealed that p.Leu189Pro and
p.Leu189Arg mutations may have interference functions, whereas
p.Gly616Arg mutations did not; however, this mutation caused the
conservative neutral amino acid at this position to be replaced by
a basic amino acid. Further experiments revealed that the missense
mutations identified in the two sporadic cases changed the
phosphorylation of downstream targets p38 and ERK1/2, and enhanced
the binding of RHOA to the MAP3K1 complex; the findings of Loke
et al (21) also
identified a p.Pro153Leu variant in a patient with gonadal
dysgenesis. Baxter et al (16) reported on two cases that had the
p.Gly616Arg variant, of which one was a female with complete
gonadal dysgenesis and another was a male with ovotesticular DSD;
furthermore, a patient with complete gonadal dysgenesis had a de
novo p.Arg339Gln missense variant and a male patient with
complex ambiguous genitalia but without gonad hypoplasia had a
p.Pro257Leu missense variant. Eggers et al (22) inferred 10% prevalence of
MAP3K1 variants amongst patients with 46, XY DSD in a
smaller cohort and this could be up to 18% if the MAP3K1
phenotypic spectrum was expanded. In addition, two MAP3K1
variants (p.Met312Leu and p.Ala1443Val) were identified in multiple
patients with a variety of phenotypes, including complete or
partial gonadal dysgenesis, hypospadias and undervirilization.
Granados et al (23)
performed clinical evaluation, endocrine evaluation and genetic
analysis of six individuals with 46, XY DSD from four unrelated
families. Three of these patients exhibited complete gonadal
dysgenesis with MAP3K1 variants (c.1760T>A, p.Leu587His;
c.2291T>G, p.Leu764Arg; c.566T>A, p.Leu189Gln). In addition,
Xue et al (24) detected a
missense mutation (c. 2117T>G; p.Leu706Arg) in exon 12 of the
MAP3K1 gene in a patient with 46, XY DSD and a missense
mutation (c.2416G>A; p.Asp806Asn) was identified in exon 14 of
the MAP3K1 gene in patients with 46, XY DSD by Das et
al (19). Notably, Pearlman
et al (20) found only one
of these individuals in pedigrees from two families of interest
exhibiting sex-limited autosomal-dominant mendelian inheritance of
46, XY DSD had micropenis and cryptorchidism (III-13). The absence
of hypospadias was found in the proband of the present study, which
is different from complete hypogonadism. The missense mutation of
MAP3K1 is a common cause of 46, XY DSD. It was speculated
that the location of missense mutations may affect the function of
genes or encoded proteins, leading to differences in clinical
phenotypes of patients. Alternatively, interactions between this
gene and other genes may also be responsible for this phenomenon,
but further research is needed.
| Table III.Reported missense mutations of the
MAP3K1 gene in 46, XY DSD. |
Table III.
Reported missense mutations of the
MAP3K1 gene in 46, XY DSD.
| First author,
year | Coding change | Protein change | Zygosity | Exon | Phenotype | Functional
study | (Refs.) |
|---|
| Eggers, 2016 | G → C | p.Asp132His | Heterozygous | 1 | DSD | No | (22) |
| Loke, 2014 | C → T | p.Pro153Leu | Heterozygous | 1 | GD | Yes | (21) |
| Pearlman,
2010; | T → C | p.Leu189Pro | Heterozygous | 2 | CGD | Yes | (20) |
| Loke, 2014; |
|
|
|
|
|
| (21) |
| Granados, 2017 |
|
|
|
|
|
| (23) |
| Pearlman,
2010; | T → G | p.Leu189Arg | Heterozygous | 2 | CGD | Yes | (20) |
| Loke, 2014 |
|
|
|
|
|
| (21) |
| Granados, 2017 | T→ A | p.Leu189Gln | Heterozygous | 2 | CGD | No | (23) |
| Eggers, 2016 | A → G | p.Gln237Arg | Heterozygous | 3 | 46, XY DSD | No | (22) |
| Baxter, 2015 | C→ T | p.Pro257Leu | Heterozygous | 3 | DSD | No | (16) |
| Eggers, 2016 | A → T | p.Met312Leu | Heterozygous | 4 | 46, XY DSD | No | (22) |
| Baxter, 2015 | G → A | p.Arg339Gln | Heterozygous | 4 | CGD | No | (16) |
| Granados, 2017 | T → A | p.Leu587His | Heterozygous | 10 | CGD | No | (23) |
| Pearlman,
2010; | G → A | p.Gly616Arg | Heterozygous | 10 | CGD | No | (20) |
| Baxter, 2015 |
|
|
|
|
|
| (16) |
| Eggers, 2016 | T → C | p.Cys691Arg | Heterozygous | 11 | CGD | No | (22) |
| Xue, 2019 | T → G | p.Leu706Arg | Heterozygous | 12 | CGD | Yes | (24) |
| Granados, 2017 | T → G | p.Leu764Arg | Heterozygous | 13 | PGD | No | (23) |
| Das, 2013 | G → A | p.Asp806Asn | Heterozygous | 14 | 46, XY DSD | No | (19) |
| The present
study | A → G | p.Gln1007Arg | Heterozygous | 14 | 46, XY DSD | No | - |
| Eggers, 2016 | C → T | p.Ala1443Val | Heterozygous | 19 | 46, XY DSD | No | (22) |
It is worth noting that in addition to 46, XY DSD,
the present proband exhibited partial GH deficiency. GH deficiency
is the most common type of restricted growth and can present as a
simple GH deficiency or with multiple pituitary hormone
deficiencies. Previous studies have reported that genetic mutations
are an important cause of idiopathic GH deficiency. Currently known
pathogenic genes of GH deficiency include GH gene,
GH-releasing hormone receptor gene, and POU1F1 gene
(25,26). To date, no patients with 46, XY
DSD have been reported to have GH deficiency and no research has
identified a causal relationship between MAP3K1 and GH
deficiency; this needs to be further explored in functional
tests.
In the gene network responsible for gonadal
development, MAP3K1 is an important component. MAP3K1
is involved in the regulation of the transcription of so many
important genes, and serves a key role in cell proliferation,
differentiation and apoptosis (27,28). Studies have revealed that variants
in the MAP3K1 gene can lead to changes in cofactor binding
and increase the phosphorylation of targets in the downstream MAPK
pathway, including ERK1/2 and p38, ultimately leading to the
occurrence of 46, XY DSD. Increased phosphorylation of ERK1/2 may
lead to decreased expression of SOX9, whereas increased
phosphorylation of p38 may increase the expression of
CTNNB1, which are important signaling molecules in
testicular or ovarian-promoting pathways (19,21,29–31). Chamberlin et al (29) showed that mutations in the
MAP3K1 gene have different effects on protein binding,
depending on the domain in which the mutation occurs. Generally,
the net effect of increased binding of the pro-kinase RHOA and
MAP3K4 proteins, and decreased binding of the inhibitory RAC1
protein results in the observed gain of function with MAP3K1
mutations. GH signaling is necessary for GH to function. Firstly,
GH is combined with GH receptor (GHR) to establish the post-GHR
process, and the most important GHR-mediated signaling pathways
include GHR/JAK2/SHC/MAPK, GHR/JAK2/STATs and GH/IRS/PI3K/Akt
pathways (32). Notably, Jin
et al (33) demonstrated
that JAK2 was required for ERK signaling in response to GH in
preadipocytes and hepatoma cells. Therefore, it is reasonable to
speculate that the MAP3K1 variant identified in the patient
assessed in the present study may cause changes in GHR-mediated
signaling pathways and ultimately lead to the occurrence of partial
GH deficiency.
The previously reported genetic pattern of
pathogenic MAP3K1 variants is autosomal dominant and
sex-limited (20,23,24). In two large families and two of 11
sporadic cases with 46, XY DSD, MAP3K1 variants were first
reported with an autosomal dominant, sex-limited pattern of
transmission (20). Phenotypic
manifestations of the large family reported a range from
hypospadias and cryptorchidism to complete hypogonadism; however,
there were eight and four unaffected female carriers, respectively.
Granados et al (23)
reported that six individuals from four unrelated families with
pathogenic MAP3K1 variants had both partial and complete
gonadal dysgenesis. In addition, 46, XX females carrying
MAP3K1 variants were not affected. Furthermore, Xue et
al (24) speculated that the
unaffected mother of a patient with 46, XY DSD may carry the same
MAP3K1 mutation; however, the mother was not genetically
tested. The present findings are consistent with the aforementioned
studies; the mother of the proband had the same variant as the
proband, but remained phenotypically healthy. These findings
indicated that the genetic pattern of the novel pathogenic
MAP3K1 variant (c.3020A>G, p.Gln1007Arg) may be based on
autosomal dominant and sex-restricted patterns.
The present study has some limitations. The patient
was diagnosed with 46, XY DSD based on clinical data, imaging
characteristics and genetic testing results; nevertheless, the
association between the phenotype and genotype of 46, XY DSD is not
known, and more patients need to be studied to supply more
comprehensive information. In addition, more functional tests are
needed to verify the findings of the present study.
In conclusion, the present study detected a novel
pathogenic MAP3K1 variant (c.3020A>G) in a patient with
46, XY DSD and partial GH deficiency. The present study expands the
mutation spectrum of MAP3K1, and will be conducive to
improved genetic diagnosis and counseling in the future. WES may
find more genetic variants that expound the heterogeneity
associated with MAP3K1 variants.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the National
Natural Science Foundation (grant nos. 81670720 and 81974124),
special funds for Taishan Scholar Project (grant no. tsqn20161071)
and the Shandong Key R&D Program (grant no. 2017GSF18129).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YC, CX and NC designed the present study. YC, JY and
XZ performed the present study and analyzed the data. YC wrote the
main manuscript. CX and NC confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The study protocol was in line with the Declaration
of Helsinki (as revised in Brazil 2013). The Ethics Committee of
Shandong Provincial Hospital affiliated to Shandong University
approved this study (approval no. 2019-147), and written informed
consent was obtained from the parents of the patient.
Patient consent for publication
Written informed consent was obtained from the
parents of the patient.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DSD
|
disorder of sex development
|
|
SRY
|
sex-determining region Y
|
|
SOX9
|
SRY-related HMG-box gene 9
|
|
MAP3K1
|
mitogen-activated protein kinase
kinase kinase 1
|
|
MR
|
magnetic resonance
|
|
GnomAD
|
Genome Aggregation Database
|
|
TOPMED
|
Trans-Omics for Precision Medicine
|
|
ExAC
|
Exome Aggregation Consortium
|
|
HGMD
|
Human Gene Mutation Database
|
|
PCR
|
polymerase chain reaction
|
|
ACMG
|
American College of Medical Genetics
and Genomics
|
|
IGF-1
|
insulin-like growth factor 1
|
|
LH
|
luteinizing hormone
|
|
FSH
|
follicle-stimulating hormone
|
|
GH
|
growth hormone
|
|
AMH
|
anti-Müllerian hormone
|
References
|
1
|
Allen L: Disorders of sexual development.
Obstet Gynecol Clin North Am. 36:25–45. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Domenice S, Arnhold IJ, Costa EM and
Mendonca BB: 46,XY Disorders of sexual development. Endotext.
Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A,
Hershman JM, Kaltsas G, Koch C, Kopp P, et al: MDText.com, Inc.;
South Dartmouth, MA: 2000
|
|
3
|
Hull CL and Fausto-Sterling A: How
sexually dimorphic are we? Review and synthesis. Am J Hum Biol.
15:112–116. 2003. View Article : Google Scholar
|
|
4
|
Wertheim-Tysarowska K, Gos M,
Sykut-Cegielska J and Bal J: Genetic analysis in inherited
metabolic disorders-from diagnosis to treatment. Own experience,
current state of knowledge and perspectives. Dev Period Med.
19:413–431. 2015.PubMed/NCBI
|
|
5
|
Berglund A, Johannsen TH, Stochholm K,
Viuff MH, Fedder J, Main KM and Gravholt CH: Incidence, prevalence,
diagnostic delay, and clinical presentation of female 46,XY
disorders of sex development. J Clin Endocrinol Metab.
101:4532–4540. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mazen I, Abdel-Hamid M, Mekkawy M,
Bignon-Topalovic J, Boudjenah R, El Gammal M, Essawi M, Bashamboo A
and McElreavey K: Identification of NR5A1 mutations and possible
digenic inheritance in 46,XY gonadal dysgenesis. Sex Dev.
10:147–151. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ostrer H: Disorders of sex development
(DSDs): An update. J Clin Endocrinol Metab. 99:1503–1509. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zheng Q, Ye J, Wu H, Yu Q and Cao J:
Association between mitogen-activated protein kinase kinase kinase
1 polymorphisms and breast cancer susceptibility: A meta-analysis
of 20 case-control studies. PLoS One. 9:e907712014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bashamboo A and McElreavey K: Human
sex-determination and disorders of sex-development (DSD). Semin
Cell Dev Biol. 45:77–83. 2015. View Article : Google Scholar
|
|
10
|
Li Q and Wang K: InterVar: Clinical
interpretation of genetic variants by the 2015 ACMG-AMP guidelines.
Am J Hum Genet. 100:267–280. 2017. View Article : Google Scholar
|
|
11
|
Suddason T and Gallagher E: A RING to rule
them all? Insights into the Map3k1 PHD motif provide a new
mechanistic understanding into the diverse roles of Map3k1. Cell
Death Differ. 22:540–548. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ono M and Harley VR: Disorders of sex
development: New genes, new concepts. Nat Rev Endocrinol. 9:79–91.
2013. View Article : Google Scholar
|
|
13
|
Jost A, Vigier B, Prépin J and Perchellet
JP: Studies on sex differentiation in mammals. Recent Prog Horm
Res. 29:1–41. 1973.PubMed/NCBI
|
|
14
|
McCann-Crosby B, Mansouri R, Dietrich JE,
McCullough LB, Sutton VR, Austin EG, Schlomer B, Roth DR, Karaviti
L, Gunn S, et al: State of the art review in gonadal dysgenesis:
Challenges in diagnosis and management. Int J Pediatr Endocrinol.
2014:42014. View Article : Google Scholar
|
|
15
|
Wolffenbuttel KP, Hersmus R, Stoop H,
Biermann K, Hoebeke P, Cools M and Looijenga LH: Gonadal dysgenesis
in disorders of sex development: Diagnosis and surgical management.
J Pediatr Urol. 12:411–416. 2016. View Article : Google Scholar
|
|
16
|
Baxter RM, Arboleda VA, Lee H, Barseghyan
H, Adam MP, Fechner PY, Bargman R, Keegan C, Travers S, Schelley S,
et al: Exome sequencing for the diagnosis of 46,XY disorders of sex
development. J Clin Endocrinol Metab. 100:E333–E344. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Foster JW, Dominguez-Steglich MA, Guioli
S, Kwok C, Weller PA, Stevanović M, Weissenbach J, Mansour S, Young
ID, Goodfellow PN, et al: Campomelic dysplasia and autosomal sex
reversal caused by mutations in an SRY-related gene. Nature.
372:525–530. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bardoni B, Zanaria E, Guioli S, Floridia
G, Worley KC, Tonini G, Ferrante E, Chiumello G, McCabe ER,
Fraccaro M, et al: A dosage sensitive locus at chromosome Xp21 is
involved in male to female sex reversal. Nat Genet. 7:497–501.
1994. View Article : Google Scholar
|
|
19
|
Das DK, Rahate SG, Mehta BP, Gawde HM and
Tamhankar PM: Mutation analysis of mitogen activated protein kinase
1 gene in Indian cases of 46,XY disorder of sex development. Indian
J Hum Genet. 19:437–442. 2013. View Article : Google Scholar
|
|
20
|
Pearlman A, Loke J, Le Caignec C, White S,
Chin L, Friedman A, Warr N, Willan J, Brauer D, Farmer C, et al:
Mutations in MAP3K1 cause 46,XY disorders of sex development and
implicate a common signal transduction pathway in human testis
determination. Am J Hum Genet. 87:898–904. 2010. View Article : Google Scholar
|
|
21
|
Loke J, Pearlman A, Radi O, Zuffardi O,
Giussani U, Pallotta R, Camerino G and Ostrer H: Mutations in
MAP3K1 tilt the balance from SOX9/FGF9 to WNT/β-catenin signaling.
Hum Mol Genet. 23:1073–1083. 2014. View Article : Google Scholar
|
|
22
|
Eggers S, Sadedin S, van den Bergen JA,
Robevska G, Ohnesorg T, Hewitt J, Lambeth L, Bouty A, Knarston IM,
Tan TY, et al: Disorders of sex development: insights from targeted
gene sequencing of a large international patient cohort. Genome
Biol. 17:2432016. View Article : Google Scholar
|
|
23
|
Granados A, Alaniz VI, Mohnach L,
Barseghyan H, Vilain E, Ostrer H, Quint EH, Chen M and Keegan CE:
MAP3K1-related gonadal dysgenesis: Six new cases and review of the
literature. Am J Med Genet C Semin Med Genet. 175:253–259. 2017.
View Article : Google Scholar
|
|
24
|
Xue M, Wang X, Li C, Zhao M, He F and Li
X: Novel pathogenic mutations in disorders of sex development
associated genes cause 46,XY complete gonadal dysgenesis. Gene.
718:1440722019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dattani MT: Growth hormone deficiency and
combined pituitary hormone deficiency: Does the genotype matter?
Clin Endocrinol (Oxf). 63:121–130. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mullis PE: Genetics of isolated growth
hormone deficiency. J Clin Res Pediatr Endocrinol. 2:52–62. 2010.
View Article : Google Scholar
|
|
27
|
Warr N, Bogani D, Siggers P, Brixey R,
Tateossian H, Dopplapudi A, Wells S, Cheeseman M, Xia Y, Ostrer H
and Greenfield A: Minor abnormalities of testis development in mice
lacking the gene encoding the MAPK signalling component, MAP3K1.
PLoS One. 6:e195722011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Charlaftis N, Suddason T, Wu X, Anwar S,
Karin M and Gallagher E: The MEKK1 PHD ubiquitinates TAB1 to
activate MAPKs in response to cytokines. EMBO J. 33:2581–2596.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chamberlin A, Huether R, Machado AZ,
Groden M, Liu HM, Upadhyay K, O V, Gomes NL, Lerario AM, Nishi MY,
et al: Mutations in MAP3K1 that cause 46,XY disorders of sex
development disrupt distinct structural domains in the protein. Hum
Mol Genet. 28:1620–1628. 2019. View Article : Google Scholar
|
|
30
|
Murakami S, Kan M, McKeehan WL and de
Crombrugghe B: Up-regulation of the chondrogenic Sox9 gene by
fibroblast growth factors is mediated by the mitogen-activated
protein kinase pathway. Proc Natl Acad Sci USA. 97:1113–1118. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Upadhyay K, Loke J, O V, Taragin B and
Ostrer H: Biallelic mutations in FLNB cause a skeletal dysplasia
with 46,XY gonadal dysgenesis by activating β-catenin. Clin Genet.
93:412–416. 2018. View Article : Google Scholar
|
|
32
|
Xu J, Keeton AB, Franklin JL, Li X,
Venable DY, Frank SJ and Messina JL: Insulin enhances growth
hormone induction of the MEK/ERK signaling pathway. J Biol Chem.
281:982–992. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jin H, Lanning NJ and Carter-Su C: JAK2,
but not Src family kinases, is required for STAT, ERK, and Akt
signaling in response to growth hormone in preadipocytes and
hepatoma cells. Mol Endocrinol. 22:1825–1841. 2008. View Article : Google Scholar
|