Introduction
Insulin-like growth factor 2 (IGF2) is a peptide
hormone that is important for embryonic development and the
regulation of metabolic processes (1,2).
IGF2 expression is tightly regulated by genomic imprinting, which
ensures spatially and temporally precise activity (3,4).
Beyond its role in development, IGF2 is involved in kidney
physiology by promoting cell proliferation, metabolic activity and
tissue repair (5–7). Emerging evidence implicates IGF2
dysregulation in the pathogenesis of several kidney disorders,
including acute kidney injury (AKI) (8), chronic kidney disease (CKD) (9) and renal malignancies (10). Through interactions with receptors
such as IGF1 receptor (IGF1R), insulin receptor isoform A (IR-A)
and IGF2 receptor (IGF2R), IGF2 activates signaling pathways,
including the PI3K/Akt and MAPK pathways, that modulate
inflammation, fibrosis and cell survival (11,12)
(Fig. 1). The present review
summarizes the structural characteristics and biological functions
of IGF2, its regulatory mechanisms and its involvement in kidney
diseases, highlighting its potential as both a diagnostic biomarker
and a therapeutic target.
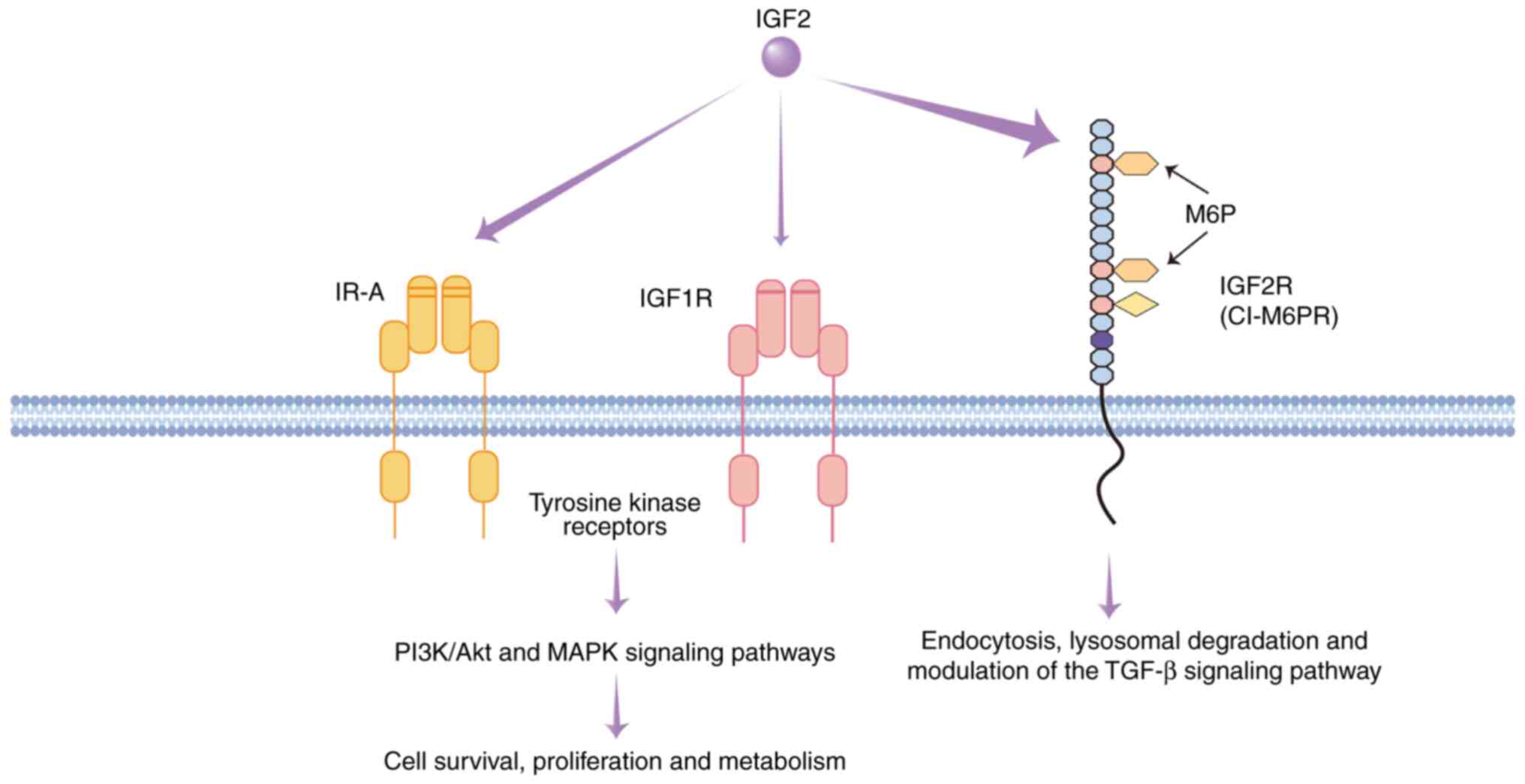 | Figure 1.IGF2-mediated signaling pathways and
the functional roles of IGF receptors. This schematic illustrates
the interactions between IGF2 and its primary receptors: IR-A,
IGF1R and IGF2R (CI-M6PR). The arrow thickness indicates the
relative binding affinity. IR-A and IGF1R are tyrosine kinase
receptors. Upon IGF2 binding, they activate the PI3K/Akt and MAPK
signaling pathways, promoting cell survival, proliferation and
metabolic regulation. By contrast, IGF2R lacks signaling capability
and primarily functions to internalize and target IGF2 for
lysosomal degradation, thus regulating its bioavailability. It also
transports M6P-tagged proteins and modulates the TGF-β pathway,
influencing extracellular matrix dynamics and tissue homeostasis.
IGF, insulin-like growth factor; IGF1R/IGF2R, IGF1/2 receptor;
CI-M6PR, cation-independent mannose-6-phosphate receptor; IR-A,
insulin receptor isoform A. |
Structure and physiological function of
IGF2
IGF2, a polypeptide hormone, has a similar action as
insulin, yet its biological activity is low (approximately 14% of
that of insulin) (13). The IGF2
gene is located on human chromosome 11p15.5 and spans ~30 kb of
genomic DNA. It encodes a 7.5-kDa peptide comprising 67 amino acid
residues and shares ~67% sequence homology with insulin-like growth
factor 1 (IGF1) (14).
IGF2 is widely expressed in embryonic and fetal
tissues, although transcript levels vary considerably across
different organs (15). The gene
is regulated by five distinct promoters (P0-P4) and consists of 10
exons, with only the final three exons encoding protein-coding
sequences (15,16). These promoters are differentially
regulated in a tissue- and developmental stage-specific manner,
giving rise to functionally distinct mRNA isoforms (16,17).
Each promoter is functionally active at specific stages of
development and in distinct tissue types. For example, the P0
promoter is predominantly active in the placenta and serves a role
in determining placental size and composition. Loss of P0 promoter
activity reduces passive diffusion across the placenta, resulting
in fetal growth retardation, although systemic levels of fetal IGF2
remain unaffected. This observation suggests that placental IGF2
expression operates independently of circulating IGF2 levels
(18).
The P1 promoter is primarily active in the adult
liver and the choroid plexus of the brain and contains an internal
ribosomal entry site (16). P2 is
mainly active in the fetal liver, whereas P3 and P4 drive
expression in non-hepatic fetal and adult tissues. Notably, P3 and
P4 are epigenetically silenced after birth but may be reactivated
in certain tumors [including Wilms' tumor (19) and rhabdomyosarcoma] (20,21).
Both P3 and P4 contain canonical TATA and CCAAT boxes recognized by
RNA polymerase II, while P2 lacks these elements (22). Transient transfection assays have
demonstrated that the activity of P2, P3 and P4 varies by cell type
and species. For example, P2 is minimally active in most cell
lines, whereas P3 exhibits the highest transcriptional activity in
hepatocyte-derived human cells (23). Overall, IGF2 transcription declines
sharply in most tissues after birth (16).
IGF2 serves an important role in tissue development
and growth, particularly during embryogenesis (24). The biological functions of IGF2 are
primarily mediated through interactions with three specific
receptors: IGF1R, IR-A and IGF2R (11,12).
IGF1R is a transmembrane tyrosine kinase receptor structurally
related to the insulin receptor (IR) (9,19,25).
Upon binding IGF2, IGF1R activates the PI3K/Akt and MAPK signaling
cascades, thereby promoting cell proliferation, differentiation and
survival, which are processes that are important for embryonic
tissue and organ formation (11,26).
IGF1R signaling also serves a central role in tumor biology by
enhancing cell survival via anti-apoptotic mechanisms, particularly
in IGF1R-dependent malignancies (27).
Compared with IGF1R, IR-A has a higher binding
affinity for IGF2 and is predominantly expressed in fetal tissues
and specific cancer types, including colorectal cancer,
osteosarcoma and thyroid cancer (23,28,29).
Upon binding to IGF2, IR-A activates the PI3K/Akt and MAPK
pathways, regulating cell metabolism, proliferation and survival,
and contributing to tumor growth and adaptability (30–32).
Under specific pathological conditions, such as tumors that
excessively secrete IGF2, notably including mesenchymal tumors and
hepatocellular carcinomas, IGF2 can induce hypoglycemia by
activating the IR-A signaling pathway. The aforementioned tumor
cells commonly highly express IGF2 mRNA (33–35).
However, defective post-translational processing leads to the
production of a high molecular weight precursor (15–25 kDa),
referred to as pro-IGF2 or ‘big IGF2’, characterized by an
uncleaved E-domain extension (36–38).
Under physiological conditions, mature IGF2 circulates primarily as
part of a 150-kDa ternary complex with IGF-binding protein
(IGFBP)-3 and the acid-labile subunit (ALS). This large complex is
unable to cross the capillary endothelium, thereby limiting its
biological activity (36,38).
By contrast, pro-IGF2 binds to IGFBP-2 or IGFBP-3 to
form a smaller (~50 kDa) binary complex. Steric hindrance from the
uncleaved E-domain prevents ALS binding, thereby impeding the
formation of the ternary complex. Due to its reduced molecular
weight, the binary complex can freely diffuse across the capillary
endothelium into the interstitial fluid (37). A clinical study demonstrated that
serum concentrations of the 50-kDa complex in patients with tumors
can be elevated >10-fold compared with normal levels, enhancing
its tissue distribution (37).
Additionally, pro-IGF2 promotes vascular leakage and tissue
penetration by stimulating tumor secretion of VEGF and MMP-9. VEGF
increases vascular permeability, while MMP-9 degrades the basement
membrane and disrupts endothelial junctions (39,40).
Furthermore, tumor-associated neovasculature is often characterized
by incomplete basement membranes and loosely connected endothelial
cells, creating a ‘leaky’ vascular phenotype that facilitates the
extravasation of small molecular complexes (41).
Once within the interstitial fluid, pro-IGF2 retains
receptor-binding affinities to both IR and IGF1R similar to mature
IGF2 but exhibits 2- to 3-fold greater molar bioactivity, leading
to prolonged receptor activation (36,37).
Consequently, in patients with extra-pancreatic tumor-induced
hypoglycemia, circulating pro-IGF2 levels are markedly elevated and
typically normalize following successful tumor resection (42).
The biological effects of ligand binding are notably
influenced by receptor subtype. For instance, in 32D clone 3 murine
hematopoietic progenitor cells, IGF2 binding to IR-A induces
pro-mitotic and anti-apoptotic signaling, whereas its binding to
IR-B promotes differentiation (43). In mouse fibroblasts expressing
exclusively IR-A, insulin binding elicits metabolic responses
(44).
In contrast to IGF1R, IGF2R exhibits distinct
structural and functional characteristics and has the highest
binding affinity for IGF2. IGF2R is a single-chain transmembrane
receptor that lacks intrinsic tyrosine kinase activity and
primarily mediates IGF2 degradation via endocytosis, thereby
suppressing cellular over-proliferation and reducing tumorigenic
potential (45–47). Additionally, IGF2R facilitates
lysosomal enzyme trafficking by recognizing mannose-6-phosphate
residues and is therefore also referred to as the
cation-independent mannose-6-phosphate receptor (46). This function is evolutionarily
conserved, and mammalian IGF2R has further acquired the capacity to
bind IGF2, thereby enhancing its role in limiting cell
proliferation (48). IGF2R also
modulates the TGF-β signaling pathway, which is involved in
extracellular matrix (ECM) synthesis, immune regulation, and the
control of cell proliferation, differentiation and development
(49,50). Collectively, these properties allow
IGF2R to maintain growth homeostasis under physiological
conditions, and inhibit tumorigenesis and progression under
pathological conditions (51).
Despite advances in deciphering the complex
structure and regulatory mechanisms of IGF2, such as its genomic
imprinting and the existence of multiple promoters and isoforms
(52,53), the role of IGF2, particularly in
renal physiology and pathology, remains to be fully elucidated. The
present review focuses on the expression, regulation and function
of IGF2 in the kidney, summarizing its contributions to renal
development and its involvement in kidney disease.
Role of IGF2 in the development and
functional maintenance of the kidney
IGF2 serves a notable role in renal development and
the maintenance of kidney function by regulating cellular
proliferation, differentiation and metabolic homeostasis (5). IGF2 expression exhibits a distinct
spatiotemporal pattern aligned with specific physiological
functions across developmental stages (23,54–56).
During embryogenesis, IGF2 is highly expressed and
is important for normal renal morphogenesis (57,58).
At this stage, IGF2 is primarily secreted by the placenta (6) and exhibits strong expression in renal
structures, particularly the ureteric bud and metanephric
mesenchyme (7). This elevated
expression facilitates nephron formation by promoting the
mesenchymal-to-epithelial transition of nephrogenic progenitors,
driving the development of renal tubules and glomeruli, and
directing mesodermal cell differentiation toward functionally
specialized nephron segments (7,24,59).
Animal studies have supported the important role of IGF2 during
this period: Disruption of IGF2 signaling leads to notably reduced
birth weight, whereas overexpression results in accelerated somatic
growth (45,60,61).
Furthermore, intrauterine growth restriction models have
demonstrated that aberrant DNA methylation of the IGF2 promoter
reduces its expression, lowers nephron endowment and increases the
risk of CKD later in life (7),
underscoring the importance of tightly regulated IGF2 expression
during renal development.
As renal development reaches completion, IGF2
expression is downregulated (62).
In the adult kidney, IGF2 is maintained at basal levels and is
primarily localized to vascular structures and interstitial cells
surrounding the glomeruli and renal tubules (57,63).
IGF2 mRNA exhibits a spatially restricted pattern: It is enriched
in the vascular components of the cortex, particularly within the
endothelium and adventitia of afferent arterioles, while in the
medulla, it is predominantly expressed in vascular and stromal
compartments, with negligible expression in tubular epithelial
cells (5). This distribution
suggests a specialized role for IGF2 in regulating renal
vasculature and glomerular function. Basal IGF2 also contributes to
renal homeostasis by supporting cellular repair and metabolic
balance (63–65). The transition from high embryonic
IGF2 expression to adult basal levels appears to be epigenetically
regulated, particularly via promoter methylation, ensuring
structural and functional stability post-development (8). Notably, a study using IGF1-deficient
transgenic mouse models showed that kidney-specific overexpression
of IGF2 selectively increased both absolute and relative kidney
weight, without altering body weight or the mass of other organs
(66). These findings support a
unique, organ-specific role for IGF2 in kidney growth, development
and homeostatic maintenance, spanning from embryonic morphogenesis
to adult physiological function.
The biological effects of IGF2 are primarily
mediated via its interaction with IGF1R and IR (67). IGF1R, the primary signaling
receptor for IGF2, is widely distributed in the kidney, with
prominent expression in renal tubules, glomeruli and mesangial
cells (68). The expression of
IGF1R is particularly high in proximal and distal tubular segments,
underscoring the role of IGF2 in regulating metabolic processes and
ion transport in these structures (5). IGF2 promotes the proliferation and
differentiation of renal precursor cells predominantly through
IGF1R activation, thus contributing to the formation of renal
tubules and glomeruli (69). In a
mouse model with P2 promoter deletion, resulting in reduced IGF2
expression, pronounced renal abnormalities were observed,
manifesting as proteinuria, podocyte depletion, glomerulosclerosis,
increased ECM deposition and glomerular basement membrane
thickening at 18 months of age (70). These findings emphasize the
importance of IGF2 in supporting renal architecture and maintaining
nephron functionality, primarily through its role in podocyte
health. The observed increase in ECM deposition is a secondary,
dysregulated consequence of podocyte injury and loss, manifesting
as mesangial expansion and glomerulosclerosis (70). Furthermore, IGF1R activation
promotes ECM synthesis in renal tubules, thereby supporting the
structural integrity and functionality of renal tissues (71).
IR is another important receptor for IGF2. Although
IR primarily regulates glucose metabolism, it is also widely
expressed in the kidney (72). In
renal tubules and glomerular endothelial cells, IR expression is
high and contributes to the regulation of renal blood flow and
tubular reabsorption functions (73). Compared with IGF1R, IR displays a
more uniform distribution across kidney regions, suggesting a
broader role in maintaining renal metabolic homeostasis (74). Additionally, IGF2 may influence
ionic transport and water-electrolyte balance in the kidney via IR
signaling. An in vitro study has shown that IGF2 expression
enhanced sodium uptake in proximal tubular brush border membrane
vesicles (65).
Unlike IGF1R and IR, IGF2R lacks intrinsic kinase
activity, and is traditionally regarded as a decoy receptor
involved in IGF2 internalization and degradation (75). However, a previous study has
revealed that IGF2R may serve an active signaling role. IGF2R
nuclear translocation has been linked to GSK3, which primes
macrophages toward an anti-inflammatory phenotype. These findings
reveal a novel immunomodulatory role for IGF2R in macrophages and
suggest that IGF2 may influence immune responses within the kidney
via IGF2R. Notably, activation of IGF1R, by either IGF1 or IGF2,
can antagonize IGF2R signaling, thereby modulating macrophage
polarization. The balance between IGF1R and IGF2R activation is
important for maintaining macrophage-mediated immune homeostasis
and appropriately shaping the immune response to infection or
inflammation (76).
Understanding the regulatory mechanisms governing
IGF2 expression is important for elucidating its roles in renal
development and disease. IGF2 expression is controlled by genomic
imprinting, with transcription occurring exclusively from the
paternal allele while the maternal allele remains epigenetically
silenced (77,78). This monoallelic expression pattern
is maintained by DNA methylation at the H19/IGF2 imprinting control
region (ICR) and its associated epigenetic regulatory elements
(79,80). Specifically, paternal-specific
methylation of the ICR, enhancer competition and CCCTC-binding
factor-mediated boundary insulation ensure the spatial and temporal
precision of IGF2 function during renal development (81).
Loss of genomic imprinting is associated with
pathological growth abnormalities. For example, Beckwith-Wiedemann
syndrome is caused by IGF2 upregulation and is characterized by
somatic overgrowth of the body, including kidney overgrowth
(61). Conversely, Silver-Russell
syndrome involves IGF2 downregulation and presents with growth
restriction of body and the kidneys are generally small in
proportion to the overall body growth restriction (82). Experimental models further
underscore the important role of imprinting in renal development.
These include the model demonstrating that CTCF mediates
methylation-sensitive enhancer-blocking activity at the H19/Igf2
locus, where it binds the unmethylated imprinting control region
(ICR) on the maternal allele to insulate Igf2 from enhancers,
thereby silencing it (79), and
the model of aberrant establishment of paternal methylation imprint
at the H19/Igf2 ICR, which disrupts the parent-of-origin-specific
expression pattern (83,84). In both cases, enhancer deletion at
the 3′ end of the H19 gene and demethylation of ICR alter IGF2
expression and impair normal kidney development (85).
Role of IGF2 in kidney disease
Above studies suggest a potential pathogenic and
therapeutic role for IGF2 in diverse renal disorders, and highlight
the need for further investigation into IGF2 signaling pathways and
receptor interactions in the kidney to advance early diagnosis,
therapeutic strategies and targeted interventions for renal
pathologies. The following section explores the effects of IGF2
administration or overexpression in experimental models of renal
disease.
IGF2 modulates renal physiology and pathology via a
complex receptor network, including IGF2R, IGF1R and IR-A, and
downstream signaling pathways such as the PI3K/Akt, MAPK and GSK3
pathways, exhibiting a biphasic regulatory pattern (76,86).
Under physiological conditions, IGF2 predominantly interacts with
IGF2R, activating the GSK3 signaling pathway and inducing the
epigenetic reprogramming of macrophages (8,76).
This promotes a metabolic shift toward oxidative phosphorylation
(OXPHOS), fosters a macrophagic anti-inflammatory phenotype
(76,87). In parallel, IGF2 signaling supports
the proliferation of renal tubular epithelial cells, thereby
contributing to podocyte cytoskeletal integrity and renal tissue
repair (70,88).
By contrast, under pathological conditions,
including chronic inflammation, hyperglycemia or loss of genomic
imprinting, IGF2 preferentially binds IGF1R and IR-A, leading to
sustained activation of the PI3K/Akt-MAPK axis. This signaling
cascade disrupts the integrity of the podocyte-slit diaphragm
complex and, in synergy with TGF-β1, stimulates collagen deposition
and promotes irreversible renal fibrosis (70,89).
Furthermore, in contexts involving biallelic IGF2 expression,
persistent pro-proliferative signaling may ensue, contributing to
proteinuria, glomerulosclerosis and the malignant transformation of
renal tissues (90). This
receptor-dependent shift in IGF2 signaling cascades represents a
central molecular mechanism that governs renal cell fate decisions
under both homeostatic and pathological states.
Role of IGF2 in AKI
AKI is a heterogeneous clinical syndrome that is
characterized by a sudden decline in the glomerular filtration rate
(GFR), commonly indicated by elevated serum creatinine (SCr) levels
and/or oliguria (91). The
incidence of AKI among hospitalized patients is ~20% and is often
associated with complications such as fluid retention, electrolyte
imbalances, uremic symptoms and drug-induced nephrotoxicity
(91).
Traditionally, ischemia-reperfusion injury,
nephrotoxic insults and sepsis have been regarded as the primary
causes of AKI (91). However,
growing evidence suggests that modifiable lifestyle factors
influence both the onset and progression of AKI. Chronic alcohol
consumption exacerbates AKI, increases the vulnerability and
severity of kidney (92,93). Cigarette smoking impairs renal
perfusion by inducing systemic inflammation, oxidative stress and
endothelial dysfunction (94).
Diets high in sodium, saturated fats and processed foods are
strongly linked to hypertension, diabetes and cardiovascular
disease, which are chronic metabolic conditions that are
established risk factors for AKI (95,96).
These findings underscore the multifactorial pathogenesis of AKI
and highlight the need for integrative prevention strategies
addressing both clinical and lifestyle-related risk factors.
At the molecular level, IGF2 may emerge as a
potential key modulator of immune response and tissue repair during
acute kidney injury (AKI). Although direct evidence elucidating the
role of IGF2 in AKI remains limited, the well-documented functions
of its related family member IGF-1 and other growth factors,
including FGF2, in renal repair processes have been established
(97,98). Mechanistically, it is proposed that
IGF2 binds to its receptor IGF2R on macrophages, activating the
GSK3β/β-catenin pathway to suppress pro-inflammatory cytokines,
such as TNF-α and IL-1β, and enhance anti-inflammatory markers,
such as arginase-1 and IL-10, thereby reshaping the immune
microenvironment to attenuate renal injury (76). Furthermore, following
ischemia-reperfusion injury, IGF2 expression is transiently
upregulated and contributes to tubular epithelial cell repair
(99). Animal studies have further
demonstrated that IGF2R agonist administration reduced tubular
necrosis by 42% and macrophage infiltration by 57%, underscoring
the protective role of IGF2 in AKI (8,76).
IGF2 signaling has also shown promise as a clinical
biomarker. The combined urinary detection of IGFBP-7 and tissue
inhibitor of metalloproteinases-2 exhibits high predictive accuracy
for AKI (area under the curve, 0.80), supporting the translational
potential of IGF2-related mechanisms (100). Mesenchymal stem and/or stromal
cells mediate part of their renoprotective effects via IGF2. Under
hypoxic or inflammatory conditions, MSCs exhibit a 3.5- to 5.2-fold
increase in IGF2 secretion, promoting macrophage polarization
toward the anti-inflammatory M2 phenotype (8). IGF2 also facilitates macrophage
metabolic reprogramming from glycolysis to mitochondrial OXPHOS,
enhancing ATP production and sustaining the anti-inflammatory
activity of the macrophages (8).
However, IGF2 is not the sole mediator of MSC-induced renal
protection. Knockdown of IGF2 partially attenuates, but does not
abolish, MSC-mediated protection, suggesting compensatory roles for
other factors such as hepatocyte growth factor, VEGF and exosomal
microRNAs (97,101). Furthermore, although hypoxia
preconditioning enhances MSC-derived IGF2 expression in
glycerol-induced AKI models, it does not significantly improve
therapeutic efficacy compared with unconditioned MSCs, highlighting
the importance of multifactorial synergy (8).
A specific subtype of AKI, aristolochic acid
nephropathy (AAN), results primarily from aristolochic acid I (AAI)
exposure, and is characterized by AKI, interstitial nephritis and
metabolic dysfunction (102,103). AA forms covalent adducts with DNA
or RNA, driving the progression of renal damage (104), with ischemic injury forming a
central pathophysiological basis (105). It is therefore plausible that AA
may interfere with IGF2 signaling through spatially specific
mechanisms. Histological analyses have demonstrated that AA
primarily targets the renal cortex, where it induces DNA adducts,
such as 7-(deoxyadenosine-N6-yl) aristolactam (dA-AAI), in proximal
tubular cells, leading to epigenetic silencing of IGF2 through
aberrant DNA methylation and disruption of ICRs (106,107). Spatial transcriptomics analysis
has revealed that AA exposure activates the TGF-β/SMAD pathway in
cortex-localized injured tubular clusters. This process promotes
the establishment of a pro-inflammatory microenvironment
characterized by macrophage infiltration and CCL5/CCR5 axis
activation (108) and may
potentially represses IGF2 transcription via SMAD3/NF-κB
co-regulation (107,109,110).
Although the intense oxidative stress induced by
aristolochic acid (AA) has been established as a core mechanism of
its nephrotoxicity, the upstream drivers of this process require
further investigation (107,111–114). Based on the well-documented role
of the xanthine oxidoreductase system as a key source of reactive
oxygen species (ROS) in renal injury (115,116), a plausible scientific hypothesis
may be proposed: AA may activate this pathway by triggering ‘purine
metabolic reprogramming’ in the renal cortex. Specifically, it may
be postulated that AA or its metabolites may upregulate the
expression or activity of xanthine dehydrogenase, leading to the
accumulation of its substrates (xanthine) and products (uric acid),
which in turn results in a burst of ROS production and ATP
depletion. This metabolic disruption may further suppress the
expression of critical repair signals like IGF2 through epigenetic
mechanisms, thereby forming a self-reinforcing ‘vicious cycle’ with
persistent inflammation and fibrosis (8,76).
Although this hypothesis still requires direct experimental
validation, studies have suggested the protective efficacy of
targeting this pathway, providing a theoretical foundation for
exploring its therapeutic potential in aristolochic acid
nephropathy (117,118).
Role of IGF2 in CKD
CKD is a progressive disorder that constitutes a
notable global health burden, affecting >10% of the global
population, ~800 million individuals (119). Over the past 2 decades, both its
incidence and mortality have steadily increased, positioning CKD as
one of the leading non-communicable causes of death worldwide
(119). IGF2 serves an important
role in renal development and functional maintenance, and its
dysregulation is increasingly implicated in CKD pathogenesis,
particularly in fibrotic remodeling and tubulointerstitial injury
(53,70,89)
(Fig. 2).
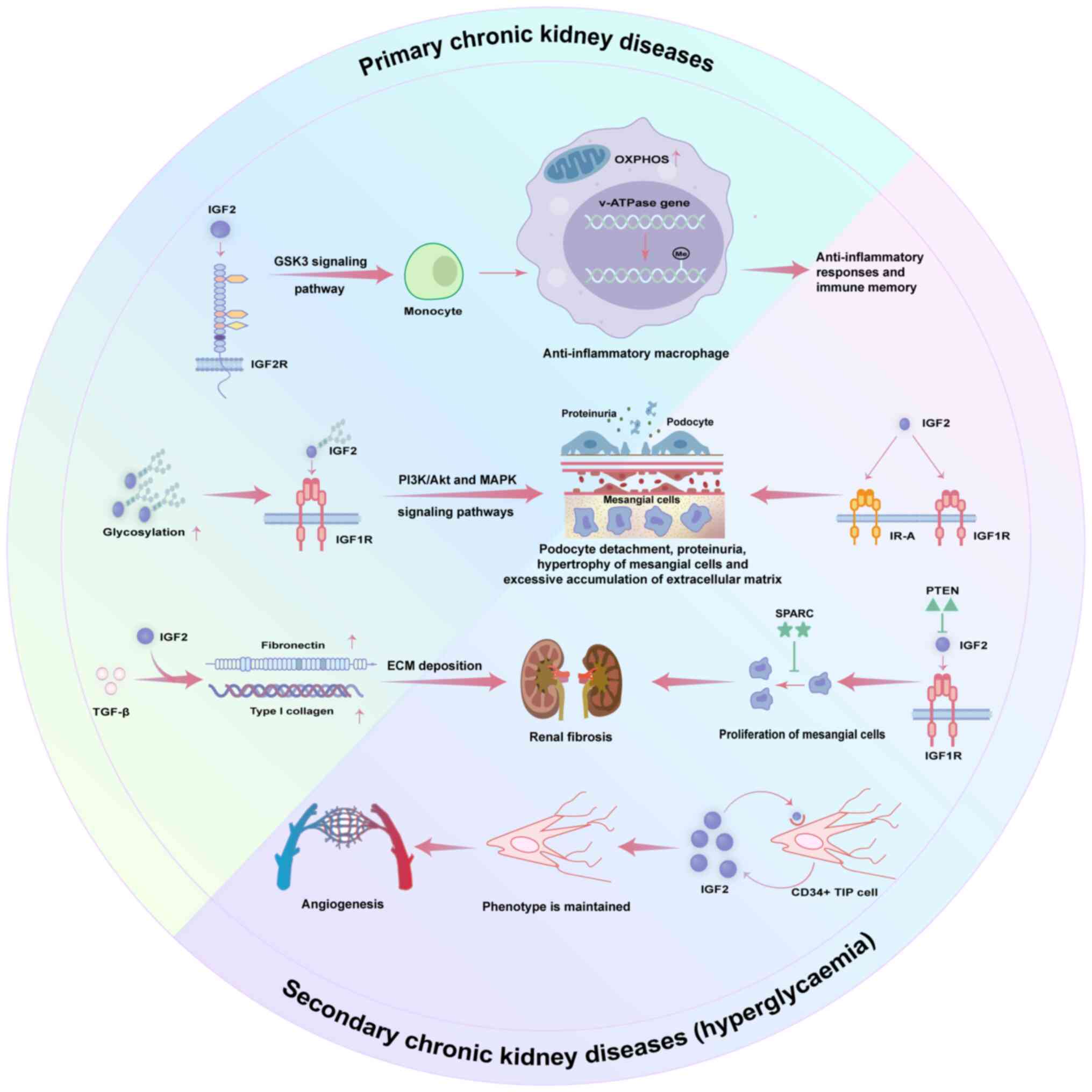 | Figure 2.Role of IGF2-mediated signaling
pathways in CKD. This figure delineates the dual roles of IGF2
signaling in CKD, encompassing both primary and
hyperglycemia-induced secondary mechanisms. In primary CKD, IGF2
binding to IGF1R activates PI3K/Akt and MAPK pathways, promoting
podocyte detachment, mesangial cell hypertrophy and excessive ECM
accumulation, ultimately compromising glomerular filtration barrier
integrity. Furthermore, IGF2 synergizes with TGF-β to upregulate
fibronectin and type I collagen, accelerating renal fibrosis.
Conversely, signaling through IGF2R activates the GSK3 pathway,
inducing anti-inflammatory macrophage differentiation and
modulating mitochondrial metabolism, which may confer protective
effects. In hyperglycemia-induced CKD, IGF2 binding to IGF1R and
IR-A drives pathological mesangial cell proliferation and ECM
accumulation. This process is exacerbated by loss of PTEN-mediated
negative feedback. Concurrently, SPARC is upregulated as a
compensatory repair mechanism. Additionally, hyperglycemia enhances
autocrine IGF2 secretion by CD34+ TIP cells, facilitating phenotype
maintenance and angiogenesis. These findings underscore the
multifaceted role of IGF2 in CKD pathophysiology, highlighting its
potential as a therapeutic target. IGF, insulin-like growth factor;
CKD, chronic kidney disease; IGF1R/IGF2R, IGF1/2 receptor; OXPHOS,
oxidative phosphorylation; v-ATPase, vacuolar-type ATPase; TIP
cell, tip endothelial cell; SPARC, secreted protein acidic and rich
in cysteine; IR-A, insulin receptor isoform A; ECM, extracellular
matrix; PTEN, phosphatase and tensin homolog on chromosome 10. |
Primary CKD
In the context of primary CKD, IGF2 acts as an
important modulator of disease progression by influencing ECM
deposition, tubular dysfunction and fibrosis via activation of the
PI3K/Akt and MAPK signaling pathways (71,89).
Clinical data reveal that serum IGF2 levels are elevated in
patients with chronic renal failure compared with healthy controls,
a finding likely attributable to impaired renal clearance. Notably,
serum IGF2 levels remain unchanged following hemodialysis, whereas
urinary excretion of IGF2 increases, suggesting a compensatory
clearance mechanism mediated by residual nephrons (120). Urinary proteomic analysis has
further identified elevated levels of glycosylated IGF2 in patients
with CKD, with this form exhibiting an inverse association with
estimated GFR, highlighting its potential utility as a biomarker
for disease progression and therapeutic monitoring (121).
Under physiological conditions, IGF2 supports
podocyte survival and cytoskeletal integrity. However, aberrant
IGF2 activation disrupts this homeostasis, leading to podocyte
detachment and glomerular filtration barrier dysfunction. Podocyte
injury thus becomes a central mechanism contributing to proteinuria
and glomerulosclerosis (71,89).
Additionally, IGF2 synergizes with TGF-β1 to enhance expression of
fibrogenic mediators such as type I collagen and fibronectin,
thereby promoting irreversible glomerular and tubulointerstitial
fibrosis (89).
IGF2 also facilitates tubular regeneration during
the early stages of CKD by promoting epithelial cell proliferation
and repair (9). This duality
underscores its context-dependent effects and highlights the
potential of IGF2 as a therapeutic target in CKD, although its
precise roles in fibrotic vs. reparative processes warrant further
exploration.
Persistent low-grade inflammation is a hallmark of
CKD, with macrophage activation and functional plasticity serving a
central role in disease progression (53). A previous study has demonstrated
that IGF2 activates GSK3 signaling through IGF2R, triggering
epigenetic reprogramming of macrophages toward an anti-inflammatory
phenotype (8,76). Specifically, IGF2 suppresses
lysosomal acidification by enhancing DNA methylation of the
vacuolar-type H-ATPase gene, thereby altering macrophage energy
metabolism. This reprogramming favors OXPHOS over glycolysis as the
dominant energy source, which contrasts with the classical Warburg
effect observed in inflammatory macrophages and tumor cells
(122–124). Additionally, this reprogramming
enables macrophages to exert anti-inflammatory responses and
develop immune memory. These findings reinforce the importance of
IGF2 as both a metabolic and immunological regulator in CKD.
Patients with CKD frequently exhibit persistent
low-grade immune activation, involving macrophage immunological
memory and phenotypic plasticity (125–127). IGF2 has been shown to enhance
this form of ‘trained immunity’, enabling macrophages to respond
more effectively to subsequent inflammatory stimuli with an
anti-inflammatory profile (8).
This process is especially relevant in CKD, where it helps
attenuate disease progression by maintaining immune homeostasis and
preventing excessive chronic inflammation (128,129).
Secondary CKD
Diabetic nephropathy (DN) is one of the most common
complications of diabetes mellitus and a notable cause of CKD
worldwide (130). Hallmark
pathological features of DN include renal hypertrophy, mesangial
expansion, excessive accumulation of ECM, tubular atrophy and
interstitial fibrosis (131). A
clinical study has demonstrated that serum levels of IGF2 are
elevated in the early stages of DN, including the microalbuminuria
phase, ~30% higher than in patients with diabetes alone, and are
positively associated with cystatin C and 24-h urinary protein
excretion (132). In advanced
stages marked by overt proteinuria, IGF2 levels further increase
≤1.5-fold compared with those of healthy controls, and show
positive associations with SCr and blood urea nitrogen levels,
indicating a potential role of IGF2 in the development of
glomerulosclerosis and interstitial fibrosis (132,133). While IGF2 serves a physiological
role in angiogenesis and tissue repair, its upregulation under
hyperglycemic conditions contributes to the pathogenesis of DN
(99,132).
IGF2 expression is markedly upregulated in diabetic
kidneys (134). Through
activation of IGF1R and IR-A, IGF2 induces abnormal glomerular cell
proliferation and promotes ECM deposition, resulting in
glomerulosclerosis and compromising glomerular filtration barrier
integrity (135). These changes
not only impair renal structure and function but also accelerate DN
progression (136,137).
In addition to its effects on glomerular cells, IGF2
regulates the balance between renal injury and repair by
interacting with key signaling pathways, notably the phosphatase
and tensin homolog on chromosome 10 (PTEN) and secreted protein
acidic and rich in cysteine (SPARC) pathways. PTEN functions as a
negative regulator of the insulin/IGF signaling pathway and serves
an important role in DN, particularly in the hyperglycemic
microenvironment (137,138). A study has demonstrated that
IGF2, PTEN and SPARC are all upregulated in renal biopsies from
patients with early-stage DN (137). When PTEN-mediated negative
feedback is impaired, IGF2 activity increases, exacerbating
pathological mesangial cell proliferation, ECM accumulation and
renal fibrosis (136). This
mechanism has been corroborated in streptozotocin-induced diabetic
rat models (139,140).
SPARC, a matricellular protein involved in tissue
remodeling and repair, is also modulated in DN. In DN, SPARC
inhibits mesangial cell hypertrophy. While its upregulation may
reflect a compensatory response to hyperglycemic injury, decreased
SPARC expression is associated with impaired renal repair
mechanisms, thereby worsening disease progression (141,142). Thus, in DN, IGF2 modulates renal
injury and repair via interactions with PTEN and SPARC,
highlighting a dual regulatory axis in disease pathophysiology.
IGF2 serves an important role in angiogenesis. In
a vitro study using CD34+ tip endothelial cell (TIP
cell) models has revealed a strong association between IGF2
expression and both the number and angiogenic capacity of these
cells. IGF2 knockdown reduces CD34+ TIP cell numbers and
inhibits vascular sprouting, supporting its role in angiogenic
regulation. These findings also suggest that IGF2 may sustain the
TIP cell phenotype via autocrine signaling (143). Therefore, targeted inhibition of
IGF2 signaling may offer a promising therapeutic strategy for
delaying DN progression by disrupting aberrant angiogenesis and
fibrotic remodeling.
Role of IGF2 in renal tumors
Renal cell carcinoma (RCC)
RCC is a prevalent renal malignancy characterized by
complex genetic and epigenetic alterations. One of the notable
molecular changes implicated in RCC is the upregulation of IGF2,
particularly through loss of imprinting (LOI), which results in
biallelic expression of the IGF2 gene. This aberrant expression
contributes to tumor initiation and progression by activating
multiple oncogenic signaling pathways (10).
A key regulatory mechanism involves the long
non-coding RNA HOXA transcript at the distal tip, which acts as a
competing endogenous RNA by sponging microRNA-615, thereby
relieving repression of IGF2 and leading to its upregulation. This
promotes renal cancer cell proliferation, migration and invasion
(144). Within the tumor
microenvironment, IGF2 interacts with IGF1R and IR-A to activate
the PI3K/Akt and MAPK pathways, which enhance tumor cell survival,
proliferation and motility (26,27).
The oncogenic potential of IGF2 is markedly enhanced in tumors with
high IR-A expression, underscoring the importance of receptor
context in determining tumor behavior (17).
Preclinical models further support the role of IGF2
in RCC progression. In mouse models, IGF2 overexpression has been
shown to accelerate tumor cell proliferation and metastasis,
suggesting that therapeutic strategies aimed at inhibiting IGF2
signaling or blocking receptor interactions may hold promise for
targeted antitumor therapy in RCC (145–148).
Wilms' tumor
Wilms' tumor, or nephroblastoma, is a pediatric
embryonal renal neoplasm arising from undifferentiated renal
precursor cells. It is associated with epigenetic dysregulation of
the IGF2 gene, particularly the loss of maternal imprinting, which
leads to biallelic IGF2 expression and abnormal cellular
proliferation (149).
During normal fetal kidney development, IGF2 is
expressed exclusively from the paternal allele. All four known IGF2
promoters (P1-P4) maintain monoallelic expression in early
development; however, imprinting at the P1 promoter begins to relax
between gestational weeks 15–17 (150). In Wilms' tumor, this relaxation
is exacerbated, ultimately resulting in LOI at all promoters and
widespread upregulation of IGF2 (150).
In addition to LOI, mutations in tripartite motif
containing 28 (TRIM28), a transcriptional co-repressor and
epigenetic regulator, have been implicated in IGF2 dysregulation.
Inactivating mutations in TRIM28 are frequently associated with
biallelic IGF2 expression, especially in tumors lacking other
common genetic alterations, suggesting that TRIM28 may drive
tumorigenesis via an independent IGF2-related mechanism (151).
Ethnic differences have been observed in the
prevalence of IGF2 imprinting abnormalities. For example, a study
has shown that LOI of IGF2 is more frequent in Wilms' tumor cases
among white children of Europe, North and South America and Oceania
compared with Japanese children, potentially reflecting
population-specific genetic or epigenetic susceptibility (152).
Collectively, the loss of IGF2 imprinting and TRIM28
mutations represent important events in the pathogenesis of Wilms'
tumor. These insights provide a compelling theoretical framework
for the development of IGF2-targeted therapies in pediatric renal
malignancies.
Therapeutic potential and treatments
targeting IGF2
As an important regulator of cell proliferation,
metabolic reprogramming and angiogenesis, IGF2 has emerged as a
promising therapeutic target, with encouraging progress reported in
both preclinical and clinical studies (153–155).
In a preclinical model, the natural compound
curcumin has demonstrated potent antitumor effects in bladder
cancer by transcriptionally suppressing IGF2, blocking IGF1R/IR
substrate 1 phosphorylation and inhibiting the downstream AKT/mTOR
signaling cascade. These inhibitory effects are reversed by
exogenous IGF2 administration or IGF1R overexpression, highlighting
the notable role of IGF2 signaling in tumor progression and
validating its therapeutic relevance (156).
Clinically, monoclonal antibodies targeting IGF1R,
such as dalotuzumab, and tyrosine kinase inhibitors, such as
linsitinib, have shown therapeutic efficacy in select sarcoma
subtypes (153). However, these
agents have shown limited benefit in breast cancer and non-small
cell lung cancer, reflecting the context-dependent nature of IGF
signaling and the heterogeneity of downstream pathway activation
(157). To overcome compensatory
mechanisms, dual-ligand neutralizing antibodies, neutralizing
antibodies with different targets have been developed. These
include dusigitumab (154), which
targets the IGF-1 receptor to block signaling from both IGF-1 and
IGF-2, and xentuzumab (155), a
dual-IGF-1/IGF-2 ligand-neutralizing antibody that directly binds
the ligands themselves. While conceptually promising, their
clinical utility has been constrained by the absence of robust
predictive biomarkers, which remains a notable barrier to broader
application (158).
In pediatric oncology, Wilms' tumor represents a
paradigm for IGF2-driven tumorigenesis (159). Using genetically engineered mouse
models harboring Wilms' tumor protein deletion and IGF2
overexpression, researchers have successfully recapitulated key
features of human WT, including impaired mesenchymal
differentiation and persistent ERK1/2 activation. These models
provide a robust platform for exploring therapeutic interventions
targeting the IGF axis (160).
With respect to angiogenesis, truncated IGF2
variants such as Des(1–6)IGF2 have been shown to promote
endothelial cell migration, tube formation and intra-ovarian
neovascularization by upregulating IL-6, urokinase plasminogen
activator surface receptor and CCL2. Notably, Des(1–6)IGF2 evades
inhibition by IGFBP-6, unlike Leu27IGF2, which preferentially binds
IGF2R and exhibits minimal angiogenic activity. These findings
implicate IGF1R and IR-A as dominant mediators of the
pro-angiogenic effects of IGF2 in pathological neovascularization
(161).
Despite these promising advances from above
experimental and mechanistic studies, several key challenges
persist. Ligand redundancy, tissue-specific pathway activation and
the lack of reliable biomarkers continue to impede precision
targeting of the IGF2 axis (76,162). Future research should focus on
developing IGF2-specific inhibitors, optimizing rational
combination therapies and identifying context-specific biomarkers,
particularly in diseases such as Wilms' tumor and
epigenetically-deregulated renal disorders, where IGF2 signaling is
pathologically upregulated.
Concluding remarks and future
perspectives
In previous years, with in-depth studies on the
biological role of IGF2 and its regulatory mechanisms, the multiple
roles of IGF2 in renal development and disease have been revealed.
Under normal physiological conditions, IGF2 serves an important
role in renal tubular and glomerular formation through precise gene
imprinting regulation (24,70).
However, under pathological conditions, aberrant expression of IGF2
accelerates the progression of renal fibrosis, DN and renal tumors
by activating specific signaling pathways (2,51,136).
Although IGF2 has been validated as a potential
biomarker and therapeutic target in renal diseases, a number of
mechanistic questions remain unresolved. Future investigations
should examine: i) The molecular interplay between IGF2 and
regulatory factors such as IGFBPs, TGF-β and VEGF; ii) the spatial
and temporal dynamics of receptor activation; and iii) the
differential roles of IGF2 in various pathological states.
Additionally, the design of IGF2-specific therapies is expected to
provide novel strategies for the treatment of kidney disease and
associated metabolic disorders.
In conclusion, IGF2 serves an increasingly
recognized role in renal biology and pathogenesis. A deeper
understanding of its regulatory mechanisms will facilitate the
development of precise diagnostic tools and targeted interventions
for renal diseases.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 82470773), Guangdong Provincial
Science and Technology Planning Project (grant no. 2023B110009),
Funding for Innovation Capacity Building Project of Guangdong
Provincial Research Institutes (grant no. KD032024001), and the
Guangdong Provincial Major Science and Technology Special Project
(grant no. KS012023323).
Availability of data and materials
Not applicable.
Authors' contributions
YS was responsible for conceptualization,
investigation, methodology, visualization, writing the original
draft, and reviewing and editing the manuscript. WHa acquired
funding, and contributed to the methodology, supervision, and
reviewing and editing of the manuscript. WL contributed to the
methodology and supervision, and reviewed and edited the
manuscript. WHu was responsible for conceptualization, the
acquisition of funding and resources, and contributed to
supervision, validation, writing the original draft, and reviewing
and editing the manuscript. Data authentication is not applicable.
All authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Efstratiadis A: Genetics of mouse growth.
Int J Dev Biol. 42:955–976. 1998.PubMed/NCBI
|
|
2
|
Eggenschwiler J, Ludwig T, Fisher P,
Leighton PA, Tilghman SM and Efstratiadis A: Mouse mutant embryos
overexpressing IGF-II exhibit phenotypic features of the
beckwith-wiedemann and simpson-golabi-behmel syndromes. Genes Dev.
11:3128–3142. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
St-Pierre J, Hivert MF, Perron P, Poirier
P, Guay SP, Brisson D and Bouchard L: IGF2 DNA methylation is a
modulator of newborn's fetal growth and development. Epigenetics.
7:1125–1132. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hoyo C, Fortner K, Murtha AP, Schildkraut
JM, Soubry A, Demark-Wahnefried W, Jirtle RL, Kurtzberg J, Forman
MR, Overcash F, et al: Association of cord blood methylation
fractions at imprinted insulin-like growth factor 2 (IGF2), plasma
IGF2, and birth weight. Cancer Causes Control. 23:635–645.
2012.PubMed/NCBI
|
|
5
|
Chin E and Bondy C: Insulin-like growth
factor system gene expression in the human kidney. J Clin
Endocrinol Metab. 75:962–968. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sferruzzi-Perri AN, Sandovici I,
Constancia M and Fowden AL: Placental phenotype and the
insulin-like growth factors: Resource allocation to fetal growth. J
Physiol. 595:5057–5093. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kuang X: Study of the relationship between
WT1 and IGF2 DNA methylation and kidney development and function in
intrauterine growth retardation rat. PhD dissertation (Shanghai).
Fudan University. 2012.(In Chinese).
|
|
8
|
Du L, Lin L, Li Q, Liu K, Huang Y, Wang X,
Cao K, Chen X, Cao W, Li F, et al: IGF-2 preprograms maturing
macrophages to acquire oxidative phosphorylation-dependent
anti-inflammatory properties. Cell Metab. 29:1363–1375.e8. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Livingstone C and Borai A: Insulin-like
growth factor-II: Its role in metabolic and endocrine disease. Clin
Endocrinol (Oxf). 80:773–781. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Matsumoto T, Kinoshita E, Maeda H, Niikawa
N, Kurosaki N, Harada N, Yun K, Sawai T, Aoki S, Kondoh T, et al:
Molecular analysis of a patient with beckwith-wiedemann syndrome,
rhabdomyosarcoma and renal cell carcinoma. Jpn J Hum Genet.
39:225–234. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
LeRoith D, Werner H, Beitner-Johnson D and
Roberts CT Jr: Molecular and cellular aspects of the insulin-like
growth factor I receptor. Endocr Rev. 16:143–163. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baserga R, Hongo A, Rubini M, Prisco M and
Valentinis B: The IGF-I receptor in cell growth, transformation and
apoptosis. Biochim Biophys Acta. 1332:F105–F126. 1997.PubMed/NCBI
|
|
13
|
Burvin R, LeRoith D, Harel H, Zloczower M,
Marbach M and Karnieli E: The effect of acute insulin-like growth
factor-II administration on glucose metabolism in the rat. Growth
Horm IGF Res. 8:205–210. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Salmon WD Jr and Daughaday WH: A
hormonally controlled serum factor which stimulates sulfate
incorporation by cartilage in vitro. J Lab Clin Med. 49:825–836.
1957.PubMed/NCBI
|
|
15
|
Engström W, Shokrai A, Otte K, Granérus M,
Gessbo A, Bierke P, Madej A, Sjölund M and Ward A: Transcriptional
regulation and biological significance of the insulin like growth
factor II gene. Cell Prolif. 31:173–189. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vu TH and Hoffman AR: Promoter-specific
imprinting of the human insulin-like growth factor-II gene. Nature.
371:714–717. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Belfiore A, Malaguarnera R, Vella V,
Lawrence MC, Sciacca L, Frasca F, Morrione A and Vigneri R: Insulin
receptor isoforms in physiology and disease: An updated view.
Endocr Rev. 38:379–431. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Constância M, Hemberger M, Hughes J, Dean
W, Ferguson-Smith A, Fundele R, Stewart F, Kelsey G, Fowden A,
Sibley C and Reik W: Placental-specific IGF-II is a major modulator
of placental and fetal growth. Nature. 417:945–948. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chao W and D'Amore PA: IGF2: Epigenetic
regulation and role in development and disease. Cytokine Growth
Factor Rev. 19:111–120. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Issa JP, Vertino PM, Boehm CD, Newsham IF
and Baylin SB: Switch from monoallelic to biallelic human IGF2
promoter methylation during aging and carcinogenesis. Proc Natl
Acad Sci USA. 93:11757–11762. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhan S, Shapiro D, Zhan S, Zhang L,
Hirschfeld S, Elassal J and Helman LJ: Concordant loss of
imprinting of the human insulin-like growth factor II gene
promoters in cancer. J Biol Chem. 270:27983–27986. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
van Dijk MA, van Schaik FM, Bootsma HJ,
Holthuizen P and Sussenbach JS: Initial characterization of the
four promoters of the human insulin-like growth factor II gene. Mol
Cell Endocrinol. 81:81–94. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Z, Huang S, Tian N, Xu Q, Zhan X,
Peng F, Wang X, Su N, Feng X, Tang X, et al: Association of the
remnant cholesterol to high-density lipoprotein cholesterol ratio
with mortality in peritoneal dialysis patients. Lipids Health Dis.
24:1072025. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Morali OG, Jouneau A, McLaughlin KJ,
Thiery JP and Larue L: IGF-II promotes mesoderm formation. Dev
Biol. 227:133–145. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pardo M, Cheng Y, Sitbon YH, Lowell JA,
Grieco SF, Worthen RJ, Desse S and Barreda-Diaz A: Insulin growth
factor 2 (IGF2) as an emergent target in psychiatric and
neurological disorders. Review. Neurosci Res. 149:1–13. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Baserga R: The IGF-I receptor in cancer
research. Exp Cell Res. 253:1–6. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wojtalla A, Salm F, Christiansen DG,
Cremona T, Cwiek P, Shalaby T, Gross N, Grotzer MA and Arcaro A:
Novel agents targeting the IGF-1R/PI3K pathway impair cell
proliferation and survival in subsets of medulloblastoma and
neuroblastoma. PLoS One. 7:e471092012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vigneri PG, Tirrò E, Pennisi MS, Massimino
M, Stella S, Romano C and Manzella L: The insulin/IGF system in
colorectal cancer development and resistance to therapy. Front
Oncol. 5:2302015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vella V, Pandini G, Sciacca L, Mineo R,
Vigneri R, Pezzino V and Belfiore A: A novel autocrine loop
involving IGF-II and the insulin receptor isoform-A stimulates
growth of thyroid cancer. J Clin Endocrinol Metab. 87:245–254.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Belfiore A, Frasca F, Pandini G, Sciacca L
and Vigneri R: Insulin receptor isoforms and insulin
receptor/insulin-like growth factor receptor hybrids in physiology
and disease. Endocr Rev. 30:586–623. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Andersen M, Nørgaard-Pedersen D, Brandt J,
Pettersson I and Slaaby R: IGF1 and IGF2 specificities to the two
insulin receptor isoforms are determined by insulin receptor amino
acid 718. PLoS One. 12:e01788852017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Louvi A, Accili D and Efstratiadis A:
Growth-promoting interaction of IGF-II with the insulin receptor
during mouse embryonic development. Dev Biol. 189:33–48. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fukuda I, Hizuka N, Ishikawa Y, Yasumoto
K, Murakami Y, Sata A, Morita J, Kurimoto M, Okubo Y and Takano K:
Clinical features of insulin-like growth factor-II producing
non-islet-cell tumor hypoglycemia. Growth Horm IGF Res. 16:211–216.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Khowaja A, Johnson-Rabbett B, Bantle J and
Moheet A: Hypoglycemia mediated by paraneoplastic production of
insulin like growth factor-2 from a malignant renal solitary
fibrous tumor-clinical case and literature review. BMC Endocr
Disord. 14:492014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rikhof B, Van Den Berg G and Van Der Graaf
WTA: Non-islet cell tumour hypoglycaemia in a patient with a
gastrointestinal stromal tumour. Acta Oncol. 44:764–766. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zapf J: Role of insulin-like growth factor
II and IGF binding proteins in extrapancreatic tumor hypoglycemia.
Horm Res. 42:20–26. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zapf J, Futo E, Peter M and Froesch ER:
Can ‘big’ insulin-like growth factor II in serum of tumor patients
account for the development of extrapancreatic tumor hypoglycemia?
J Clin Invest. 90:2574–2584. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dynkevich Y, Rother KI, Whitford I,
Qureshi S, Galiveeti S, Szulc AL, Danoff A, Breen TL, Kaviani N,
Shanik MH, et al: Tumors, IGF-2, and hypoglycemia: insights from
the clinic, the laboratory, and the historical archive. Endocr Rev.
34:798–826. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wu XH, Wang LH, Song JF, Wang WP and Shan
BE: Effect of IGF-II stimulation on the expression of matrix
metalloproteinase-9 in human ovarian cancer cell line SKOV3. Prog
Obstet Gynecol. 12:413–415. 2003.(In Chinese).
|
|
40
|
Han Y, Cui J, Tao J, Guo L, Guo P, Sun M,
Kang J, Zhang X, Yan C and Li S: CREG inhibits migration of human
vascular smooth muscle cells by mediating IGF-II endocytosis. Exp
Cell Res. 315:3301–3311. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhu M, Zhuang J, Li Z, Liu Q, Zhao R, Gao
Z, Midgley AC, Qi T, Tian J, Zhang Z, et al:
Machine-learning-assisted single-vessel analysis of nanoparticle
permeability in tumour vasculatures. Nat Nanotechnol. 18:657–666.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou L, Liu Y, Xu T, Dong L, Yang X and
Wang C: Malignant solitary fibrous tumor of the kidney with IGF2
secretion and without hypoglycemia. World J Surg Oncol. 22:1792024.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sciacca L, Prisco M, Wu A, Belfiore A,
Vigneri R and Baserga R: Signaling differences from the A and B
isoforms of the insulin receptor (IR) in 32D cells in the presence
or absence of IR substrate-1. Endocrinology. 144:2650–2658. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Escribano O, Gómez-Hernández A,
Díaz-Castroverde S, Nevado C, García G, Otero YF, Perdomo L, Beneit
N and Benito M: Insulin receptor isoform A confers a higher
proliferative capability to pancreatic beta cells enabling glucose
availability and IGF-I signaling. Mol Cell Endocrinol. 409:82–91.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ghosh P, Dahms NM and Kornfeld S: Mannose
6-phosphate receptors: New twists in the tale. Nat Rev Mol Cell
Biol. 4:202–212. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
El-Shewy HM and Luttrell LM: Insulin-like
growth factor-2/mannose-6 phosphate receptors. Vitam Horm.
80:667–697. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang Y, MacDonald RG, Thinakaran G and Kar
S: Insulin-like growth factor-II/cation-independent mannose
6-phosphate receptor in neurodegenerative diseases. Mol Neurobiol.
54:2636–2658. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nadimpalli SK and Amancha PK: Evolution of
mannose 6-phosphate receptors (MPR300 and 46): Lysosomal enzyme
sorting proteins. Curr Protein Pept Sci. 11:68–90. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Massagué J: The transforming growth
factor-beta family. Annu Rev Cell Biol. 6:597–641. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Leksa V, Godar S, Schiller HB, Fuertbauer
E, Muhammad A, Slezakova K, Horejsi V, Steinlein P, Weidle UH,
Binder BR and Stockinger H: TGF-beta-induced apoptosis in
endothelial cells mediated by M6P/IGFII-R and mini-plasminogen. J
Cell Sci. 118:4577–4586. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Livingstone C: IGF2 and cancer. Endocr
Relat Cancer. 20:R321–R339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sélénou C, Brioude F, Giabicani E, Sobrier
ML and Netchine I: IGF2: Development, genetic and epigenetic
abnormalities. Cells. 11:18862022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhu Y, Chen L and Song B, Cui Z, Chen G,
Yu Z and Song B: Insulin-like growth factor-2 (IGF-2) in fibrosis.
Biomolecules. 12:15572022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wolf E, Hoeflich A and Lahm H: What is the
function of IGF-II in postnatal life? Answers from transgenic mouse
models. Growth Horm IGF Res. 8:185–193. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Barroca V, Lewandowski D, Jaracz-Ros A and
Hardouin SN: Paternal insulin-like growth factor 2 (Igf2) regulates
stem cell activity during adulthood. eBioMedicine. 15:150–162.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang MJ, Chen F, Liu QG, Liu CC, Yao H, Yu
B, Zhang HB, Yan HX, Ye Y, Chen T, et al: Insulin-like growth
factor 2 is a key mitogen driving liver repopulation in mice. Cell
Death Dis. 9:262018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wolf E, Kramer R, Blum WF, Föll J and Brem
G: Consequences of postnatally elevated insulin-like growth
factor-II in transgenic mice: Endocrine changes and effects on body
and organ growth. Endocrinology. 135:1877–1886. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Blackburn A, Schmitt A, Schmidt P, Wanke
R, Hermanns W, Brem G and Wolf E: Actions and interactions of
growth hormone and insulin-like growth factor-II: Body and organ
growth of transgenic mice. Transgenic Res. 6:213–222. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Avner ED and Sweeney WE Jr: Polypeptide
growth factors in metanephric growth and segmental nephron
differentiation. Pediatr Nephrol. 4:372–377. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jelinic P and Shaw P: Loss of imprinting
and cancer. J Pathol. 211:261–268. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sun FL, Dean WL, Kelsey G, Allen ND and
Reik W: Transactivation of Igf2 in a mouse model of
beckwith-wiedemann syndrome. Nature. 389:809–815. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lui JC, Finkielstain GP, Barnes KM and
Baron J: An imprinted gene network that controls mammalian somatic
growth is down-regulated during postnatal growth deceleration in
multiple organs. Am J Physiol Regul Integr Comp Physiol.
295:R189–R196. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Hirschberg R and Adler S: Insulin-like
growth factor system and the kidney: Physiology, pathophysiology,
and therapeutic implications. Am J Kidney Dis. 31:901–919. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Svensson J, Tivesten A, Sjögren K,
Isaksson O, Bergström G, Mohan S, Mölne J, Isgaard J and Ohlsson C:
Liver-derived IGF-I regulates kidney size, sodium reabsorption, and
renal IGF-II expression. J Endocrinol. 193:359–366. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Feld S and Hirschberg R: Growth hormone,
the insulin-like growth factor system, and the kidney. Endocr Rev.
17:423–480. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Moerth C, Schneider MR, Renner-Mueller I,
Blutke A, Elmlinger MW, Erben RG, Camacho-Hübner C, Hoeflich A and
Wolf E: Postnatally elevated levels of insulin-like growth factor
(IGF)-II fail to rescue the dwarfism of IGF-I-deficient mice except
kidney weight. Endocrinology. 148:441–451. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
LeRoith D, Holly JMP and Forbes BE:
Insulin-like growth factors: Ligands, binding proteins, and
receptors. Mol Metab. 52:1012452021. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Rabkin R and Schaefer F: New concepts:
Growth hormone, insulin-like growth factor-I and the kidney. Growth
Horm IGF Res. 14:270–276. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kie JH, Kapturczak MH, Traylor A, Agarwal
A and Hill-Kapturczak N: Heme oxygenase-1 deficiency promotes
epithelial-mesenchymal transition and renal fibrosis. J Am Soc
Nephrol. 19:1681–1691. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Hale LJ, Welsh GI, Perks CM, Hurcombe JA,
Moore S, Hers I, Saleem MA, Mathieson PW, Murphy AJ, Jeansson M, et
al: Insulin-like growth factor-II is produced by, signals to and is
an important survival factor for the mature podocyte in man and
mouse. J Pathol. 230:95–106. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Grotendorst GR, Rahmanie H and Duncan MR:
Combinatorial signaling pathways determine fibroblast proliferation
and myofibroblast differentiation. FASEB J. 18:469–479. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Elchebly M, Payette P, Michaliszyn E,
Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J,
Chan CC, et al: Increased insulin sensitivity and obesity
resistance in mice lacking the protein tyrosine phosphatase-1B
gene. Science. 283:1544–1548. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Daza-Arnedo R, Rico-Fontalvo J,
Aroca-Martínez G, Rodríguez-Yanez T, Martínez-Ávila MC,
Almanza-Hurtado A, Cardona-Blanco M, Henao-Velásquez C,
Fernández-Franco J, Unigarro-Palacios M, et al: Insulin and the
kidneys: A contemporary view on the molecular basis. Front Nephrol.
3:11333522023. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Singh S, Sharma R, Kumari M and Tiwari S:
Insulin receptors in the kidneys in health and disease. World J
Nephrol. 8:11–22. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Enguita-Germán M and Fortes P: Targeting
the insulin-like growth factor pathway in hepatocellular carcinoma.
World J Hepatol. 6:716–737. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Wang X, Lin L, Lan B, Wang Y, Du L, Chen
X, Li Q, Liu K, Hu M, Xue Y, et al: IGF2R-initiated proton
rechanneling dictates an anti-inflammatory property in macrophages.
Sci Adv. 6:eabb73892020. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Rainier S, Dobry CJ and Feinberg AP: Loss
of imprinting in hepatoblastoma. Cancer Res. 55:1836–1838.
1995.PubMed/NCBI
|
|
78
|
Honda S, Arai Y, Haruta M, Sasaki F, Ohira
M, Yamaoka H, Horie H, Nakagawara A, Hiyama E, Todo S and Kaneko Y:
Loss of imprinting of IGF2 correlates with hypermethylation of the
H19 differentially methylated region in hepatoblastoma. Br J
Cancer. 99:1891–1899. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Bell AC and Felsenfeld G: Methylation of a
CTCF-dependent boundary controls imprinted expression of the Igf2
gene. Nature. 405:482–485. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Thorvaldsen JL, Duran KL and Bartolomei
MS: Deletion of the H19 differentially methylated domain results in
loss of imprinted expression of H19 and Igf2. Genes Dev.
12:3693–3702. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Tremblay KD, Saam JR, Ingram RS, Tilghman
SM and Bartolomei MS: A paternal-specific methylation imprint marks
the alleles of the mouse H19 gene. Nat Genet. 9:407–413. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Chang S and Bartolomei MS: Modeling human
epigenetic disorders in mice: Beckwith-wiedemann syndrome and
silver-russell syndrome. Dis Model Mech. 13:dmm0441232020.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Han L, Lee DH and Szabó PE: CTCF is the
master organizer of domain-wide allele-specific chromatin at the
H19/Igf2 imprinted region. Mol Cell Biol. 28:1124–1135. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Matsuzaki H, Kuramochi D, Okamura E,
Hirakawa K, Ushiki A and Tanimoto K: Recapitulation of gametic DNA
methylation and its post-fertilization maintenance with reassembled
DNA elements at the mouse Igf2/H19 locus. Epigenetics Chromatin.
13:22020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Leighton PA, Saam JR, Ingram RS, Stewart
CL and Tilghman SM: An enhancer deletion affects both H19 and Igf2
expression. Genes Dev. 9:2079–2089. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Neirijnck Y, Papaioannou MD and Nef S: The
insulin/IGF system in mammalian sexual development and
reproduction. Int J Mol Sci. 20:44402019. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Huang SCC, Smith AM, Everts B, Colonna M,
Pearce EL, Schilling JD and Pearce EJ: Metabolic reprogramming
mediated by the mTORC2-IRF4 signaling axis is essential for
macrophage alternative activation. Immunity. 45:817–830. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Bridgewater DJ, Ho J, Sauro V and Matsell
DG: Insulin-like growth factors inhibit podocyte apoptosis through
the PI3 kinase pathway. Kidney Int. 67:1308–1314. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Tziastoudi M, Theoharides TC, Nikolaou E,
Efthymiadi M, Eleftheriadis T and Stefanidis I: Key genetic
components of fibrosis in diabetic nephropathy: An updated
systematic review and meta-analysis. Int J Mol Sci. 23:153312022.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Liao J, Song S, Gusscott S, Fu Z,
VanderKolk I, Busscher BM, Lau KH, Brind'Amour J and Szabó PE:
Establishment of paternal methylation imprint at the H19/Igf2
imprinting control region. Sci Adv. 9:eadi20502023. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Levey AS and James MT: Acute kidney
injury. Ann Intern Med. 167:ITC66–ITC80. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zhan Z, Chen J, Zhou H, Hong X, Li L, Qin
X, Fu H and Liu Y: Chronic alcohol consumption aggravates acute
kidney injury through integrin β1/JNK signaling. Redox Biol.
77:1033862024. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Yamamoto R, Li Q, Otsuki N, Shinzawa M,
Yamaguchi M, Wakasugi M, Nagasawa Y and Isaka Y: A dose-dependent
association between alcohol consumption and incidence of
proteinuria and low glomerular filtration rate: A systematic review
and meta-analysis of cohort studies. Nutrients. 15:15922023.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Orth SR: Smoking and the kidney. J Am Soc
Nephrol. 13:1663–1672. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Chen Q and Ou L: Meta-analysis of the
association between the dietary inflammatory index and risk of
chronic kidney disease. Eur J Clin Nutr. 79:7–14. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Danziger J, Chen KP, Lee J, Feng M, Mark
RG, Celi LA and Mukamal KJ: Obesity, acute kidney injury, and
mortality in critical illness. Crit Care Med. 44:328–334. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Tan X, Tao Q, Yin S, Fu G, Wang C, Xiang
F, Hu H, Zhang S, Wang Z and Li D: A single administration of FGF2
after renal ischemia-reperfusion injury alleviates post-injury
interstitial fibrosis. Nephrol Dial Transplant. 38:2537–2549. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Tsao T, Wang J, Fervenza FC, Vu TH, Jin
IH, Hoffman AR and Rabkin R: Renal growth hormone-insulin-like
growth factor-I system in acute renal failure. Kidney Int.
47:1658–1668. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Zhang L, Ma X and Wang JQ: Research
progress on the correlation between insulin-like growth factor II,
its binding protein 2 and diabetic nephropathy. Clin Med China.
32:954–957. 2016.(In Chinese).
|
|
100
|
Kashani K, Al-Khafaji A, Ardiles T,
Artigas A, Bagshaw SM, Bell M, Bihorac A, Birkhahn R, Cely CM,
Chawla LS, et al: Discovery and validation of cell cycle arrest
biomarkers in human acute kidney injury. Crit Care. 17:R252013.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Livingston MJ, Shu S, Fan Y, Li Z, Jiao Q,
Yin XM, Venkatachalam MA and Dong Z: Tubular cells produce FGF2 via
autophagy after acute kidney injury leading to fibroblast
activation and renal fibrosis. Autophagy. 19:256–277. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Das S, Thakur S, Korenjak M, Sidorenko VS,
Chung FFL and Zavadil J: Aristolochic acid-associated cancers: A
public health risk in need of global action. Nat Rev Cancer.
22:576–591. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Vanherweghem JL, Depierreux M, Tielemans
C, Abramowicz D, Dratwa M, Jadoul M, Richard C, Vandervelde D,
Verbeelen D, Vanhaelen-Fastre R, et al: Rapidly progressive
interstitial renal fibrosis in young women: Association with
slimming regimen including Chinese herbs. Lancet. 341:387–391.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Lebeau C, Arlt VM, Schmeiser HH, Boom A,
Verroust PJ, Devuyst O and Beauwens R: Aristolochic acid impedes
endocytosis and induces DNA adducts in proximal tubule cells.
Kidney Int. 60:1332–1342. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Wen YJ, Qu L and Li XM: Ischemic injury
underlies the pathogenesis of aristolochic acid-induced acute
kidney injury. Transl Res. 152:38–46. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Komatsu M, Funakoshi T, Aki T and Unuma K:
Aristolochic acid-induced DNA adduct formation triggers acute DNA
damage response in rat kidney proximal tubular cells. Toxicol Lett.
406:1–8. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Chen J, Li H, Bai Y, Luo P, Cheng G, Ding
Z, Xu Z, Gu L, Wong L, Pang X, et al: Spatially resolved
multi-omics unravels region-specific responses, microenvironment
remodeling and metabolic reprogramming in aristolochic acid
nephropathy. Innov Med. 2:1000662024. View Article : Google Scholar
|
|
108
|
Ratnayake D, Nguyen PD, Rossello FJ,
Wimmer VC, Tan JL, Galvis LA, Julier Z, Wood AJ, Boudier T, Isiaku
AI, et al: Macrophages provide a transient muscle stem cell niche
via NAMPT secretion. Nature. 591:281–287. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Li H, Li P, Li S, Zhang X, Dong X and Yang
M: Mechanism of transforming growth factor- β1 induce renal
fibrosis based on transcriptome sequencing analysis. J Zhejiang
Univ (Med Sci). 52:594–604. 2023.(In Chinese). View Article : Google Scholar
|
|
110
|
Chen J, Luo P, Wang C, Yang C, Bai Y, He
X, Zhang Q, Zhang J, Yang J, Wang S and Wang J: Integrated
single-cell transcriptomics and proteomics reveal cellular-specific
responses and microenvironment remodeling in aristolochic acid
nephropathy. JCI Insight. 7:e1573602022. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Youl ENH, Husson C, El Khattabi C, El Mere
S, Declèves AE, Pochet S, Nortier J and Antoine MH:
Characterization of cytotoxic effects of aristolochic acids on the
vascular endothelium. Toxicol In Vitro. 65:1048112020. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Yamamoto S, Yamamoto M, Nakamura J, Mii A,
Yamamoto S, Takahashi M, Kaneko K, Uchino E, Sato Y, Fukuma S, et
al: Spatiotemporal ATP dynamics during AKI predict renal prognosis.
J Am Soc Nephrol. 31:2855–2869. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Furuhashi M: New insights into purine
metabolism in metabolic diseases: Role of xanthine oxidoreductase
activity. Am J Physiol Endocrinol Metab. 319:E827–E834. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Johnson TA, Jinnah HA and Kamatani N:
Shortage of cellular ATP as a cause of diseases and strategies to
enhance ATP. Front Pharmacol. 10:982019. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Dissanayake LV, Zietara A, Levchenko V,
Spires DR, Burgos Angulo M, El-Meanawy A, Geurts AM, Dwinell MR,
Palygin O and Staruschenko A: Lack of xanthine dehydrogenase leads
to a remarkable renal decline in a novel hypouricemic rat model.
iscience. 25:1048872022. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Chung HY, Baek BS, Song SH, Kim MS, Huh
JI, Shim KH, Kim KW and Lee KH: Xanthine dehydrogenase/ xanthine
oxidase and oxidative stress. Age (Omaha). 20:127–140. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Dong Y, Chen S, He H, Sun ZR, Jiang LX, Gu
YQ, Zhang Y, Feng F, Chen C, Fan ZC, et al: Skullcapflavone II, a
novel NQO1 inhibitor, alleviates aristolochic acid I-induced liver
and kidney injury in mice. Acta Pharmacol Sin. 44:1429–1441. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Li C, Wang X, Bi Y, Yu H, Wei J and Zhang
Y, Han L and Zhang Y: Potent inhibitors of organic anion
transporters 1 and 3 from natural compounds and their protective
effect on aristolochic acid nephropathy. Toxicol Sci. 175:279–291.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Kovesdy CP: Epidemiology of chronic kidney
disease: An update 2022. Kidney Int Suppl (2011). 12:7–11. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Yang DL and Luo NP: Changes of serum TGF-α
and IGF-II levels in patients with chronic renal failure before
receiving homedialysis and their clinical significance. J Pract Med
& Pharm. 22:683–685. 2005.(In Chinese).
|
|
121
|
Lohia S, Latosinska A, Zoidakis J,
Makridakis M, Mischak H, Glorieux G, Vlahou A and Jankowski V:
#4547 Identification of glycosylated igf2 in human urinary
peptidome and its association with CKD. Nephrol Dial Transplant. 38
(Suppl 1):gfad063c_4547. 2023. View Article : Google Scholar
|
|
122
|
Pearce EL and Pearce EJ: Metabolic
pathways in immune cell activation and quiescence. Immunity.
38:633–643. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Kelly B and O'Neill LA: Metabolic
reprogramming in macrophages and dendritic cells in innate
immunity. Cell Res. 25:771–784. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
O'Neill LAJ and Pearce EJ:
Immunometabolism governs dendritic cell and macrophage function. J
Exp Med. 213:15–23. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Netea MG, Joosten LA, Latz E, Mills KH,
Natoli G, Stunnenberg HG, O'Neill LA and Xavier RJ: Trained
immunity: A program of innate immune memory in health and disease.
Science. 352:aaf10982016. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Netea MG and van der Meer JW: Trained
immunity: An ancient way of remembering. Cell Host Microbe.
21:297–300. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Paust S and von Andrian UH: Natural killer
cell memory. Nat Immunol. 12:500–508. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Mihai S, Codrici E, Popescu ID, Enciu AM,
Albulescu L, Necula LG, Mambet C, Anton G and Tanase C:
Inflammation-related mechanisms in chronic kidney disease
prediction, progression, and outcome. J Immunol Res.
2018:21803732018. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng
J, Li Y, Wang X and Zhao L: Inflammatory responses and
inflammation-associated diseases in organs. Oncotarget.
9:7204–7218. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Thomas S and Karalliedde J: Diabetic
kidney disease. Medicine. 50:704–710. 2022. View Article : Google Scholar
|
|
131
|
Nishi S, Ueno M, Hisaki S, Iino N, Iguchi
S, Oyama Y, Imai N, Arakawa M and Gejyo F: Ultrastructural
characteristics of diabetic nephropathy. Med Electron Microsc.
33:65–73. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Jia PW and Wang AJ: Correlation of IGF-2
and IGFBP-2 with Diabetic Nephropathy Progress Indicators. J
Kunming Med Univ. 36:55–59. 2015.(In Chinese).
|
|
133
|
Kuang J, Yang HZ, Dong H, et al: IGF and
IGFBP and their relationship with nephropathy in type-2 diabetes
mellitus. Chin J Pract Intern Med. 26:433–436. 2006.(In
Chinese).
|
|
134
|
Devanathan N and Kimble-Hill AC:
Systematic survey of the role of IGF in the link between diabetes
and cancer. Indiana Univ J Undergrad Res. 4:17–26. 2018.PubMed/NCBI
|
|
135
|
Pricci F, Pugliese G, Romano G, Romeo G,
Locuratolo N, Pugliese F, Mene P, Galli G, Casini A, Rotella CM and
Di Mario U: Insulin-like growth factors I and II stimulate
extracellular matrix production in human glomerular mesangial
cells. Comparison with transforming growth factor-beta.
Endocrinology. 137:879–885. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Mahimainathan L, Das F, Venkatesan B and
Choudhury GG: Mesangial cell hypertrophy by high glucose is
mediated by downregulation of the tumor suppressor PTEN. Diabetes.
55:2115–2125. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Sireesha M, Sambasivan V, Kumar VK, Radha
S, Raj AY and Qurratulain H: Relevance of insulin-like growth
factor 2 in the etiopathophysiology of diabetic nephropathy:
Possible roles of phosphatase and tensin homolog on chromosome 10
and secreted protein acidic and rich in cysteine as regulators of
repair. J Diabetes. 1:118–124. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Zhang LM, Wu BB, Mei CL, Fu LL and Wang
WJ: Effects of secreted protein acidic and rich in cysteine peptide
on change of ultra microstructure and extracellular matrix
secretion of human mesangial cells cultured in vitro. Acad J
Sec Mil Med Univ. 28:36–39. 2007.(In Chinese).
|
|
139
|
Bloomgarden ZT: Diabetic nephropathy.
Diabetes Care. 31:823–827. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Bhattacharya S, Mandal SK, Bandyopadhyay
R, Chakrabarti S, Basu AK and Pal S: A study on nephropathy in type
2 diabetes mellitus: Histology and its correlation with clinical
and biochemical parameters. J Indian Med Assoc. 105:592594–596.
2007.PubMed/NCBI
|
|
141
|
Taneda S, Pippin JW, Sage EH, Hudkins KL,
Takeuchi Y, Couser WG and Alpers CE: Amelioration of diabetic
nephropathy in SPARC-null mice. J Am Soc Nephrol. 14:968–980. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Reed MJ, Puolakkainen P, Lane TF,
Dickerson D, Bornstein P and Sage EH: Differential expression of
SPARC and thrombospondin 1 in wound repair: Immunolocalization and
in situ hybridization. J Histochem Cytochem. 41:1467–1477. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Dallinga MG, Yetkin-Arik B, Kayser RP,
Vogels IMC, Nowak-Sliwinska P, Griffioen AW, van Noorden CJF,
Klaassen I and Schlingemann RO: IGF2 and IGF1R identified as novel
tip cell genes in primary microvascular endothelial cell
monolayers. Angiogenesis. 21:823–836. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Wang Q, Wu G, Zhang Z, Tang Q, Zheng W,
Chen X, Chen F, Li Q and Che X: Long non-coding RNA HOTTIP promotes
renal cell carcinoma progression through the regulation of the
miR-615/IGF-2 pathway. Int J Oncol. 53:2278–2288. 2018.PubMed/NCBI
|
|
145
|
Qian SB, Li XY, Yu YJ, Wu Y, Shen HB, Xu D
and Qi J: Effect of targeted induction of IGF2 promoter hP4
methylation on imprinting status of renal cancer cells. Int J Urol
Nephrol. 39:1000–1004. 2019.(In Chinese).
|
|
146
|
Nonomura N, Nishimura K, Miki T, Kanno N,
Kojima Y, Yokoyama M and Okuyama A: Loss of imprinting of the
insulin-like growth factor II gene in renal cell carcinoma. Cancer
Res. 57:2575–2577. 1997.PubMed/NCBI
|
|
147
|
Oda H, Kume H, Shimizu Y, Inoue T and
Ishikawa T: Loss of imprinting of igf2 in renal-cell carcinomas.
Int J Cancer. 75:343–346. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Jiang A, Wu X, Wang D, Wang A, Dong K, Liu
B, Qu L, Luo P, Wang J, Tong Q and Wang L: A new thinking:
Deciphering the aberrance and clinical implication of IGF axis
regulation pattern in clear cell renal cell carcinoma. Front
Immunol. 13:9355952022. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Steenman MJ, Rainier S, Dobry CJ, Grundy
P, Horon IL and Feinberg AP: Loss of imprinting of IGF2 is linked
to reduced expression and abnormal methylation of H19 in Wilms'
tumour. Nat Genet. 7:433–439. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Taniguchi T, Schofield AE, Scarlett JL,
Morison IM, Sullivan MJ and Reeve AE: Altered specificity of IGF2
promoter imprinting during fetal development and onset of Wilms
tumour. Oncogene. 11:751–756. 1995.PubMed/NCBI
|
|
151
|
Wegert J, Fischer AK, Palhazi B, Treger
TD, Hilgers C, Ziegler B, Jung H, Jüttner E, Waha A, Fuchs J, et
al: TRIM28 inactivation in epithelial nephroblastoma is frequent
and often associated with predisposing TRIM28 germline variants. J
Pathol. 262:10–21. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Haruta M, Arai Y, Watanabe N, Fujiwara Y,
Honda S, Ohshima J, Kasai F, Nakadate H, Horie H, Okita H, et al:
Different incidences of epigenetic but not genetic abnormalities
between Wilms tumors in japanese and caucasian children. Cancer
Sci. 103:1129–1135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Iams WT and Lovly CM: Molecular pathways:
Clinical applications and future direction of insulin-like growth
factor-1 receptor pathway blockade. Clin Cancer Res. 21:4270–4277.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Haluska P, Menefee M, Plimack ER,
Rosenberg J, Northfelt D, LaVallee T, Shi L, Yu XQ, Burke P, Huang
J, et al: Phase I dose-escalation study of MEDI-573, a bispecific,
antiligand monoclonal antibody against IGFI and IGFII, in patients
with advanced solid tumors. Clin Cancer Res. 20:4747–4757. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Rihawi K, Ong M, Michalarea V, Bent L,
Buschke S, Bogenrieder T, Anthoney A, De Bono JS and Twelves C:
Phase I dose escalation study of 3-weekly BI 836845, a fully human,
affinity optimized, insulin-like growth factor (IGF) ligand
neutralizing antibody, in patients with advanced solid tumors. J
Clin Oncol. 32 (15 Suppl):S26222014. View Article : Google Scholar
|
|
156
|
Tian B, Zhao Y, Liang T, Ye X, Li Z, Yan
D, Fu Q and Li Y: Curcumin inhibits urothelial tumor development by
suppressing IGF2 and IGF2-mediated PI3K/AKT/mTOR signaling pathway.
J Drug Target. 25:626–636. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Anderson PM, Bielack SS, Gorlick RG,
Skubitz K, Daw NC, Herzog CE, Monge OR, Lassaletta A, Boldrini E,
Pápai Z, et al: A phase II study of clinical activity of SCH 717454
(robatumumab) in patients with relapsed osteosarcoma and ewing
sarcoma. Pediatr Blood Cancer. 63:1761–1770. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Arango-Argoty G, Bikiel DE, Sun GJ,
Kipkogei E, Smith KM, Carrasco Pro S, Choe EY and Jacob E:
AI-driven predictive biomarker discovery with contrastive learning
to improve clinical trial outcomes. Cancer Cell. 43:875–890.e8.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Ogawa O, Eccles MR, Szeto J, McNoe LA, Yun
K, Maw MA, Smith PJ and Reeve AE: Relaxation of insulin-like growth
factor II gene imprinting implicated in Wilms' tumour. Nature.
362:749–751. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Hu Q, Gao F, Tian W, Ruteshouser EC, Wang
Y, Lazar A, Stewart J, Strong LC, Behringer RR and Huff V: Wt1
ablation and Igf2 upregulation in mice result in Wilms tumors with
elevated ERK1/2 phosphorylation. J Clin Invest. 121:174–183. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Alders L, Pirlet E, Gesquiere E and
Bronckaers A: The role of IGF-2 and its variants in enhancing
endothelial migration and angiogenesis. Front Cell Dev Biol.
13:15987052025. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Yang Y and Yee D: Targeting insulin and
insulin-like growth factor signaling in breast cancer. J Mammary
Gland Biol Neoplasia. 17:251–261. 2012. View Article : Google Scholar : PubMed/NCBI
|














