Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of
the most malignant tumors of the digestive system. Due to its deep
anatomical location and occult clinical manifestations, there are
no effective methods for early diagnosis, resulting in a high
mortality rate (1). Despite
advances in the diagnosis and treatment of pancreatic cancer, with
a 5-year survival rate of 9%, PDAC is predicted to become the
second leading cause of cancer-related mortality worldwide by 2030
(2).
KRAS mutations are considered to initiate PDAC and
the frequency of KRAS mutations in PDAC ranges from 90–95%
(3). The dominance of KRAS
mutations suggests that targeted therapy against the Ras signaling
network may be an effective treatment modality for PDAC. To date,
several targeted therapies for PDAC have been approved (4). The first approved was EGFR inhibitor
erlotinib, which in combination with gemcitabine had a survival
benefit compared to gemcitabine alone, but was effectively
abandoned by the community after negative data emerged on
EGFR-targeted therapy for KRAS mutant colorectal cancer (5). Then, the TRK kinase inhibitors
larotrectinib and entrectinib were approved for solid tumors
containing the NTRK-fusion gene. However, the NTRK-fusion gene
occurs in only 0.5% of PDAC and the majority of PDAC without the
NTRK-fusion gene has not been systematically evaluated (6). Finally, the PARP inhibitor olaparib
can extend progression free survival but not overall survival in
patients of late-stage PDAC with germline mutations in BRCA1 or
BRCA2 (~7.5%) (7). In conclusion,
their clinical activity has been disappointing so far. Therefore,
there is an urgent need to identify new therapeutic modalities for
pancreatic cancer.
It is well known that the vast majority of mutations
in Ras are missense mutations in three hotspot residues, G12, G13
and Q61. The order of frequency observed at the G12 is G12D, G12V,
G12C, G12A, G12S and G12R and G12C mutation prevalent in lung
cancer and G12D being the most common in PDAC. In fact, KRAS G12D
(KRASG12D) is one of the most important tumor
therapeutic targets (8).
A previous study demonstrated that wild-type KRAS
(KRASWT) has a survival advantage in PDAC and patients
with KRASWT have a longer overall survival time
(9). Patients with mutated KRAS
have been shown to have worse survival and a shorter overall
survival following gemcitabine-based first-line chemotherapy,
regardless of age, sex, tumor stage, tumor morphology, or
chemotherapy regimen. KRASWT can antagonize the effects
of mutated KRASG12D, resulting in inefficient cellular
transformation (10). In
addition, the increased dose of mutated KRASG12D is
accompanied by the deletion of KRASWT in PDAC (11). This suggests that
KRASWT may exert a potential tumor suppressive effect
through certain pathways, but the mechanism has not yet been
elucidated.
In view of the characteristics of KRAS wild-type and
mutant genes in cancer, the present study overexpressed these two
genes. It focused on the differences in biological behavior between
KRASWT and KRASG12D and explored the pathway
and mechanism in pancreatic cancer and provides theoretical basis
for the study of the KRAS gene.
Materials and methods
Cells and lentiviruses
A PANC-1 human pancreatic ductal adenocarcinoma cell
line was provided by the Sichuan Institute of Medical Imaging, the
lentivirus was purchased from Shanghai GeneChem Co., Ltd., and
KRASWT and KRASG12D cells were constructed by
the authors.
Lentiviral infection
Lentiviruses expressing KRASWT and
KRASG12D were generated and purchased from Shanghai
GeneChem Co., Ltd. Lentiviruses were infected into cells at a
multiplicity of infection (MOI) of 200 for KRASG12D and
50 for KRASWT. Briefly, cell suspensions of
3–5×104/ml were prepared in 96-well plates (100 µl per
well) and culture was performed for 18–24 h. According to cell MOI
and virus titer, KRASWT and
KRASG12D-overexpressing viruses were added to the wells.
Culture was performed at 37°C for 12–16 h, after which the medium
was replaced with conventional medium. Additional culture was
performed for a further 3–4 days and the infection efficiency was
observed in >10 fields of view under a fluorescence microscope
(magnification, ×20; Leica DMIL LED; Leica Microsystems, Inc.). The
medium was then replaced with medium containing 2 µg/ml purinomycin
for further culture (37°C, 5% CO2) for over a week and
follow-up experiments were carried out.
Cell culture
Cells were cultured in DMEM (Hyclone; Cytiva)
containing 10% fetal bovine serum(Gibco; Thermo Fisher Scientific,
Inc.), 100 U/ml penicillin, 100 µg/ml streptomycin in a humidified
incubator at 37.5°C with 5% CO2. Trypsin (0.25%) was
used for digestion and subculture at a rate of 1:3.
KRASWT and KRASG12D cells were cultured and
subcultured with DMEM containing 2 µg/ml purinomycin, which was
replaced with conventional DMEM during the subsequent
experiments.
Reverse transcription-quantitative
(RT-q) PCR
The expression of the KRAS gene in transfected
KRASWT and KRASG12D cells was detected using RT-qPCR. When cell
density was ~80%, total RNA was extracted by TRIzol®
(cat. no. 15596026; Invitrogen; Thermo Fisher Scientific, Inc.),
cDNA was synthesized (RevertAid First Strand cDNA Synthesis Kit;
cat. no. K1622; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol and qPCR (Power Up™ SYBR™ Green; cat. no.
A25742; Applied Biosystems; Thermo Fisher Scientific, Inc.) was
performed. All steps were carried out according to the
manufacturers' protocols. qPCR was conducted using the Light Cycler
96 (Roche Diagnostics) under the following conditions: 2 min at
50°C, 2 min at 95°C, followed by 40 cycles at 95°C for 15 sec, 57°C
for 15 sec and 72°C for 1 min. Fold change in expression was
calculated using the standard 2-ΔΔCq formula (12). GAPDH was used as an internal
loading control and the experiment was repeated three times. The
primer sequences were as follows, GAPDH:
5′-ACTAGGCGCTCACTGTTCTCT-3′ forward and 5′-GGAATTTGCCATGGGTGGAA-3′
reverse; KRASWT: 5′-GCCTGCTGAAAATGACTGA-3′ forward and
5′-CTCCTCTTGACCTGCTGTG-3′ reverse; KRASG12D:
5′-ACACAAAACAGGCTCAGGA-3′ forward and 5′-GTCGGATCTCTCTCACCAA-3′
reverse.
CCK-8 assay
Cell suspensions of 2×104/ml were
prepared in 96-well plates (100 µl per well) and five repeats in
each group were detected at 24, 48, 72, 96 and 120 h, respectively.
CCK-8 reagent (10 µl) was added to each well and incubated at 37°C
for 1 h. Absorbance was then determined at 450 nm by SpectraMax
Paradigm microplate reader (Molecular Devices. Inc.).
Colony formation assay
A total of 200 cells/well were inoculated in a
6-well plate and cultured at 37°C with 5% CO2 for 2
weeks. When there were visible colonies (>50 cells), cells were
washed twice with PBS, fixed with 4% paraformaldehyde for 15 min at
room temperature and stained with 0.1% crystal violet for 15 min at
room temperature. The dying solution was washed with clear water
and dried naturally. The colonies were scanned and their number was
counted.
Wound healing assay
Cell suspensions of 1×105/well were
prepared in a 6-well plate and cultured in a medium containing 10%
FBS. The cells were scratched when the monolayer fusion was ~90%.
Horizontal lines were drawn at the 6-well plate, cells were washed
with PBS and then a position selected where the scratches were
clear, followed by further culture with serum-free medium (37°C, 5%
CO2). Images of the same positions were captured at 0,
24, 48, 72, 96 and 120 h respectively.
Transwell assay
Cell suspensions of 2×105/ml were
prepared in serum-free medium. A total of 500 µl medium with 10%
FBS was added to the lower Transwell chamber and 100 µl cell
suspension was added to the upper chamber, followed by culture for
48 h (37°C, 5% CO2). The Transwell chamber was cleaned
with PBS, fixed with 4% paraformaldehyde for 15 min at room
temperature and stained with 0.1% crystal violet for 15 min at room
temperature.
Tumor formation in nude mice
A total of 30 male nude mice (BALB-/c-nu, specific
pathogen-free; Beijing Hufukang Biotechnology Co., Ltd.) aged 4–6
weeks (16–18 g) were randomly divided into three groups, termed
PANC-1, KRASWT and KRASG12D and raised under
the same conditions (room temperature ~26–28°C; relative humidity
~40–60% with ventilation at ~10–15 times/h and a 10/14 h light/dark
cycle). Cell suspensions of 3–5×106/ml were prepared in
each group, a total of 100 µl of cell suspension was collected and
inoculated into the underarm skin of nude mice. A subcutaneous mass
was observed ~1 week after inoculation, the long (a) and short
diameter (b) of the tumor were measured (every other day, a total
of 12 times) and the tumor volume was calculated according to the
formula ab2/2. Following sacrifice by cervical
dislocation (mortality ascertained by the observation of
respiratory, heartbeat, pupil and nervous reflex), the tumors were
removed and stored in liquid nitrogen. The present study was
approved by the Ethics Committee of North Sichuan Medical College
(approval no. 2022035) and all procedures were carried out in
accordance with relevant guidelines and regulations.
Western blotting
Cells were lysed with lysate (1 ml protein lysate
mixed with 10 µl protease inhibitors) and the lysates were
centrifuged at 1,3500 × g at 4°C for 10 min. The protein
concentration was detected using a BCA protein kit, according to
the manufacturer's instructions. Protein lysates (30 g) were
separated via 10% SDS-PAGE and transferred to PVDF membranes, which
were blocked with 5% skimmed milk for 1 h at room temperature. The
membranes were subsequently incubated overnight at 4°C with the
following primary antibodies: Anti-e-cadherin (1:5,000; cat. no.
60335-1; ProteinTech Group, Inc.), anti-α-E-catenin (1:3,000; cat.
no. 66221-1; ProteinTech Group, Inc.), anti-MMP-9 (1:1,000; cat.
no. 13667; CST), anti-MMP-3 (1:1,000; cat. no. 14351; CST),
anti-STAT3 (1:1,000; cat. no. 22785, ZenBio) and
anti-phosphorylated (p-) STAT3 (1:2,000; cat. no. 9145; CST).
Following the primary antibody incubation, the membranes were
washed three times with PBS for 15 min and incubated with
HRP-conjugated goat anti-rabbit (1:5,000; cat. no. BA1039; Wuhan
Boster Biological Technology, Ltd.) or anti-mouse (1:5,000; cat.
no. BA1038; Wuhan Boster Biological Technology, Ltd.) secondary
antibodies for 2 h at room temperature, then washed with PBS 3
times for 15 min. Protein bands were visualized with an ECL
development kit (MilliporeSigma) using an enhanced
chemiluminescence detection system (FUSION Fx; Vilber Lourmat).
Immunohistochemistry
Tumor tissues were fixed in 10% paraformaldehyde for
>12 h at room temperature, were paraffin-embedded and sectioned
(3–5 µm), and the sections were then placed into xylene for
dewaxing. A descending alcohol series was added for dehydration.
Sections were placed in citrate buffer (pH 6.0) for antigen
retrieval (95°C for 15 min) and the endogenous peroxidase was
blocked using an endogenous peroxidase blocker (cat. no. SP-9000;
OriGene Technologies, Inc.) at 25°C for 10 min. Sections were
blocked with goat serum (cat. no. SP-9000; OriGene Technologies,
Inc.) at 25°C for 10 min and then incubated with a primary antibody
(α-E-catenin; 1:500; cat. no. 66221-1; ProteinTech Group, Inc.;
e-cadherin; 1:2,000; cat. no. 60335-1; ProteinTech Group, Inc.;
MMP-9; 1:300, cat. no. 13667; CST) for 1 h at 37°C, washed with PBS
three times and incubated with goat anti-rabbit (1:5,000; cat. no.
BA1039; Wuhan Boster Biological Technology, Ltd.) or anti-mouse
(1:5,000; cat. no. BA1038; Wuhan Boster Biological Technology,
Ltd.) secondary antibodies at 25°C for 15 min. Subsequently,
sections were incubated with 3,3′-diaminobenzidine substrate for
5–10 min at room temperature, stained for 30 sec with hematoxylin
and differentiated for 2 sec with 1% hydrochloric acid alcohol at
room temperature followed by gradient alcohol series for
dehydration and xylene clearing before being sealed with neutral
gum and >10 fields of view were observed under a light
microscope (magnification, ×5; ECLIPSE 80i; Nikon Corp.).
Statistical analysis
All statistical analysis was performed by SPSS 19.0
(IBM Corp.) and GraphPad Prism 7 (GraphPad Software, Inc.) and the
data are expressed as the mean ± standard deviation. One-way
analysis of variance was used for multiple group comparisons and
the post-hoc test was performed by the LSD method. P<0.05 was
considered to indicate a statistically significant difference.
Results
KRASWT gene inhibits the
proliferation of continuous tumor-cell line (PANC-1)
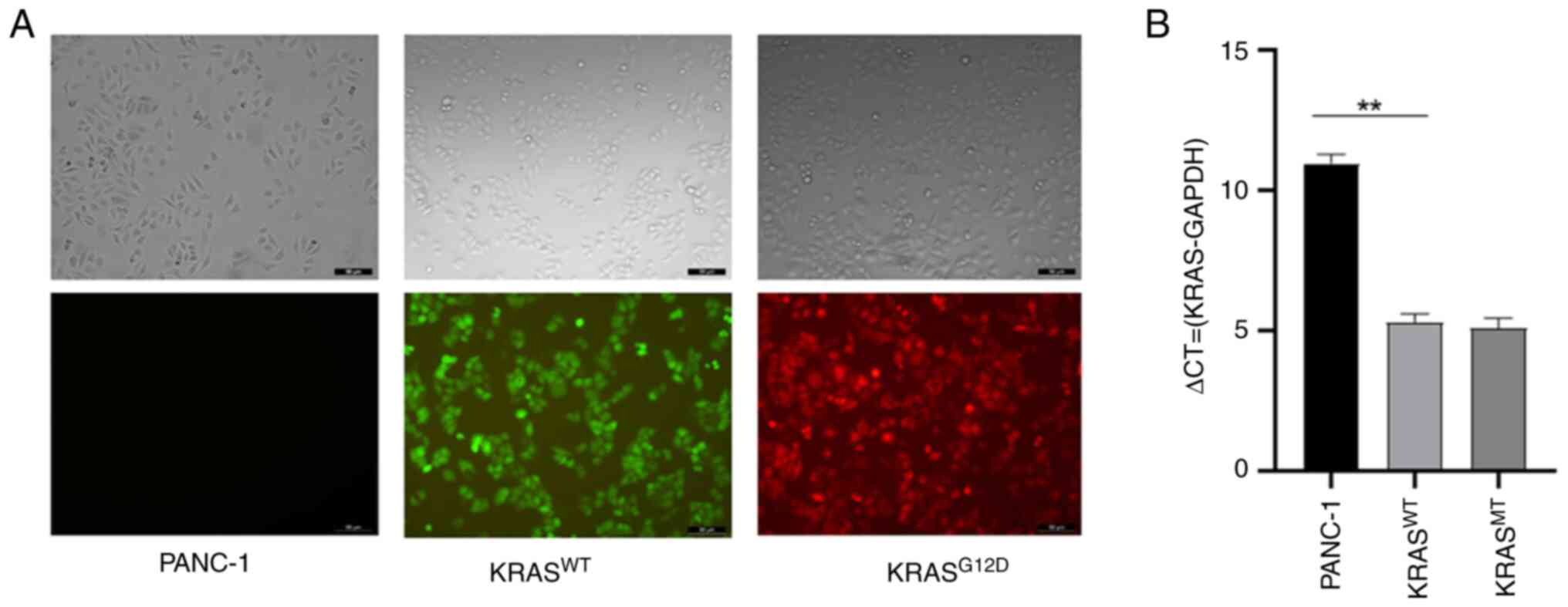
The transfection effect is shown in Fig. 1A and the verification of
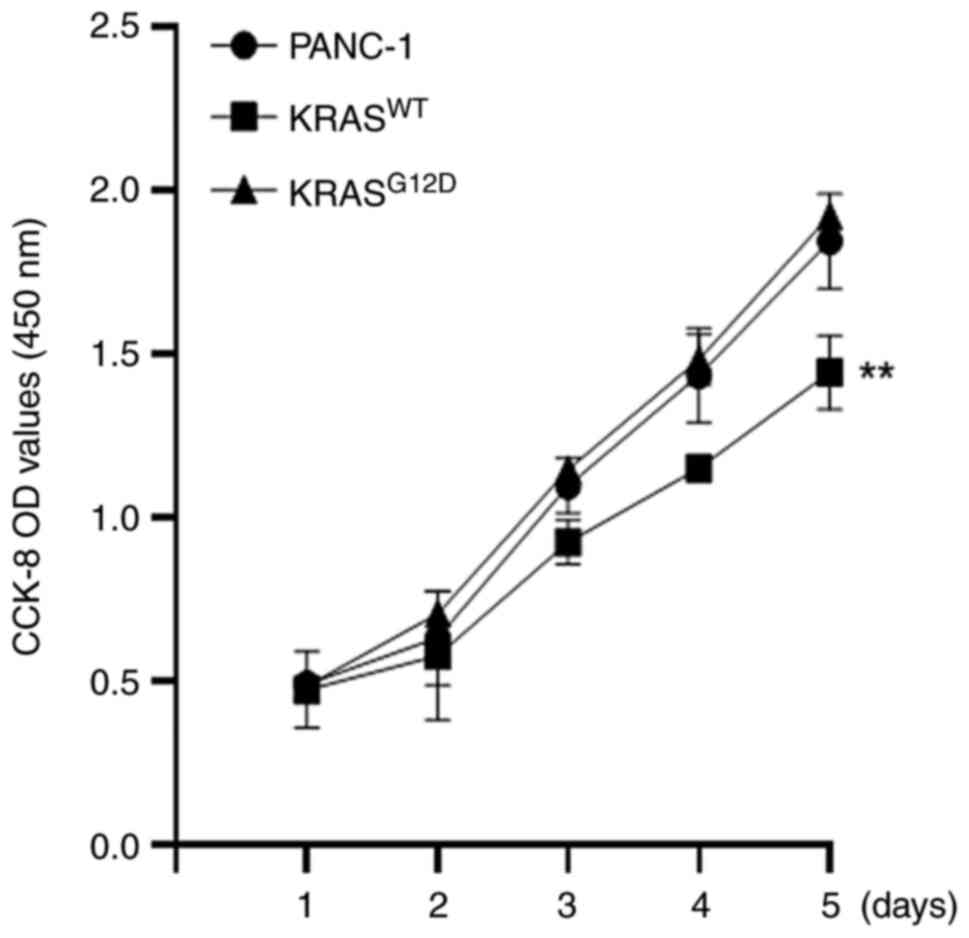
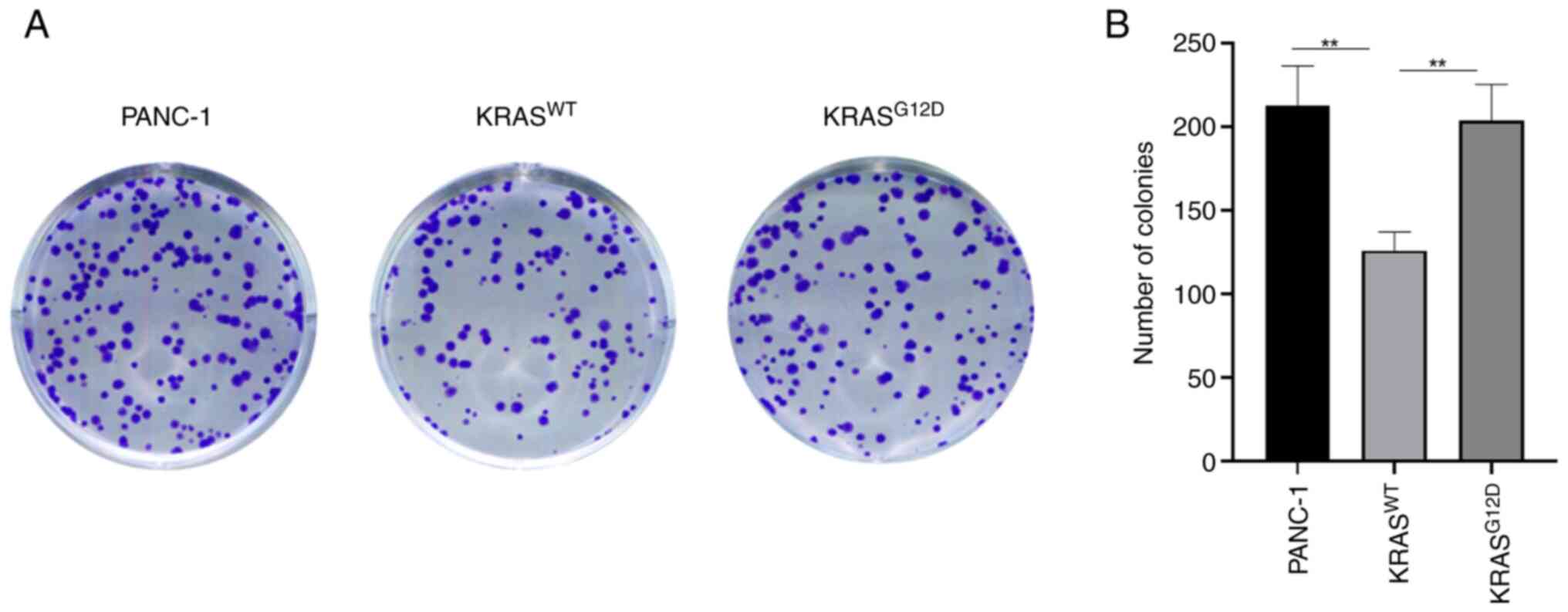
transfection effect was performed using RT-qPCR assay (Fig. 1B). CCK-8 and clone formation
assays were used to detect the proliferation ability of pancreatic
cancer following the overexpression of the KRAS gene. The CCK-8
assay results showed that, compared with the PANC-1, the
proliferation of KRASWT was reduced and the
proliferation of KRASG12D was not significantly changed
(Fig. 2), which was consistent
with the results of the colony formation assay (Fig. 3). These results indicated that the
KRASWT gene could inhibit the proliferation of
pancreatic cancer cells.
KRASWT gene inhibits the
migration and invasion of PANC-1
Next, the effect of the KRAS gene on the invasion
and migration of pancreatic cancer cells was investigated.
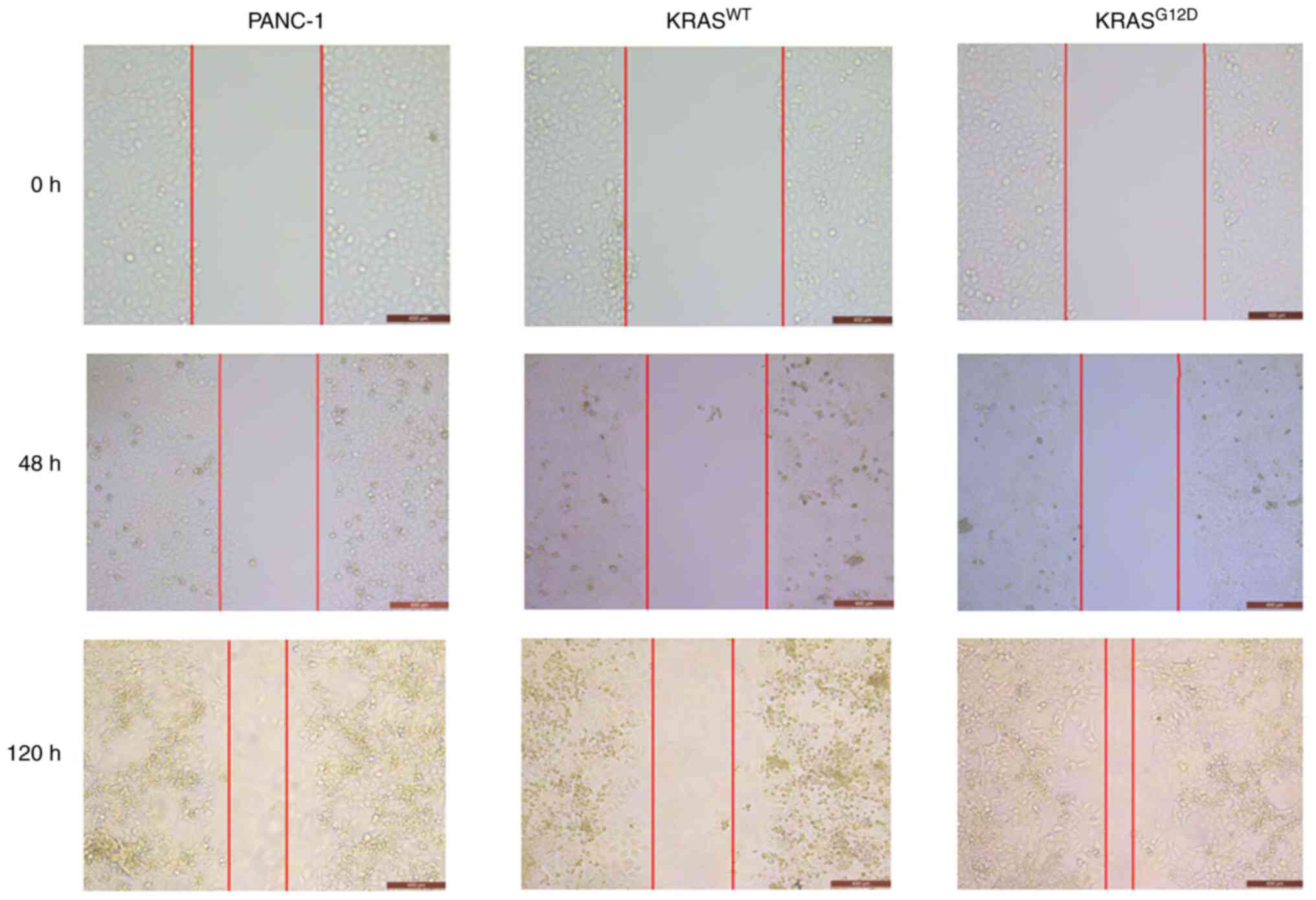
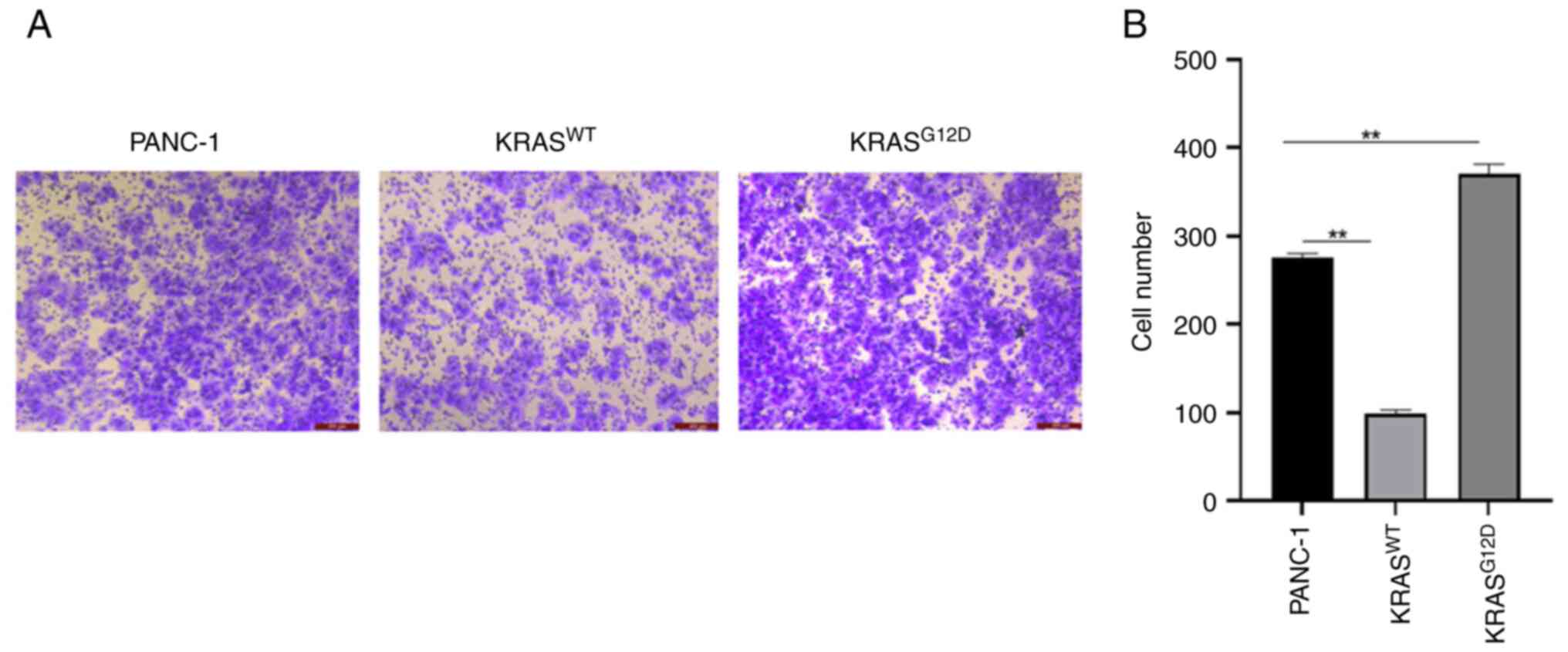
Transwell and wound healing assays were performed to detect cell
invasion and migration capacity. As shown in Fig. 4, the migration of
KRASG12D cells was enhanced and that of
KRASWT cells was weakened, as compared with those of
PANC-1 cells after 48 h and the difference was more pronounced
after 120 h. Similarly, a Transwell assay revealed an enhanced
invasion of KRASG12D cells and decreased invasion of
KRASWT cells (Fig. 5).
The results suggested that the KRASWT gene can inhibit
the migration and invasion of pancreatic cancer.
KRASWT gene may inhibit the
migration and invasion of PANC-1 through the Wnt/β-catenin
pathway
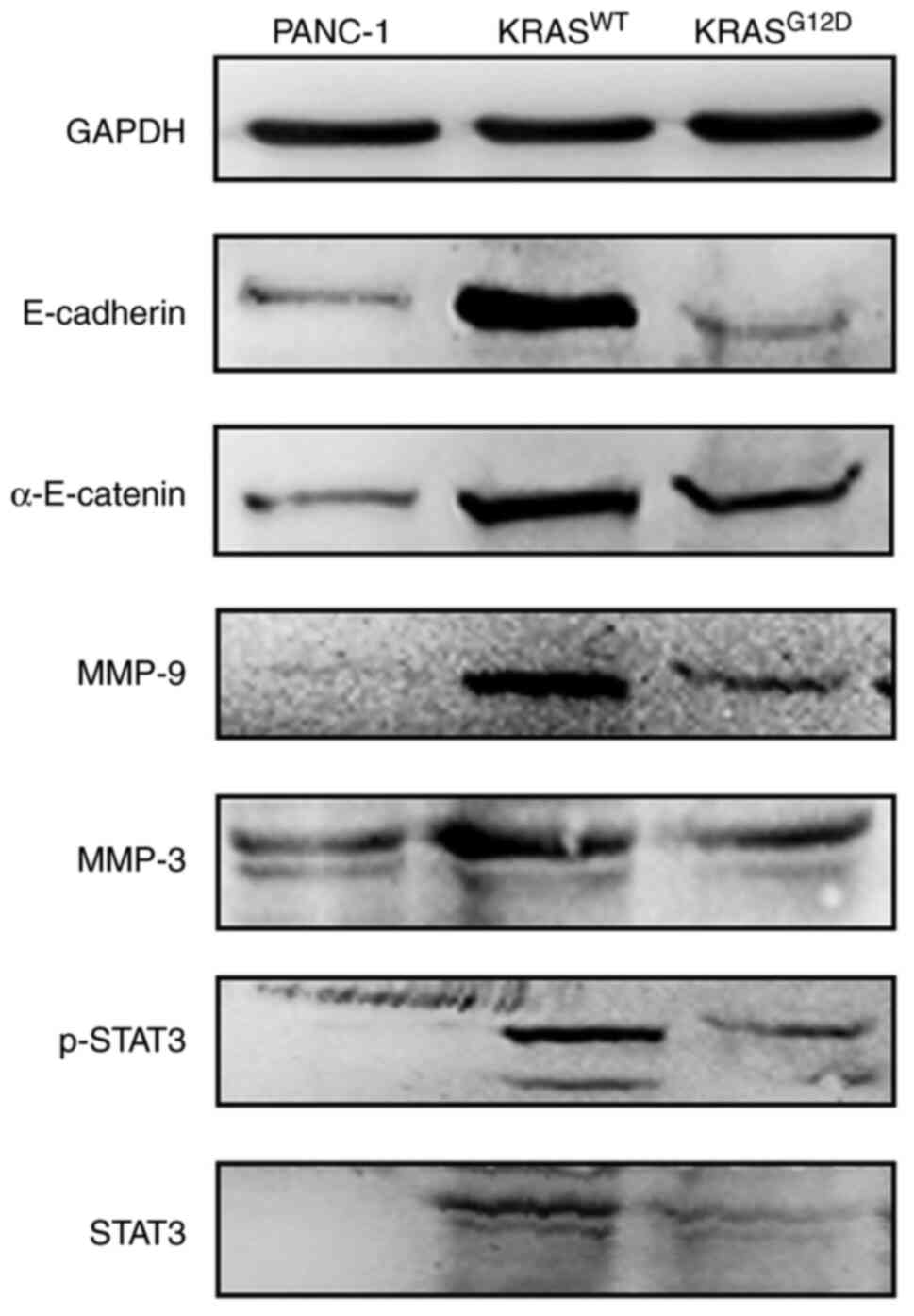
In order to explore the mechanism through which KRAS
regulates pancreatic cancer cell invasion and migration, some
signaling pathway molecules that serve key roles in cell invasion
and migration were examined. E-cadherin and α-E-catenin, which are
key regulators of the Wnt/β-catenin pathway, as well as MMP-3,
MMP-9, STAT3 and p-STAT3 were selected for western blotting. As
shown in Fig. 6, compared with
PANC-1, all the expressions of proteins of KRASWT were
upregulated and that of KRASG12D was not significantly
changed. These data suggested that the inhibition of migration and
invasion of PANC-1 cells by the KRASWT gene may be
mediated by the Wnt/β-catenin pathway.
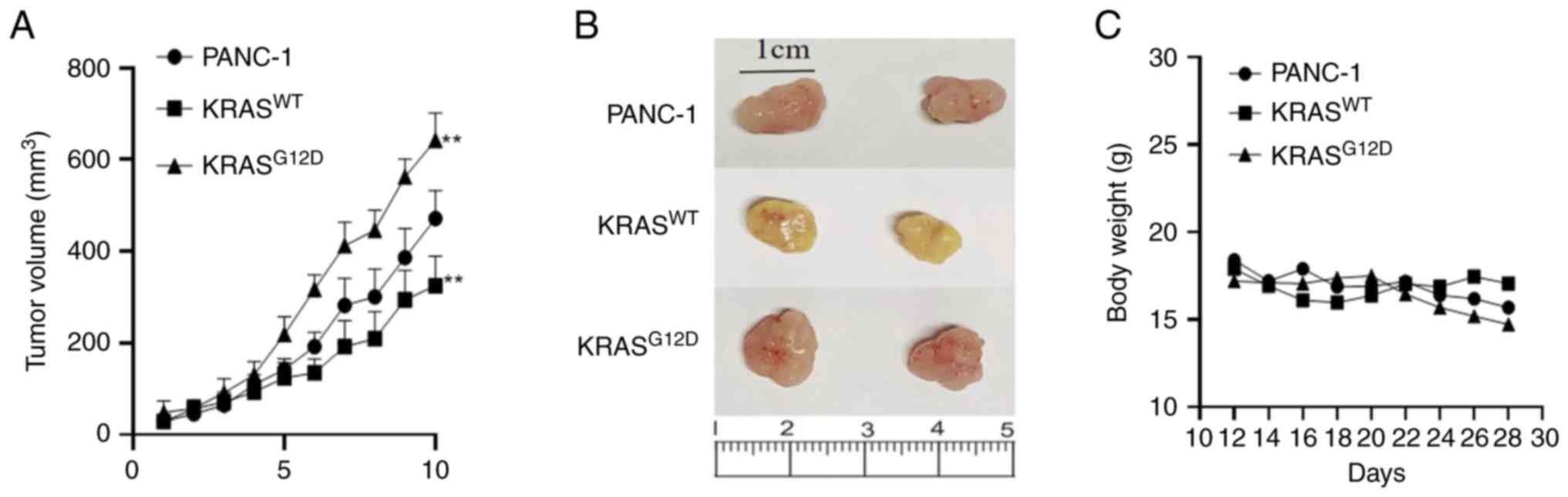
KRASWT gene inhibits tumor
growth in nude mice
In order to explore the different effects of KRAS on
pancreatic cancer proliferation, migration and invasion in
vivo and in vitro, tumor growth curves were drawn
according to the average tumor volume measured at each monitoring
point. The results showed that, compared with PANC-1, the average
tumor volume in the KRASWT was decreased, while that of
KRASG12D was increased, indicating that the
KRASWT gene can inhibit the proliferation of pancreatic
cancer in vivo, while the mutant KRAS gene can promote the
proliferation of pancreatic cancer (Fig. 7).
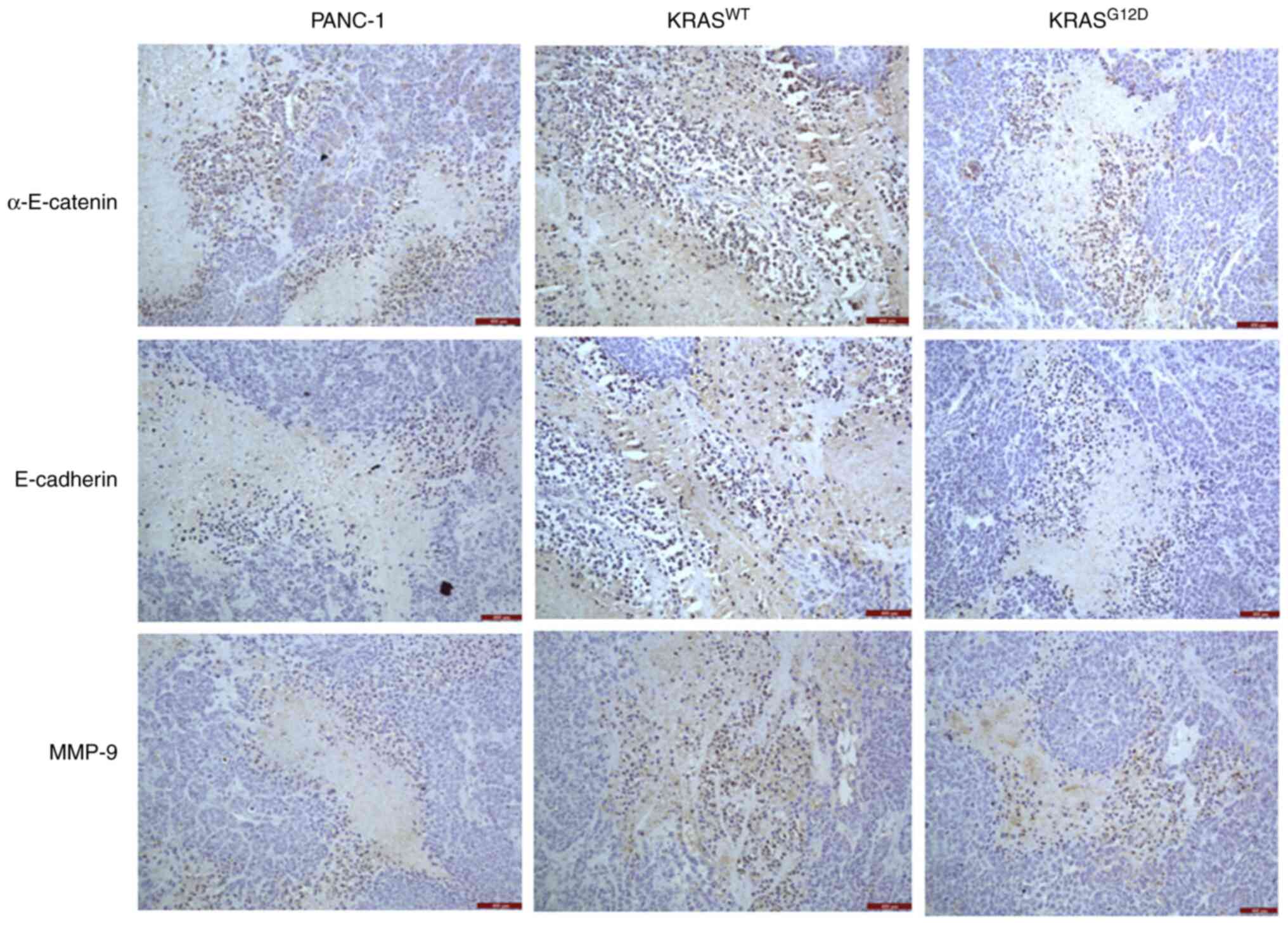
KRASWT gene inhibits
protein expression in nude mouse tumors
Immunohistochemistry was performed on tumors from
nude mice and the results showed that, compared with PANC-1, the
expression of E-cadherin, α-E-catenin and MMP-9 of
KRASG12D exhibited no significant changes. However, the
expression of E-cadherin, α-E-catenin and MMP-9 in
KRASWT was significantly upregulated (Fig. 8), which suggested that the
inhibition of the migration and invasion of PANC-1 cells by
KRASWT gene may be mediated by the Wnt/β-catenin
pathway.
Discussion
KRAS mutations are considered to be the driving
event in the development of pancreatic cancer. Studies (13,14) have shown that KRAS mutations can
be detected in 90% of pancreatic ductal intraepithelial neoplasia
(PANIN), 45–60% of pancreatic ductal papillary myxomas and >90%
of pancreatic adenocarcinoma, with 84% of the mutations leading to
the substitution of a single amino acid at the G12 site, among
which G12D is the most common (42%).
The G12D mutant subtype is associated with a
decreased overall survival. Windon et al (9) suggested that KRASWT may
have a survival advantage in PDAC, as patients with
KRASG12D exhibited a worse survival rate and shorter
overall survival time following gemcitabine-based first-line
chemotherapy (11.6 vs. 5.6 months, P=0.03). Ambrogio et al
(10) suggested that
KRASWT can antagonize the effect of KRASG12D
and the loss of KRASWT can accelerate cell proliferation
and tumor progression through the increase of the GTPase level of
KRASG12D. KRASWT can also disrupt the
inhibitory response of KRASG12D to MEK. According to
Mueller et al (15),
KRASWT is absent to varying degrees during tumor
development and is associated with a high mRNA expression of the
KRASG12D gene. Mueller et al also performed a
microdissection of 19 patients with low-grade pancreatic
intraepithelial neoplasia and deep sequencing of KRAS exon 2,
confirming that KRASG12D significantly increases
metastatic potential, which explains the high incidence of early
progression and metastasis in PDAC. The above studies show that
KRASWT exerts different degrees of tumor suppression.
Therefore, the KRASWT gene was directly overexpressed in
the present study to investigate the differences in biological
behaviors and the mechanism of action.
The results of CCK-8 and clone formation assay were
consistent. As compared with PANC-1, the proliferation of
KRASWT cells was weakened, while no significant
difference was observed in KRASG12D, indicating that the
KRASWT gene can reduce the proliferation of PANC-1
(Figs. 2 and 3). At the same time, a tumor formation
model of pancreatic cancer was established in nude mice (Fig. 7). In vivo, the average
tumor volume in the KRASWT group was smaller than PANC-1
group and KRASG12D group, which was consistent with the
results of the in vitro CCK-8 and clone formation assays,
indicating that KRASWT gene can reduce the proliferation
of pancreatic cancer cells in vivo and in vitro. In
addition, it was found that the tumor volume of the
KRASG12D group was larger than that of the PANC-1 group,
indicating that the mutant KRAS gene can promote the proliferation
of pancreatic cancer in vivo. However, the promotion effect
was not obvious in vitro.
Wound healing and Transwell assays were then
performed to detect migration and invasion. As shown in Figs. 4 and 5, as compared with PANC-1 group, the
migration and invasion of KRASG12D group was enhanced,
while that of KRASWT group was significantly weakened,
suggesting that the KRASWT gene could inhibit the
migration and invasion of pancreatic cancer.
Epithelial-to-mesenchymal transition (EMT) is a
progressive process of phenotype conversion from epithelial to
mesenchymal (16,17). Among them, E-cadherin ensures the
integrity of epithelial phenotypes by influencing cell polarity and
tissue integrity to form stable adhesion (18), which is a key event in the EMT
process (19,20). α-E-catenin also plays an important
role in the regulation and coordination of intracellular adhesion
and is a key regulator of the Wnt/β-catenin pathway (21). α-E-catenin is a major sensor of
mechanical force in adherens junctions and its cytoplasmic domain
is connected to the actin cytoskeleton by β-catenin and
α-E-catenin. α-E-catenin can bind to E-cadherin to form
intercellular adhesion and mediate the invasion and migration of
tumor cells (22–24). E-cadherin and α-E-catenin were
therefore selected to verify whether the Wnt/β-catenin pathway
plays a role in inhibiting migration and invasion. According to
western blotting (Fig. 6) and
immunohistochemistry (Fig. 8), as
compared with PANC-1, there was no significant difference in the
expression of E-cadherin and α-E-catenin in KRASG12D.
The upregulated expression of E-cadherin and α-E-catenin in
KRASWT indicated that the KRASWT gene may
play a potential role in inhibiting the invasion and migration of
pancreatic cancer through the Wnt/β-catenin pathway. A previous
study revealed that the expression levels of β-catenin and RAS are
increased in gemcitabine-resistant pancreatic cancer cells
(25); inhibition of
Wnt/β-catenin and RAS/ERK pathways may provide a therapeutic
strategy for gemcitabine-resistant pancreatic cancer. In fact,
Wnt/β-catenin signaling and RAS/ERK pathways not only dominate
gemcitabine resistance, it is possible that Wnt/β-catenin signaling
directly responds to mutations in the RAS gene, which the present
study confirmed.
In the process of tumor development, the destruction
of basement membrane is an important step for tumor invasion and
metastasis (26). In addition to
degrading various protein components of extracellular matrix, some
MMPs, MMP-3, MMP-9, MMP-14 and MMP-2 can also induce EMT or its
related processes (27). A
previous study demonstrated that MMP-9 is highly expressed in
non-small cell lung, cervical and ovarian cancer, among others, and
has become one of the important factors related to cancer
metastasis and invasion (28).
MMP-3 protein breaks down a variety of extracellular matrix
molecules and cleaves various adhesion molecules, growth factors
and other MMPs (29). MMP-3 can
activate MMP-1 and other family members and its overexpression is
associated with the growth and metastasis of various cancers,
including breast cancer (30).
When stimulated by external signals, STAT3 migrates to the nucleus
in the form of activated p-STAT3, which stimulates cell growth and
angiogenesis, activates the transcription of target genes and
regulates cell proliferation, differentiation and metastasis
(31,32). Studies have shown that
STAT3/p-STAT3 is highly expressed in a variety of tumors and its
activation product, p-STAT3, is associated with tumor grade and
patient prognosis (33,34). As a major mediator of the JAK/STAT
pathway, STAT3 may be an important regulator of tumor progression
by enhancing aggressiveness or promoting EMT (35,36).
These studies indicate that the high expression of
MMP-3, MMP-9, STAT3 and p-STAT3 is an important manifestation of
tumor invasion and metastasis. The present results showed that, as
compared with PANC-1 group, the migration and invasion of
KRASWT group was weakened. However, western blotting and
immunohistochemistry results showed that the expression of MMP-3,
MMP-9, STAT3 and p-STAT3 in KRASWT group were
upregulated, which was inconsistent with previous reports. The
present study hypothesized that the inhibition of pancreatic cancer
cell invasion and migration by the KRASWT gene may not
be due to the degradation of extracellular matrix or the inhibition
of the function of the JAK/STAT pathway, it is also possible that
the inhibitory effect of Wnt/β-catenin pathway on the invasion and
migration of pancreatic cancer cells was greater compared with that
of extracellular matrix or JAK/STAT pathway and the reasons need to
be further studied.
In conclusion, the present study investigated the
biological effects of KRASWT on the proliferation and
migration of pancreatic cancer cells, it was found that
KRASWT could inhibit the proliferation of pancreatic
cancer in vivo and in vitro and mutated KRAS could
enhance the proliferation of pancreatic cancer in vivo, but
this promotion was not obvious in vitro. Meanwhile,
KRASWT can inhibit the migration and invasion of
pancreatic cancer, which may be achieved through the Wnt/β-catenin
pathway. The present study provided a theoretical and experimental
basis for the strategy of KRAS gene therapy for pancreatic
cancer.
Acknowledgements
Not applicable.
Funding
Funding was provided by the Science and Technology Innovation
project of Sichuan Province (grant no. 2021033) and the Nanchong
City School cooperation project (grant nos. 22SXCXTD0002 and
22SXQT0100).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
In the planning and execution of this paper, the
authors XH and CZ designed the study, XH completed the main writing
and most of the experiments, CZ and RZ completed the feeding, data
collection and statistical analysis of the body weight and tumor
data of the nude mice., BM and JY participated in the data
statistics. XH and BM confirm the authenticity of all raw data. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of North Sichuan Medical College (approval no. 2022035)
and all procedures were carried out in accordance with relevant
guidelines and regulations.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Eguchi H, Kobayashi S, Gotoh K, Noda T and
Doki Y: Characteristics of early-onset pancreatic cancer and its
association with familial pancreatic cancer and hereditary
pancreatic cancer syndromes. Ann Gastroenterol Surg. 4:229–233.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Moore A and Donahue T: Pancreatic cancer.
JAMA. 322:14262019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shibata H, Komura S, Yamada Y, Sankoda N,
Tanaka A, Ukai T, Kabata M, Sakurai S, Kuze B, Woltjen K, et al: In
vivo reprogramming drives Kras-induced cancer development. Nat
Commun. 9:20812018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Luo J: KRAS mutation in pancreatic cancer.
Semin Oncol. 48:10–18. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Moore MJ, Goldstein D, Hamm J, Figer A,
Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, et al:
Erlotinib plus gemcitabine compared with gemcitabine alone in
patients with advanced pancreatic cancer: A phase III trial of the
National Cancer Institute of Canada Clinical Trials Group. J Clin
Oncol. 25:1960–1966. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Drilon A, Laetsch TW, Kummar S, DuBois SG,
Lassen UN, Demetri GD, Nathenson M, Doebele RC, Farago AF, Pappo
AS, et al: Efficacy of Larotrectinib in TRK fusion-positive cancers
in adults and children. N Engl J Med. 378:731–739. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang L, Guo Z, Wang F and Fu L: KRAS
mutation: From undruggable to druggable in cancer. Signal Transduct
Target Ther. 6:3862021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Haigis KM: KRAS alleles: The devil is in
the detail. Trends Cancer. 3:686–697. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Windon AL, Loaiza-Bonilla A, Jensen CE,
Randall M, Morrissette JJD and Shroff SG: A KRAS wild type
mutational status confers a survival advantage in pancreatic ductal
adenocarcinoma. J Gastrointest Oncol. 9:1–10. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ambrogio C, Kohler J, Zhou ZW, Wang H,
Paranal R, Li J, Capelletti M, Caffarra C, Li S, Lv Q, et al: KRAS
dimerization impacts MEK inhibitor sensitivity and oncogenic
activity of mutant KRAS. Cell. 172:857–868. e152018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kent OA: Increased mutant KRAS gene dosage
drives pancreatic cancer progression: Evidence for wild-type KRAS
as a tumor suppressor? Hepatobiliary Surg Nutr. 7:403–405. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Perets R, Greenberg O, Shentzer T,
Semenisty V, Epelbaum R, Bick T, Sarji S, Ben-Izhak O, Sabo E and
Hershkovitz D: Mutant KRAS circulating tumor DNA is an accurate
tool for pancreatic cancer monitoring. Oncologist. 23:566–572.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hobbs GA, Baker NM, Miermont AM, Thurman
RD, Pierobon M, Tran TH, Anderson AO, Waters AM, Diehl JN, Papke B,
et al: Atypical KRASG12R mutant is impaired in PI3K
signaling and macropinocytosis in pancreatic cancer. Cancer Discov.
10:104–123. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mueller S, Engleitner T, Maresch R,
Zukowska M, Lange S, Kaltenbacher T, Konukiewitz B, Öllinger R,
Zwiebel M, Strong A, et al: Evolutionary routes and KRAS dosage
define pancreatic cancer phenotypes. Nature. 554:62–68. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Santamaria PG, Moreno-Bueno G, Portillo F
and Cano A: EMT: Present and future in clinical oncology. Mol
Oncol. 11:718–738. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Georgakopoulos-Soares I, Chartoumpekis DV,
Kyriazopoulou V and Zaravinos A: EMT factors and metabolic pathways
in cancer. Front Oncol. 10:4992020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sommariva M and Gagliano N: E-Cadherin in
pancreatic ductal adenocarcinoma: A multifaceted actor during EMT.
Cells. 9:10402020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Loh CY, Chai JY, Tang TF, Wong WF, Sethi
G, Shanmugam MK, Chong PP and Looi CY: The E-Cadherin and
N-Cadherin switch in epithelial-to-mesenchymal transition:
Signaling, therapeutic implications and challenges. Cells.
8:11182019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Christgen M, Bartels S, van Luttikhuizen
JL, Bublitz J, Rieger LU, Christgen H, Stark H, Sander B, Lehmann
U, Steinemann D, et al: E-cadherin to P-cadherin switching in
lobular breast cancer with tubular elements. Mod Pathol.
33:2483–2498. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang B, Li X, Liu L and Wang M: β-Catenin:
Oncogenic role and therapeutic target in cervical cancer. Biol Res.
53:332020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu XP, Pokutta S, Torres M, Swift MF,
Hanein D, Volkmann N and Weis WI: Structural basis of
αE-catenin-F-actin catch bond behavior. Elife. 9:e608782020.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Terekhova K, Pokutta S, Kee YS, Li J,
Tajkhorshid E, Fuller G, Dunn AR and Weis WI: Binding partner- and
force-promoted changes in αE-catenin conformation probed by native
cysteine labeling. Sci Rep. 9:153752019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rogers CD, Sorrells LK and Bronner ME: A
catenin-dependent balance between N-cadherin and E-cadherin
controls neuroectodermal cell fate choices. Mech Dev. 152:44–56.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ryu WJ, Han G, Lee SH and Choi KY:
Suppression of Wnt/β-catenin and RAS/ERK pathways provides a
therapeutic strategy for gemcitabine-resistant pancreatic cancer.
Biochem Biophys Res Commun. 549:40–46. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kelppe J, Thoren H, Haglund C, Sorsa T and
Hagstrom J: MMP-7, −8, −9, E-cadherin and beta-catenin expression
in 34 ameloblastoma cases. Clin Exp Dent Res. 7:63–69. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li Y, He J, Wang F, Wang X, Yang F, Zhao
C, Feng C and Li T: Role of MMP-9 in epithelial-mesenchymal
transition of thyroid cancer. World J Surg Oncol. 18:1812020.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao Y, Qiao X, Wang L, Tan TK, Zhao H,
Zhang Y, Zhang J, Rao P, Cao Q, Wang Y, et al: Matrix
metalloproteinase 9 induces endothelial-mesenchymal transition via
Notch activation in human kidney glomerular endothelial cells. BMC
Cell Biol. 17:212016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liutkevicius V, Lesauskaite V,
Liutkeviciene R, Vaiciulis P and Uloza V: Matrix Metalloproteinases
(MMP-2,-3,-9) Gene polymorphisms in cases of benign vocal fold
lesions and laryngeal carcinoma. In Vivo. 34:267–274. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Suhaimi SA, Chan SC and Rosli R: Matrix
Metallopeptidase 3 Polymorphisms: Emerging genetic markers in human
breast cancer metastasis. J Breast Cancer. 23:1–9. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen XK, Gu CL, Fan JQ and Zhang XM:
P-STAT3 and IL-17 in tumor tissues enhances the prognostic value of
CEA and CA125 in patients with lung adenocarcinoma. Biomed
Pharmacother. 125:1098712020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liang B, Li SY, Gong HZ, Wang LX, Lu J,
Zhao YX and Gu N: Clinicopathological and prognostic roles of STAT3
and its phosphorylation in glioma. Dis Markers. 2020:88338852020.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Song M, Wang C, Yang H, Chen Y, Feng X, Li
B and Fan H: P-STAT3 inhibition activates endoplasmic reticulum
stress-induced splenocyte apoptosis in chronic stress. Front
Physiol. 11:6802020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li Y, Wang Y, Shi Z, Liu J, Zheng S, Yang
J, Liu Y, Yang Y, Chang F and Yu W: Clinicopathological and
prognostic role of STAT3/p-STAT3 in breast cancer patients in
China: A meta-analysis. Sci Rep. 9:112432019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu HW, Lee PM, Bamodu OA, Su YK, Fong IH,
Yeh CT, Chien MH, Kan IH and Lin CM: Enhanced Hsa-miR-181d/p-STAT3
and Hsa-miR-181d/p-STAT5A ratios mediate the anticancer effect of
garcinol in STAT3/5A-Addicted glioblastoma. Cancers (Basel).
11:18882019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Susman S, Pirlog R, Leucuta D, Mitre AO,
Padurean VA, Melincovici C, Moldovan I, Crișan D and Florian SI:
The role of p-Stat3 Y705 immunohistochemistry in glioblastoma
prognosis. Diagn Pathol. 14:1242019. View Article : Google Scholar : PubMed/NCBI
|