Introduction
Ketamine (Ket), commonly known as K powder, induces
a strong sense of euphoria and exhibits notable hallucinogenic
properties, leading to a large number of individuals, predominantly
young individuals, that misuse this substance for recreational
purposes (1). Studies have
identified that long-term Ket abuse can cause damage to the nervous
(2), cardiovascular (3) and urinary systems (4). The most common complications of
urinary system damage are categorized as lower urinary tract
symptoms (LUTS), characterized by frequent urination, urgency,
suprapubic discomfort and hematuria. Symptoms of Ket usage that
bear similarity to interstitial cystitis are attributed to
Ket-induced cystitis (KIC) (5);
however, the pathogenesis of this condition remains ambiguous.
Previous studies suggest that Ket and its metabolites in urine
exert direct toxic effects on bladder urothelial cells, resulting
in chronic inflammation and fibrosis of the bladder mucosa
(6,7).
Aldehyde dehydrogenase 2 (ALDH2) is an important
enzyme in aldehyde metabolism and plays an important role in
antioxidant protection (8).
Studies have revealed that inhibiting ALDH2 expression can promote
the inflammatory response in septic rats, thereby aggravating
kidney damage (9,10). ALDH2 activation can also mitigate
the hepatotoxic effects of acetaminophen and mediate cell damage
caused by resultant oxidative products (11). Additionally, ALDH2 may exert
protective effects against myocardial damage associated with
pulmonary hypertension (12).
These findings suggest that ALDH2 plays an important role in
mediating oxidative stress and regulating inflammation during the
consequent organ damage. Previous studies have revealed that ALDH2
expression is closely associated with urinary system diseases;
ALDH2 has demonstrated anti-inflammatory protective effects in
diseases such as renal ischemia-reperfusion injury (13), acute pyelitis (14), LUTS (15) and bladder tumors (16).
Our previous study identified that ALDH2 is
upregulated in mice following Ket administration and that ALDH2
deficiency in mice aggravates KIC (17). However, the effects of ALDH2 on KIC
in vitro remain to be fully elucidated. While our previous
investigation on ALDH2 knockout mice demonstrated that ALDH2
deficiency exacerbated KIC pathology, the specific cellular
mechanisms and signaling pathways involved remain undefined.
Previous studies have also indicated that ALDH2 mitigates
lipopolysaccharide-induced damage to myocardial H9C2 cells through
the cyclic GMP-AMP synthase/stimulator of interferon genes protein
pathway (18) and protects against
acute kidney injury via autophagy regulation in HK-2 cells
(19). The present study aimed to
elucidate the molecular mechanisms through which ALDH2 protected
bladder epithelial cells from Ket-induced injury, specifically by
examining the oxidative stress-mediated crosstalk between the NF-κB
and mTOR pathways.
Materials and methods
Cell culture and reagents
The immortalized human bladder epithelial cell line
SV-HUC-1 was purchased from The Cell Bank of Type Culture
Collection of The Chinese Academy of Sciences. Cells were cultured
in 89% F12K medium (cat. no. LA1320; Beijing Solarbio Science &
Technology Co., Ltd.) supplemented with 10% heat-inactivated fetal
bovine serum (cat. no. A511-001; Lonsera; Shanghai Shuangru
Biotechnology Co., Ltd.) and 1% penicillin-streptomycin (cat. no.
BL505A; Biosharp Life Sciences) at 37°C with 5% CO2. The
growth of the cells was observed for 24 h and when the cell density
reached >80%, they were passaged. SV-HUC-1 cells were used at
passages 2 and 3. Cells were authenticated by short tandem repeat
profiling and tested negative for mycoplasma contamination.
According to previous studies, the stimulatory effect of Ket on
cells is both concentration- and time-dependent (20,21).
Based on these previous KIC-related studies, the present study
selected 0.5, 1, 1.5 and 2 mM as test concentrations for Ket
treatment. To test the effects of differing Ket concentrations,
SV-HUC-1 cells were initially incubated in 6-well plates. Upon
reaching 80% confluency, cells were exposed to equal volumes of Ket
at different working concentrations (0, 0.5, 1, 1.5 and 2 mM) for
48 h at 37°C. Ket hydrochloride was purchased from Fujian Gutian
Pharmaceutical Co., Ltd. After 48 h of incubation with Ket, cells
were collected for ALDH2 protein semi-quantitative analysis using
western blotting.
Transfection of cells for ALDH2
knockdown and overexpression
SV-HUC-1 cells were transfected with siRNA sequences
targeting ALDH2 (si-ALDH2) in order to establish a model of ALDH2
knockdown; cells were also transfected with an appropriate
non-targeting negative control (NC) siRNA (si-NC). All siRNA
sequences used in this study are listed in Table I. The siRNA sequences used were
designed and synthesized by Jiangsu Saisuofei Biotechnology Co.,
Ltd. siRNA sequences were transfected into SV-HUC-1 cells using
Lipo8000™ Transfection Reagent (cat. no. C0533; Beyotime
Biotechnology) according to the manufacturers' instructions.
Reverse transcription-quantitative PCR (RT-qPCR) was used to screen
for knockdown efficiency of small interfering RNA (siRNA)
sequences. The siRNA sequence with the highest knockdown efficiency
was identified to be si-ALDH2-1272 (Fig. S1, Fig. S2, Fig. S3, Fig. S4, Fig. S5).
 | Table I.siRNA sequences. |
Table I.
siRNA sequences.
| Oligo name | Sequence
(5′-3′) | RNA concentration,
ng/µl |
|---|
|
ALDH2-human-1272 |
| 260.85 |
|
Forward |
CCUGAAGUUCAAGACCAUATT |
|
|
Reverse |
UAUGGUCUUGAACUUCAGGTT |
|
|
ALDH2-human-1500 |
| 245.37 |
|
Forward |
GGCAUACACUGAAGUGAAATT |
|
|
Reverse |
UUUCACUUCAGUGUAUGCCTT |
|
|
ALDH2-human-493 |
| 250.71 |
|
Forward |
GGAGACUUCUUCAGCUACATT |
|
|
Reverse |
UGUAGCUGAAGAAGUCUCCTT |
|
| siRNA FAM marker
negative control |
| 263.19 |
|
Forward |
UUCUCCGAACGUGUCACGUTT |
|
|
Reverse |
ACGUGACACGUUCGGAGAATT |
|
To induce ALDH2 overexpression (OE-ALDH2), the
coding DNA sequence (CDs) region of the ALDH2 mRNA sequence was
cloned and inserted into the pcDNA3.1-EGFP (Genecefe Bio)
overexpression vector. Empty plasmid was used as the NC. PCR
conditions: Incubation at 42°C for 40 min, heating at 85°C for 5
min and storing at 4°C. cDNA was stored at −20°C. The
overexpression vector containing the ALDH2 CDs sequence was
transfected into SV-HUC-1 cells using Lipo8000 Transfection Reagent
(cat. no. C0533; Beyotime Biotechnology) according to the
manufacturers' instructions.
The present study subsequently used western blotting
to detect the protein expression of ALDH2 in transfected cells to
verify the successful transfection of si-ALDH2 and OE-ALDH2
compared with their control groups (Fig. S6). According to the results of the
aforementioned investigation into the effects of Ket dosage on
ALDH2 expression, 1 mM Ket was selected as the optimal
concentration for treatment of SV-HUC-1 cells. Additionally, 10 uM
of the mTOR activator MHY1485 (Act; cat. no. HY-B0795;
MedChemExpress) was used to treat cells; this helps cells resist
apoptosis and oxidative stress damage, allowing them to survive in
harsh environments. Subsequently, the present study established a
number of treatment groups, including: i) NC groups comprising
cells that did not undergo transfection (NC, NC + Act, NC + Ket and
NC + Ket + Act groups); ii) ALDH2 knockdown groups (si-ALDH2,
si-ALDH2 + Act, si-ALDH2 + Ket and si-ALDH2 + Ket + Act groups);
and iii) ALDH2 overexpression groups (OE-ALDH2, OE-ALDH2 + Act,
OE-ALDH2 + Ket and OE-ALDH2 + Ket + Act groups). After
transfection, cells in each group were incubated with equal volumes
of complete culture medium for 48 h at 37°C before observation
under a light microscope and collection for subsequent
experiments.
RT-qPCR screening for si-ALDH2
sequence knockdown efficiency
The mRNA sequence of ALDH2 was obtained from the
Reference Sequence database curated by the National Center for
Biotechnology Information (22)
and the primers used in RT-qPCR were designed and synthesized by
Sangon Biotech Co., Ltd., using the Primer Premier 5.0 software
(Premier Biosoft Inc.). The housekeeping gene GAPDH was used as an
internal reference.
Total RNA was extracted from SV-HUC-1 cells using
Trizol reagent (cat. no. R1100; Beijing Solarbio Technology Co.,
Ltd.). Subsequently, 50% by volume chloroform (cat. no. C2432;
Merck KGaA) was added to cells for the RNA extraction, and the
upper aqueous phase was collected after centrifugation. An equal
volume of isopropanol (cat. no. W292907; Merck KGaA) was added to
the supernatant before RNA was precipitated by centrifugation at
11,300 × g for 15 min at 4°C. Extracted RNA was washed with 70%
alcohol, dried and suspended in enzyme water. Subsequently, RNA
concentration and purity were determined using an
ultra-micronucleic acid detector (cat. no. FC-1100; Hangzhou
Suizhen Biotechnology Co., Ltd.). A total of 200 ng RNA was reverse
transcribed using a reverse transcription kit (cat. no. RN05004M;
Mona Biotechnology, Co., Ltd.) according to the manufacturer's
protocol, and the resulting cDNA was stored at −20°C until
subsequent use in the qPCR protocol.
First, RNA concentration was measured, followed by
cDNA synthesis. Each reaction for the qPCR process included: 5 µl
SYBR-Green Premix Taq (cat. no. RN04006M; Mona), 1 µl cDNA (100
ng/µl), 0.3 µl forward primer (10 µmol/l), 0.3 µl reverse primer
(10 µmol/l) and 3.4 µl water. The thermocycling conditions were as
follows: Initial denaturation was performed at 95°C for 30 sec,
followed by 40 cycles of denaturation at 95°C for 5 sec, annealing
at 60°C for 30 sec and extension at 72°C for 15 sec. qPCR was
performed on the ABI 7500 qPCR instrument (Thermo Fisher
Scientific, Inc.). Relative protein expression was normalized
against the expression of GAPDH from control group cells. Protein
expression levels were analyzed using SPSS software (Version 23.0;
IBM Corp.) via the 2−ΔΔCq method (23). The formulas used for
semi-quantification of protein expression were as follows: ΔCq=(Cq
gene-Cq GAPDH) and ΔΔCq=(ΔCq treatment-ΔCq control). Primer
sequences are provided in Table
II.
| Table II.Reverse transcription-quantitative
PCR primer sequences. |
Table II.
Reverse transcription-quantitative
PCR primer sequences.
| Primer | Sequence
(5′-3′) | Sequence length,
bp |
|---|
| ALDH2 |
| 135 |
|
Forward |
AACCTGTGGGGGTGTGCGG |
|
|
Reverse |
GGGCGGTGAGGGGTGTCTG |
|
| GAPDH |
| 151 |
|
Forward |
CGGATTTGGTCGTATTG |
|
|
Reverse |
GAAGATGGTGATGGGATT |
|
Cell Counting Kit-8 (CCK-8)
determination
After transfection, the cytotoxicity of Ket in
SV-HUC-1 cells was evaluated via CCK-8 assay (cat. no. M006;
Zhejiang Meisen Cell Technology Co., Ltd.). A total of 20 µl CCK-8
solution was added to each well and cells were incubated with CCK-8
reagent for 2.5 h at 37°C with 5% CO2. The absorbance of
cells was determined at 450 nm by microplate reader.
Flow cytometry detection of cell
apoptosis
After digestion for 2 min with EDTA-free trypsin at
room temperature, cells were collected by centrifugation at 25 × g
for 5 min at room temperature. Subsequently, cells were resuspended
in PBS (cat. no. F002; Zhejiang Meisen Cell Technology Co., Ltd)
that had been pre-chilled at 4°C, centrifuged again as in the first
step and washed. Cells were resuspended in 300 µl binding buffer
and incubated with 5 µl annexin V-FITC (cat. no. 401006; BestBio)
at room temperature for 15 min in the dark. Cells were subsequently
stained with 10 µl PI (cat. no. C1052; Beyotime Biotechnology),
mixed and incubated in the dark at room temperature for 10 min.
Finally, cells were detected by flow cytometry (FACSVerse; BD
Biosciences) and analyzed using FlowJo software (version 7.6, BD
Biosciences).
Fluorescent probe detection of
intracellular reactive oxygen species (ROS) levels
The fluorescent probe 2′,7′-dichlorofluorescein
diacetate (DCFH-DA) from the Reactive Oxygen Species Assay Kit
(cat. no. S0033S; Beyotime Biotechnology) was diluted in serum-free
medium to reach a final concentration of 10 µmol/l at a dilution of
1:1,000. The cell culture medium was removed from cell samples of
all transfection treatments and cells were subsequently seeded in
12-well plates. A total of 1 ml diluted DCFH-DA was added to each
12-well plate and cells were incubated with the probes at 37°C for
20 min. Excess DCFH-DA was removed by washing the cells with
serum-free cell culture thrice. DAPI was diluted in serum-free
culture medium and cells were incubated with DAPI at 37°C for 10
min. Cells were washed three times with serum-free culture medium
and subsequently seeded in complete culture medium. Finally, cells
were observed and imaged using a fluorescence microscope (Olympus
Corporation). The average fluorescence intensity of ROS was
detected using Image J software (National Institutes of
Health).
Western blot analysis
Transfected and untransfected cell samples were
lysed in RIPA buffer (cat. no. P0013B; Beyotime Biotechnology)
containing protease inhibitors (cat. no. S1873; Beyotime
Biotechnology) and the protein concentration was subsequently
quantified using the BCA method. Equal amounts of protein (25 µg)
were loaded and separated on 12% gels by SDS-PAGE and then
transferred onto PVDF membranes (cat. no. IPVH00010;
MilliporeSigma). The membranes were rinsed three times with TBST
(0.1% Tween-20) for 5 min each time, and then slowly shaken with 5%
BSA (cat. no. P0023B; Beyotime Biotechnology) at room temperature
for 2 h. After blocking, the membranes were incubated with primary
antibodies at 4°C overnight, washed with TBST (0.1% Tween-20) and
subsequently incubated with secondary antibodies at 37°C for 1 h.
The primary antibodies used were as follows: ALDH2 (1:1,000; cat.
no. 18818; CST Biological Reagents Co., Ltd.), caspase-3 (1:1,000;
cat. no. 9662; CST Biological Reagents Co., Ltd.), nuclear factor
erythroid 2-related factor 2 (Nrf2; 1:1,000; cat. no. 12721; CST
Biological Reagents Co., Ltd.), mTOR (1:1,000; cat. no. AF6308;
Affinity Biosciences), phosphorylated (p)-mTOR (1:1,000; cat. no.
AF3308; Affinity Biosciences), cyclooxygenase-2 (COX-2; 1:1,000;
cat. no. 12282; CST Biological Reagents Co., Ltd.), NF-κB (1:1,000;
cat. no. 12540; CST Biological Reagents Co., Ltd.), p-NF-κB (1:800;
cat. no. 3033; CST Biological Reagents Co., Ltd.), IL-1 (1:2,000;
cat. no. ab283818; Abcam), IL-6 (1:2,000; cat. no. ab229381; Abcam)
and GAPDH (1:5,000; cat. no. 2118; CST Biological Reagents Co.,
Ltd.). Goat anti-rabbit HRP-conjugated antibodies were used as
secondary antibodies (1:10,000; cat. no. AP307P; MilliporeSigma).
GAPDH was used as an internal loading control for densitometric
analysis, whereas phosphorylated proteins were normalized to their
total protein counterparts. ECL luminescent liquid (cat. no.
P0018AS; Beyotime Biotechnology) was added to the membrane, and the
protein bands on the membrane were visualized. The bands were
semi-quantified using MD ImageQuant software (version 5.2;
Molecular Dynamics, Inc.).
Co-immunoprecipitation (CO-IP)
verification of the protein interaction between mTOR and NF-κB
Cells were lysed in 2 ml of IP lysis buffer [150 mM
NaCl, 50 mM Tris (pH 7.5), 1 mM EDTA, 0.5%NP40, 10% glycerol and
protease inhibitor cocktail] supplemented with protease inhibitors.
The lysates were cleared by centrifugation at 12,000 × g for 15 min
at 4°C. The supernatant was used as the total protein, and a
portion was reserved as an input control. Equal amounts of protein
(25 µg) were taken and then incubated with 2 µg of rabbit IgG (cat.
no. 2729; CST Biological Reagents Co., Ltd.) or 50 µl of anti-mTOR
(1:1,00; cat. no. 2983; CST Biological Reagents Co., Ltd.) antibody
overnight at 4°C with gentle agitation. Following this, 50 µl of
Protein A/G magnetic beads (cat. no. P2108; Beyotime Biotechnology)
were added and incubated for 2 h at 4°C. The beads were washed
three times with lysis buffer at 1,000 × g for 1 min at 4°C, and
the immunoprecipitated proteins were eluted by boiling in SDS
loading buffer and subjected to SDS-PAGE followed by immunoblotting
(mTOR and NF-κB antibodies, as described in in the western blot
analysis).
Statistical analysis
All statistical analyses in the present study were
conducted on GraphPad software (version 8.0; Dotmatics) and data
are presented as the mean ± SD. Comparisons between groups were
performed using one-way ANOVA followed by Tukey's post hoc test.
P<0.05 was considered to indicate a statistically significant
difference. All experiments were repeated independently at least
three times to obtain three biological replicates, with triplicate
technical replicates performed for each sample.
Results
ALDH2 protein expression in SV-HUC-1
cells
Relative ALDH2 expression levels in the 0.5, 1 and
1.5 mM induction groups were significantly higher compared with the
0 mM control group (P<0.05). However, no significant difference
in ALDH2 expression was observed in the 2 mM induction group
compared with the 0 mM group (P>0.05), as shown in Fig. 1. The results of the present study
revealed that ALDH2 expression level increased following Ket
stimulation; however, ALDH2 expression decreased at Ket
concentrations >1.5 mM due to significant reductions in cell
viability. Furthermore, caspase-3 protein expression increased as
Ket concentration, and therefore SV-HUC-1 cell apoptosis,
increased. Caspase-3 protein expression was shown to be Ket
concentration-dependent. CCK-8 cell viability assays demonstrated
that cell viability steadily decreased with increasing Ket
concentration. Based on these experimental results, 1 mM was
selected as the optimal working concentration of Ket for subsequent
experiments.
ALDH2 protects bladder cells from
apoptotic damage
To explore differences in bladder endothelial cell
viability under different treatment conditions, the present study
ascertained cell viability in NC, ALDH2 knockdown and OE-ALDH2
groups; these groups were subdivided into additional treatment
groups comprising cells administered Ket, Act and combination
treatments, as well as control groups that were administered no
additional treatments. The results of the present study identified
that cells in the si-ALDH2 + Ket group demonstrated a notable
decrease in cell viability levels compared with the si-ALDH2 group.
Similarly, cell viability rates were notably lower in the si-ALDH2
+ Ket group compared with the NC + Ket group. Following treatment
with Act, cell viability rates markedly increased across all
groups. However, the present study observed an increase in cell
viability in the OE-ALDH2 + Ket group compared with the NC + Ket
group. This increase was notably more pronounced following the
addition of Act between si-ALDH2 + Ket and si-ALDH2 + Ket + Act
(Fig. 2). There was little
difference in cell viability between the NC, si-ALDH2 and OE-ALDH2
groups. Summarily, these results indicated that ALDH2 expression
was likely associated with maintaining cell viability in
Ket-stimulated SV-HUC-1 cells. Furthermore, the addition of an mTOR
activator was shown mitigate the effects of Ket on cell
viability.
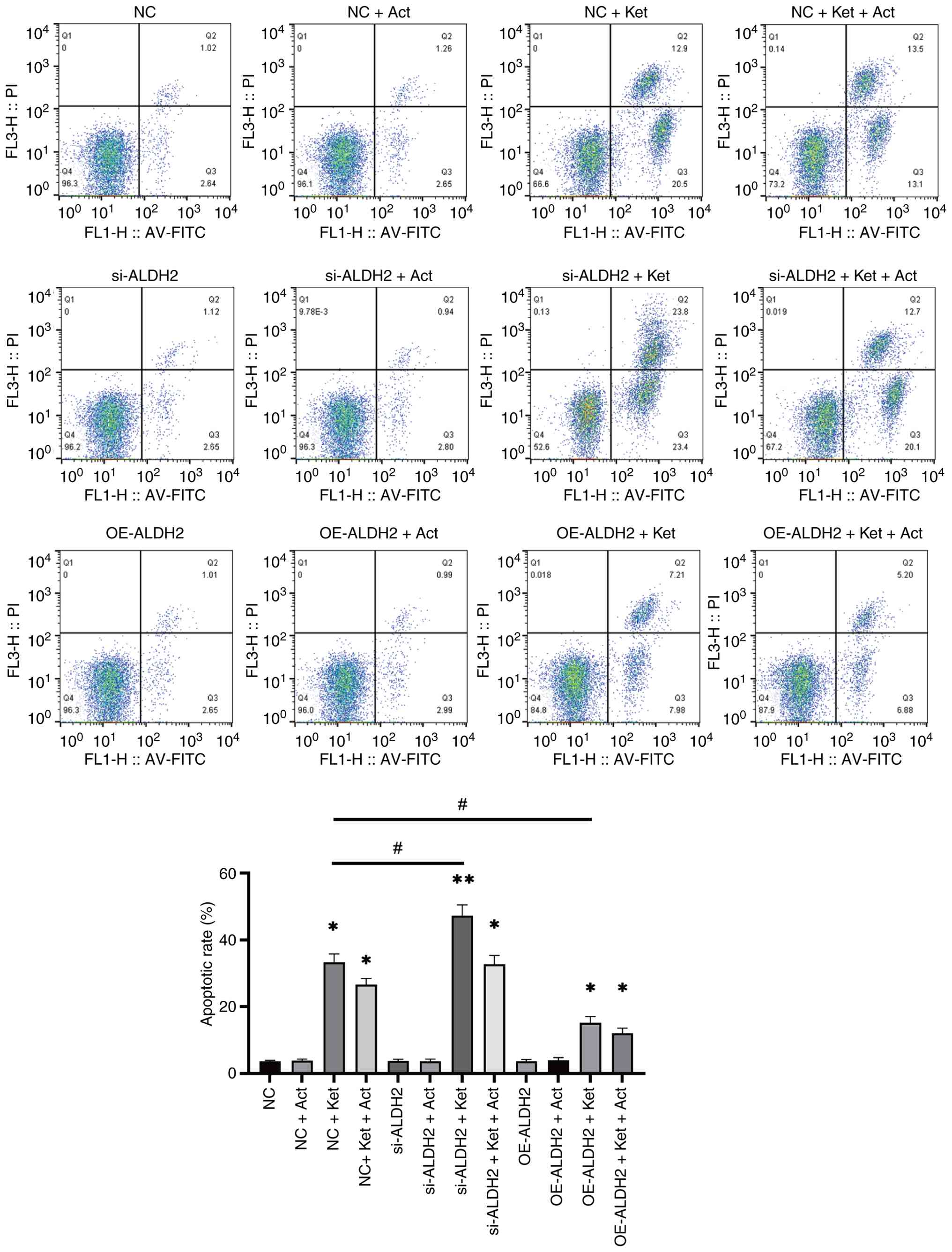
ALDH2 upregulation reduces cell
injury
To further clarify the effects of ALDH2 expression
on KIC, the present study used flow cytometry to detect apoptosis
in bladder epithelial cells. The results of the present study
revealed a significant increase in apoptosis in the si-ALDH2 + Ket
group compared with the si-ALDH2 group (Fig. 3). The increase in apoptotic rate
observed in the si-ALDH2 + Ket group was also significant compared
with the NC + Ket group, which indicated that ALDH2 knockdown
promoted apoptosis in KIC. However, apoptotic rate was
significantly reduced in the OE-ALDH2 + Ket group compared with the
NC + Ket group. This indicated that ALDH2 upregulation may have
reduced the occurrence of cell injury. Furthermore, apoptosis
appeared to be partially suppressed in bladder cells following
co-treatment with Act. These findings suggested that ALDH2
upregulation inhibited apoptosis in bladder endothelial cells.
ALDH2 upregulation mitigates oxidative
stress
Apoptosis is largely associated with intracellular
oxidative stress (24). Therefore,
the present study used fluorescent probes to detect the
intracellular ROS levels of various treatment groups. As indicated
in Fig. 4, the fluorescence
intensity of ROS in the si-ALDH2 + Ket group was significantly
greater than in the NC + Ket and si-ALDH2 groups. However, after
the addition of Act, the fluorescence intensity of ROS decreased
significantly. Furthermore, the fluorescence intensity of ROS in
the OE-ALDH2 + Ket group was markedly lower than in the si-ALDH2 +
Ket and NC + Ket groups. Furthermore, no significant differences
were observed in ROS fluorescence intensity between the NC, NC-Act,
si-ALDH2, si-ALDH2 + Act, OE-ALDH2 and OE-ALDH2 + Act groups. These
findings suggested that ALDH2 upregulation reduced oxidative stress
damage in bladder endothelial cells
ALDH2 upregulation prevents
inflammation by regulating the mTOR/NF-κB pathway
To further explore the mechanism by which ALDH2
reduces inflammation in the bladder, the present study analyzed the
expression of components of the mTOR/NF-κB signaling pathway to
elucidate the specific role of ALDH2. Consistent with the results
of apoptosis and ROS analyses, the results of western blot analysis
(Fig. 5) demonstrated that
phosphorylation levels of the nuclear transcription factor NF-κB
were highest in the si-ALDH2 + Ket group at a relative expression
level of 1.69±0.06; this was significantly higher than the relative
expression of p-NF-κB observed in the NC + Ket group at 1.39±0.07.
However, this expression was notably reduced in the OE-ALDH2 + Ket
group 1.05±0.07. These results show that decreased ALDH2 expression
leads to increased inflammation, while upregulation of ALDH2
expression leads to decreased inflammation. It indicates that
inflammatory levels were negatively associated with ALDH2
expression. The expression levels of COX-2 (3.51±0.48 vs.
2.48±0.29), IL-1 (4.00±037 vs. 3.29±033) and IL-6 (5.23±0.82 vs.
2.99±0.11) were also significantly elevated in the si-ALDH2 + Ket
group compared with the NC + Ket group. Additionally, compared with
all other treatment groups in the present study, the si-ALDH2 + Ket
treatment group demonstrated the highest expression levels of these
proteins, which are all located downstream of NF-κB (25). However, the expression levels of
the aforementioned proteins were significantly reduced in the
OE-ALDH2 + Ket group compared with the si-ALDH2 + Ket and NC + Ket
groups. After co-treatment with Act, the expression levels of the
inflammatory proteins COX-2, IL-1 and IL-6 in each transfection
group notably decreased. Furthermore, mTOR phosphorylation in the
OE-ALDH2 + Ket group was significantly elevated compared with the
NC + Ket group. mTOR phosphorylation levels were also significantly
lower in the si-ALDH2 + Ket group compared with the NC + Ket group.
Nrf2 protein expression showed a similar trend. Compared with the
NC + Ket group, significantly reduced expression levels of Nrf2
were observed in the si-ALDH2 + Ket group, which demonstrated the
lowest Nrf2 expression levels among all treatment groups.
Additionally, significant upregulation of Nrf2 was observed in the
OE-ALDH2 + Ket group compared with the NC + Ket group, while, there
were no statistical significances between cells not treated with
Ket (NC, NC + Act, si-ALDH2, si-ALDH2 + Act, OE-ALDH2 and OE-ALDH2
+ Act). These results suggested that ALDH2 may have modulated the
degree of inflammation in bladder epithelial cells by promoting
mTOR phosphorylation and inhibiting NF-κB expression. Additionally,
the results of the CO-IP assay in the present study indicated an
indirect or functional interaction between mTOR and NF-κB (Fig. S7).
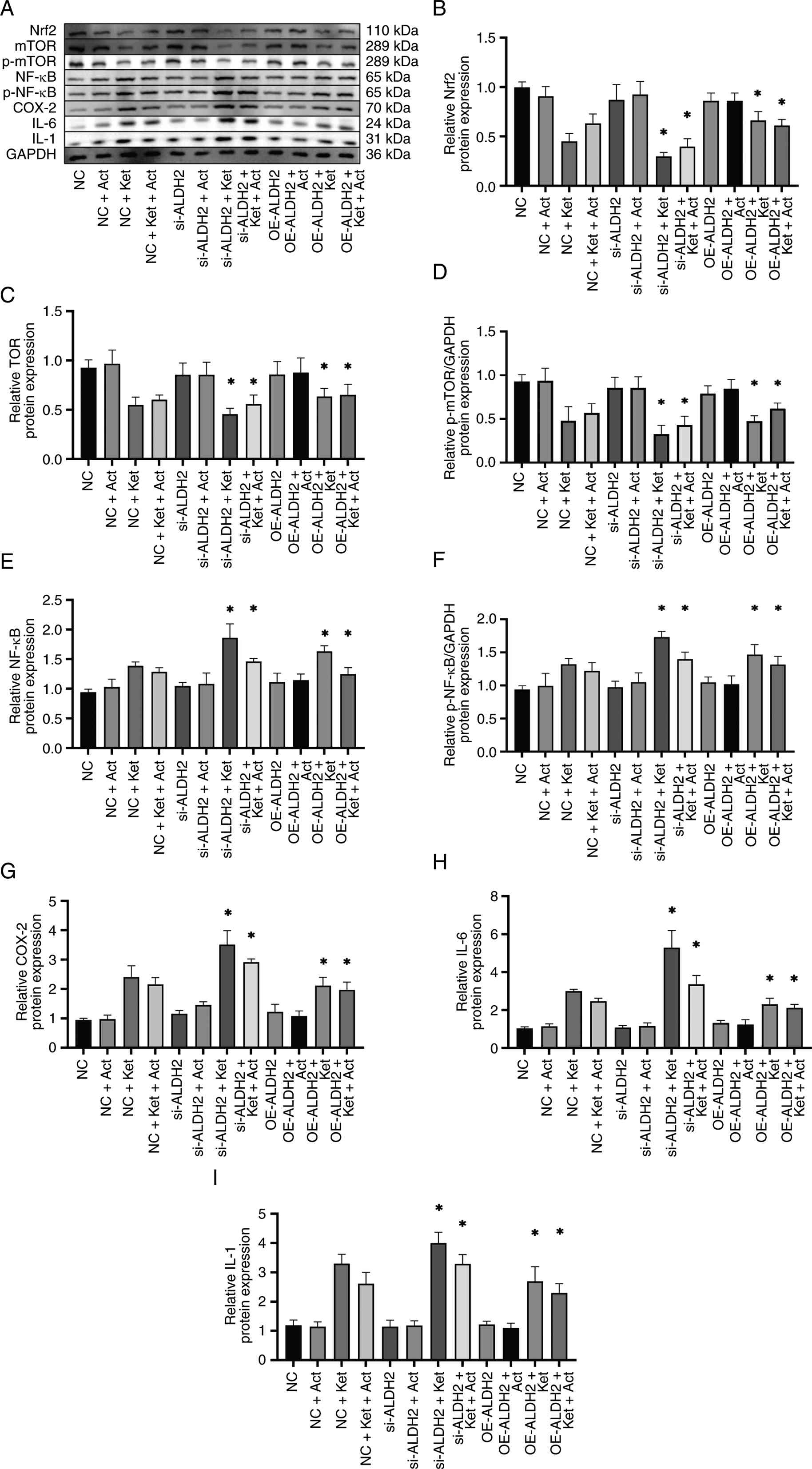 | Figure 5.Effects of ALDH2 expression on
inflammation in Ket-induced SV-HUC-1 cells. (A) Western blotting
was used to detect the protein expression levels and its
semi-quantification of (B) Nrf2, (C) mTOR, (D) p-mTOR, (E) NF-κB,
(F) p-NF-κB, (G) COX-2, (H) IL-6 and (I) IL-1 in each treatment
group. Blots were normalized against GAPDH, with p-mTOR and p-NF-κB
expression also normalized against total mTOR and NF-κB. Results of
western blotting were semi-quantified. *P<0.05 vs. NC + Ket
group. Data are expressed as the mean ± SD of 3 biological
replicates. NC, negative control; ALDH2, aldehyde dehydrogenase 2;
Act, mTOR activator MHY1485; Ket, ketamine; si-ALDH2, small
interfering RNA sequences targeting ALDH2; OE-ALDH2, ALDH2
overexpression; Nrf2, nuclear factor erythroid 2-related factor 2;
p-, phosphorylated-; COX-2, cyclooxygenase-2. |
Discussion
Currently, symptomatic methods remain the primary
approach for treating KIC, including steroidal and non-steroidal
anti-inflammatory drugs, oral anticholinergics and intravesical
instillations of drugs, such as hyaluronic acid. However, the
outcomes of such treatments are often poor for patients with late
stage KIC, underscoring the notable importance of studying KIC
pathogenesis (26). A previous
study has reported that bladder urothelial denudation, lamina
propria fibrosis and inflammation are common pathological features
of KIC (27). Ket and its
metabolites in urine exert direct toxic effects on the bladder
urothelium, generating free radicals of ROS and reactive nitrogen
species. These free radicals react with polyunsaturated fatty acids
in the cell membrane to form lipid peroxides, which damage bladder
cells and disrupt the bladder mucosal barrier. These lipid
peroxides include 4-hydroxy-2-nonenal and malondialdehyde, which
can disrupt the mitochondrial respiratory chain and the activity of
mitochondrial antioxidant enzymes. Consequently, oxidative stress
is considered to be an important factor in the occurrence and
development of KIC (17).
ALDH2 can convert aldehydes into their corresponding
non-toxic acid forms (28), as
well as reduce inflammation and inhibit apoptosis. A study reported
by Zhang and Fu (29) revealed
that ALDH2 inhibited tumor occurrence and progression, reduced the
production of carcinogenic aldehydes and served as a potential
target for tumor treatment. Another study by Zhong et al
(30) also observed that increased
ALDH2 expression reduced oxidative stress damage during
ischemia-reperfusion injury and inhibited renal cell apoptosis.
Furthermore, a study reported by Ramakrishnan et al
(31) also observed that ALDH2 was
upregulated in muscle-invasive bladder cancer as part of a
protective anticancer response. Based on the notable antioxidant
and anti-inflammatory effects of ALDH2 in urinary tract tumors
(16) and inflammation, the
aforementioned findings suggest that ALDH2 may represent a novel
target for the prevention and treatment of KIC.
The results of our previous in vivo study
(17) in Ket-induced mice
demonstrated that inflammation and fibrosis in ALDH2 knockout mice
were markedly increased compared with wild-type mice. To further
support our previous hypothesis that ALDH2 exerts protective
effects against bladder injury, the present study established a
model of Ket-induced bladder endothelial cell injury to study the
effects of ALDH2 gene expression on markers of KIC. The present
study observed that exposure to low concentrations of Ket
upregulated ALDH2 protein expression in SV-HUC-1 cells, which was
consistent with our previous in vivo observations on ALDH2
expression in mice, indicating that this upregulation might be
protective. However, at higher concentrations of Ket, cell damage
exceeds its own compensatory capacity, leading to a decrease in the
protective expression of ALDH2. With the decrease in ALDH2
expression, apoptosis protein Caspase-3 was further activated and
cell viability decreased significantly, demonstrating that ALDH2
may play a role in protecting against oxidative stress. Other
studies, such as a study by Ji et al (32) on a mouse model of heart failure,
identified that heat shock factor protein 1 regulated ALDH2
expression and delayed the onset of heart failure. Furthermore,
another study reported by Wohlfart et al (33) identified that ALDH2 was upregulated
in glyoxalase 1−/− zebrafish in a compensatory manner,
therefore increasing the antioxidant and detoxification capacity of
reactive carbonyl substances.
Upon direct stimulation of bladder mucosal tissue by
toxic products, oxidative stress-induced damage results in cellular
production of several ROS. Oxidative stress can activate the
important inflammatory transcription factor NF-κB. NF-κB, as an
upstream core transcription factor, mediates the activation of the
caspase cascade further promoting the expression of inflammatory
proteins (COX-2 and inducible nitric oxide synthase) and the
release of inflammatory mediators (IL-1β and IL-6), and inhibiting
the expression of the anti-oxidative protein Nrf2 (34). A study reported by Tsai et
al (35) observed that ALDH2
transcriptional activation reduced ROS production and inhibited the
NF-κB pathway, thereby reducing vascular smooth muscle cell
apoptosis and preventing the formation of abdominal aortic
aneurysms. Another study revealed that ALDH2 protects the kidneys
against ischemia-reperfusion injury by inhibiting the
IκBα/NF-κB/IL-17C pathway (36).
The experimental results of the present study revealed that in
oxidative stress damage induced by Ket, ALDH2 overexpression
reduced ROS production and inhibited activation of the NF-κB
pathway, reducing the production of inflammatory factors. However,
ALDH2 knockdown resulted in an imbalance in oxidation-antioxidant
levels, such as increased ROS levels and decreased cell viability,
thus activating the NF-κB pathway and increasing the production of
pro-inflammatory and pro-fibrotic factors.
mTOR is a serine/threonine kinase that regulates
various cellular functions, including the cellular stress response,
metabolism, survival and growth (37). Research has increasingly focused on
the role of mTOR in a number of important pathophysiological
processes in the human body, including inflammation, injury,
proliferation, tissue repair and tumorigenesis (38). A study reported by Zahid et
al (39) on alcohol-induced
metabolic diseases demonstrated that transcriptional inhibition of
ALDH2 expression resulted in alcohol-induced chronic oxidative
stress to modulate the mTOR pathway, thus promoting the onset and
progression of hepatocellular carcinoma. Another study has revealed
that ALDH2 regulates autophagy via the Akt/mTOR pathway, thereby
reducing oxidative stress and mitigating renal ischemia-reperfusion
injury (40). The present study
demonstrated that in oxidative stress damage induced by Ket, ALDH2
knockdown induced ROS accumulation, which inhibited mTOR via the
oxidative stress pathway. Reduced mTOR expression has been shown to
impair cellular antioxidant and metabolic homeostasis, which
ultimately amplifies the inflammatory and fibrotic responses.
Functional interception of ROS activity via methods such as mTOR
activation can partially reverse this pathological process
(41). Notably, the results of the
co-IP assay performed in the present study demonstrated that mTOR
and NF-κB are likely associated through functional interactions or
indirect pathways, rather than via direct protein-protein
interactions.
The results of the present study supported our
hypothesis that ALDH2 was likely responsible for reducing the
oxidative stress-induced damage to bladder tissue caused by Ket and
its metabolites, and that this mitigation of oxidative stress was
achieved via regulation of the NF-κB and mTOR pathways to reduce
bladder epithelial cell inflammation. Knockdown of the ALDH2
gene was also shown to aggravate KIC. As these results were
consistent with the results of our previous in vivo study in
mice, we hypothesize that ALDH2 deficiency represents an important
risk factor for KIC and other lower urinary tract diseases.
Currently, our research into the effects of ALDH2 on KIC has
focused on the use of mouse and cell models; however, in future
studies we will conduct clinical analyses on patients with KIC,
including measurements of daily Ket dosage, and serum and urinary
Ket concentration.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present work was supported by the Jiangsu University Medical
Education Collaborative Innovation Fund (grant no. JDYY2023139) and
the Wuxi Municipal Health Commission Scientific Research Youth
Project Fund (grant no. Q202462).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
XX, YY and MZ designed the study, performed
experiments, analyzed data and drafted the manuscript. PG
contributed towards performing experiments. MZ was also involved in
revising the manuscript critically for important intellectual
content and giving final approval of the version to be published.
Furthermore, MZ was responsible for experimental platforms, funding
and laboratory conditions. XX and MZ confirm the authenticity of
all the raw data. All authors have read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang JW, Kivovich V and Gordon L: Ketamine
abuse syndrome: Hepatobiliary and urinary pathology among
adolescents in flushing, NY. Pediatr Emerg Care. 33:e24–e26. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ezquerra-Romano I, Lawn W, Krupitsky E and
Morgan CJA: Ketamine for the treatment of addiction: Evidence and
potential mechanisms. Neuropharmacology. 142:72–82. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Orhurhu VJ, Vashisht R, Claus LE and Cohen
SP: Ketamine Toxicity. StatPerals [Internet]: StatPearls Publishing
Treasure Island, FL: 2025
|
|
4
|
Jalil R and Gupta S: Illicit ketamine and
its bladder consequences: Is it irreversible? BMJ Case Rep.
2012:bcr20120072442012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Noorzurani R, Vicknasingam B and Narayanan
S: Illicit ketamine induced frequency of micturition in a young
Malay woman. Drug Alcohol Rev. 29:334–336. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Juan YS, Lee YL, Long CY, Wong JH, Jang
MY, Lu JH, Wu WJ, Huang YS, Chang WC and Chuang SM: Translocation
of NF-κB and expression of cyclooxygenase-2 are enhanced by
ketamine-induced ulcerative cystitis in rat bladder. Am J Pathol.
185:2269–228. 20155 View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim A, Yu HY, Heo J, Song M, Shin JH, Lim
J, Yoon SJ, Kim Y, Lee S, Kim SW, et al: Mesenchymal stem cells
protect against the tissue fibrosis of ketamine-induced cystitis in
rat bladder. Sci Rep. 6:308812016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li R, Zhao Z, Sun M, Luo J and Xiao Y:
ALDH2 gene polymorphism in different types of cancers and its
clinical significance. Life Sci. 147:59–66. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hu JF, Wang HX, Li HH, Hu J, Yu Y and Gao
Q: Inhibition of ALDH2 expression aggravates renal injury in a rat
sepsis syndrome model. Exp Ther Med. 14:2249–2254. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Peng J, Wang S, Pan X, Wu M, Zhan X, Wang
D, Zhu G, Wang W, Tang H, An N and Pei J: Identification of ALDH2
as a novel target for the treatment of acute kidney injury in
kidney transplantation based on WGCNA and machine learning
algorithms and exploration of its potential mechanism of action
using animal experiments. Front Immunol. 16:15368002025. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wimborne HJ, Hu J, Takemoto K, Nguyen NT,
Jaeschke H, Lemasters JJ and Zhong Z: Aldehyde dehydrogenase-2
activation decreases acetaminophen hepatotoxicity by prevention of
mitochondrial depolarization. Toxicol Appl Pharmacol.
396:1149822020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xia G, Fan F, Liu M, Wang S, Wu J, Shen C,
Han S, Wang C, Jia J, Zou Y, et al: Aldehyde dehydrogenase 2
deficiency blunts compensatory cardiac hypertrophy through
modulating Akt phosphorylation early after transverse aorta
constriction in mice. Biochim Biophys Acta. 1862:1587–1593. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kucuk A, Kabadere S, Tosun M, Koken T,
Kinaci MK, Isikli B and Erkasap N: Protective effects of
doxycycline in ischemia/reperfusion injury on kidney. J Physiol
Biochem. 65:183–191. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tang S, Huang T, Jing H, Huang Z, Chen H,
Fan Y, Zhong J and Zhou J: Aldehyde dehydrogenase-2 acts as a
potential genetic target for renal fibrosis. Life Sci.
239:1170152019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wong SY, Woo J, Leung JC and Leung PC:
Depressive symptoms and lifestyle factors as risk factors of lower
urinary tract symptoms in Southern Chinese men: A prospective
study. Aging Male. 13:113–119. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu S, Chen J, Dong P, Zhang S, He Y, Sun
L, Zhu J, Cheng Y, Li X, Tang A, et al: Global gene expression
profiling identifies ALDH2, CCNE1 and SMAD3 as potential prognostic
markers in upper tract urothelial carcinoma. BMC Cancer.
14:8362014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xi XJ, Chen SH and Mi H: Aldh2 gene
reduces oxidative stress in the bladder by regulating the NF-κB
pathway in a mouse model of ketamine-induced cystitis. Exp Ther
Med. 20:1112020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu H, Hu Q, Ren K, Wu P, Wang Y and Lv C:
ALDH2 mitigates LPS-induced cardiac dysfunction, inflammation, and
apoptosis through the cGAS/STING pathway. Mol Med. 29:1712023.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu T, Guo J, Wei M, Wang J, Yang K, Pan C,
Pang J, Xue L, Yuan Q, Xue M, et al: Aldehyde dehydrogenase 2
protects against acute kidney injury by regulating autophagy via
the Beclin-1 pathway. JCI Insight. 6:e1381832021.PubMed/NCBI
|
|
20
|
Xi XJ, Zeng JJ, Lu Y, Chen SH, Jiang ZW,
He PJ and Mi H: Extracellular vesicles enhance oxidative stress
through P38/NF-kB pathway in ketamine-induced ulcerative cystitis.
J Cell Mol Med. 24:7609–7624. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shen CH, Wang ST, Lee YR, Liu SY, Li YZ,
Wu JD, Chen YJ and Liu YW: Biological effect of ketamine in
urothelial cell lines and global gene expression analysis in the
bladders of ketamine-injected mice. Mol Med Rep. 11:887–895. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
O'Leary NA, Wright MW, Brister JR, Ciufo
S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B,
Ako-Adjei D, et al: Reference sequence (RefSeq) database at NCBI:
Current status, taxonomic expansion, and functional annotation.
Nucleic Acids Res. 44:D733–D745. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Üremiş N and Üremiş MM:
Oxidative/nitrosative stress, apoptosis, and redox signaling: Key
players in neurodegenerative diseases. J Biochem Mol Toxicol.
39:e701332025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lawrence T: The nuclear factor NF-kappaB
pathway in inflammation. Cold Spring Harb Perspect Biol.
1:a0016512009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhou J, Scott C, Miab ZR and Lehmann C:
Current approaches for the treatment of ketamine-induced cystitis.
Neurourol Urodyn. 42:680–689. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jhang JF, Hsu YH and Kuo HC: Possible
pathophysiology of ketamine-related cystitis and associated
treatment strategies. Int J Urol. 22:816–825. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Matsumoto A: Fundamental Properties of
aldehyde dehydrogenase 2 (ALDH2) and the importance of the ALDH2
polymorphism. Nihon Eiseigaku Zasshi. 71:55–68. 2016.(In Japanese).
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang H and Fu L: The role of ALDH2 in
tumorigenesis and tumor progression: Targeting ALDH2 as a potential
cancer treatment. Acta Pharm Sin B. 11:1400–1411. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhong Z, Hu Q, Fu Z, Wang R, Xiong Y,
Zhang Y, Liu Z, Wang Y and Ye Q: Increased expression of aldehyde
dehydrogenase 2 reduces renal cell apoptosis during
ischemia/reperfusion injury after hypothermic machine perfusion.
Artif Organs. 40:596–603. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ramakrishnan S, Granger V, Rak M, Hu Q,
Attwood K, Aquila L, Krishnan N, Osiecki R, Azabdaftari G, Guru K,
et al: Inhibition of EZH2 induces NK cell-mediated differentiation
and death in muscle-invasive bladder cancer. Cell Death Differ.
26:2100–2114. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ji E, Jiao T, Shen Y, Xu Y, Sun Y, Cai Z,
Zhang Q and Li J: Molecular Mechanism of HSF1-Upregulated ALDH2 by
PKC in ameliorating pressure overload-induced heart failure in
mice. Biomed Res Int. 2020:34816232020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wohlfart DP, Lou B, Middel CS, Morgenstern
J, Fleming T, Sticht C, Hausser I, Hell R, Hammes HP, Szendrödi J,
et al: Accumulation of acetaldehyde in aldh2.1(−/-) zebrafish
causes increased retinal angiogenesis and impaired glucose
metabolism. Redox Biol. 50:1022492022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yeo D, Hwang SJ, Kim WJ, Youn HJ and Lee
HJ: The aqueous extract from Artemisia capillaris inhibits acute
gastric mucosal injury by inhibition of ROS and NF-kB. Biomed
Pharmacother. 99:681–687. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tsai SH, Hsu LA, Tsai HY, Yeh YH, Lu CY,
Chen PC, Wang JC, Chiu YL, Lin CY and Hsu YJ: Aldehyde
dehydrogenase 2 protects against abdominal aortic aneurysm
formation by reducing reactive oxygen species, vascular
inflammation, and apoptosis of vascular smooth muscle cells. FASEB
J. 34:9498–9511. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen Y, Xiong Y, Luo J, Hu Q, Lan J, Zou
Y, Ma Q, Yao H, Liu Z, Zhong Z and Ye Q: Aldehyde dehydrogenase 2
protects the kidney from ischemia-reperfusion injury by suppressing
the IκBα/NF-κB/IL-17C pathway. Oxid Med Cell Longev.
2023:22640302023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Marafie SK, Al-Mulla F and Abubaker J:
mTOR: Its critical role in metabolic diseases, cancer, and the
aging process. Int J Mol Sci. 25:61412024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Omolekan TO, Chamcheu JC, Buerger C and
Huang S: PI3K/AKT/mTOR signaling network in human health and
diseases. Cells. 13:15002024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zahid KR, Yao S, Khan ARR, Raza U and Gou
D: mTOR/HDAC1 crosstalk mediated suppression of ADH1A and ALDH2
links alcohol metabolism to hepatocellular carcinoma onset and
progression in silico. Front Oncol. 9:10002019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lin D, Xiang T, Qiu Q, Leung J, Xu J, Zhou
W, Hu Q, Lan J, Liu Z, Zhong Z, et al: Aldehyde dehydrogenase 2
regulates autophagy via the Akt-mTOR pathway to mitigate renal
ischemia-reperfusion injury in hypothermic machine perfusion. Life
Sci. 253:1177052020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Szwed A, Kim E and Jacinto E: Regulation
and metabolic functions of mTORC1 and mTORC2. Physiol Rev.
101:1371–1426. 2021. View Article : Google Scholar : PubMed/NCBI
|