Introduction
Reactive oxygen species (ROS) are mainly comprised
of hydrogen peroxide (H2O2), superoxide
anions (O2•−) and hydroxyl radicals
(•OH), which affect numerous cellular processes,
including metabolism, differentiation and cell proliferation and
death, by regulating critical signaling pathways (1,2).
Unlike other ROS, H2O2 is capable of freely
diffusing through biological membranes the width of several cells
prior to reacting with specific molecular targets. ROS are mostly
generated during the process of mitochondrial respiration and by
specific oxidases, including nicotine adenine diphosphate (NADPH)
oxidase and xanthine oxidase (3).
The main metabolic pathways use superoxide dismutases, which
metabolize O2•− to
H2O2(4).
Further metabolism by catalase or glutathione (GSH) peroxidase
yields O2 and H2O (5). Oxidative stress occurs through an
increase in ROS levels and/or a decrease in cellular antioxidants,
and leads to cell death (6–8). Exogenous H2O2 is
frequently used as a representative ROS in modeling and inducing
oxidative stress.
Three major groups of mitogen-activated protein
kinases (MAPKs) exist: extracellular signal-regulated kinase
(ERK1/2), c-Jun N-terminal kinase/stress-activated protein kinase
(JNK/SAPK) and p38 (9). MAPKs are
involved in crucial signaling pathways in cell proliferation,
differentiation and cell death in response to various signals
produced by growth factors, hormones and cytokines, as well as
genotoxic and oxidative stressors (9,10).
Each MAPK pathway has comparatively varied upstream activators and
unambiguous substrates (11).
Abundant evidence has demonstrated that JNK and p38 are activated
by ROS or mild oxidative shifts in the intracellular
thiol/disulfide redox state, initiating processes associated with
apoptosis (12,13). ROS provoke ERK phosphorylation and
also stimulate the ERK pathway (14). In the majority of instances, ERK
activation has a pro-survival effect rather than a pro-apoptotic
effect (15). In addition, MAPK
pathways are also activated by the direct inhibition of MAPK
phosphatases by the ROS. Since the differing and opposing effects
on MAPKs are caused by various ROS in the cells, the correlation
between ROS and MAPKs requires further clarification, particularly
with regard to the signaling associated with cell survival and
death.
Cultured normal human cells are invaluable
biological models for mechanistic studies of oxidative stress.
H2O2-induced cytotoxicity in normal
fibroblast cells in vitro may be of interest in
toxicological research with regard to the toxic potential of
exogenous H2O2 in human pulmonary fibroblasts
(HPFs) since HPFs are closely involved in lung inflammation,
fibrosis and cancer. However, the toxicological mechanism of the
effects of exogenous H2O2 on normal HPFs
remains unknown with regard to MAPKs. The present study
investigated the effects of the well-known antioxidants N-acetyl
cysteine (NAC) and propyl gallate (PG), as well as the MAPK
inhibitors, on H2O2-treated HPFs in relation
to cell growth and death and the ROS and GSH levels.
Materials and methods
Cell culture
HPFs purchased from PromoCell GmbH (Heidelberg,
Germany) were maintained in a humidified incubator at 37°C with 5%
CO2. The HPFs were cultured in RPMI-1640 supplemented
with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v)
penicillin-streptomycin (GIBCO BRL, Grand Island, NY, USA). The
HPFs were grown in 100-mm plastic tissue culture dishes (Nunc,
Roskilde, Denmark) and harvested with trypsin-EDTA solution while
in the logarithmic growth phase. The HPFs between passages four and
eight were used. The study was approved by the Ethics Committee of
Chonbuk National University, Jeonju, Republic of Korea.
Reagents
H2O2, NAC and PG were
purchased from Sigma-Aldrich Chemical Company (St. Louis, MO, USA).
The NAC was dissolved in buffer [20 mM HEPES (pH 7.0)], while the
PG was dissolved in ethanol at 200 mM as a stock solution. JNK
inhibitors (SP600125), MEK inhibitors (PD98059) and p38 inhibitors
(SB203580) were purchased from Calbiochem (San Diego, CA, USA). All
the inhibitors were dissolved in DMSO at 10 mM as stock solutions.
The HPFs were pretreated with 2 mM NAC, 400 μM PG or 10
μM MAPK inhibitors for 1 h prior to treatment with
H2O2. Ethanol (0.2%) and DMSO (0.2%) were
used as control vehicles and did not affect cell growth or
death.
Cell growth and cell number assays
The changes in cell growth in the HPFs were
indirectly determined by measuring the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma-Aldrich Chemical Company) dye absorbance. In brief,
4×104 cells per well were seeded in 96-well microtiter
plates (Nunc). Following exposure to 50 μM
H2O2, with or without 2 mM NAC, 400 μM
PG or 10 μM MAPK inhibitors for 24 h, 20 μl MTT
solution (2 mg/ml in PBS) was added to each well of the 96-well
plates. The plates were incubated for an additional 4 h at 37°C.
The media in the plates were withdrawn by pipetting and 200
μl DMSO was added to each well to solubilize the formazan
crystals. The optical density was measured at 570 nm using a
microplate reader (Synergy™ 2; BioTek Instruments Inc., Winooski,
VT, USA).
Annexin V-fluorescein isothiocyanate
(FITC) staining for cell death detection
Apoptosis was determined by staining cells with
annexin (Invitrogen Corporation, Camarillo, CA, USA; Ex/Em = 488
nm/519 nm). In brief, 1×106 cells were incubated in a
60-mm culture dish (Nunc) with 50 μM
H2O2, with or without 2 mM NAC, 400 μM
PG or 10 μM MAPK inhibitors for 24 h. The cells were washed
twice with cold PBS, then resuspended in 500 μl binding
buffer (10 mM HEPES/NaOH, pH 7.4; 140 mM NaCl; 2.5 mM
CaCl2) at a concentration of 1×106 cells/ml.
Annexin V-FITC (5 μl) was then added to the cells, which
were analyzed with a FACStar flow cytometer (Becton Dickinson,
Franklin Lakes, NJ, USA).
Measurement of mitochondrial membrane
potential (MMP; Δψm)
MMP (Δψm) levels were measured with a
rhodamine 123 fluorescent dye (Sigma-Aldrich Chemical Company;
Ex/Em = 485 nm/ 535 nm). In brief, 1×106 cells were
incubated in a 60-mm culture dish (Nunc) with 50 μM
H2O2, with or without 2 mM NAC, 400 μM
PG or 10 μM MAPK inhibitors for 24 h. The cells were washed
twice with PBS and incubated with the rhodamine 123 (0.1
μg/ml) at 37°C for 30 min. Rhodamine 123 staining intensity
was determined with a FACStar flow cytometer (Becton Dickinson).
Rhodamine 123-negative cells indicated the loss of MMP
(Δψm) in cells.
Detection of intracellular ROS
levels
Intracellular ROS such as
H2O2, •OH and ONOO•
were detected using an oxidation-sensitive fluorescent probe dye,
2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA;
Ex/Em = 495 nm/529 nm; Invitrogen Molecular Probes, Eugene, OR,
USA). H2DCFDA is poorly selective for
O2•−. By contrast, dihydroethidium (DHE;
Ex/Em = 518 nm/605 nm; Invitrogen Molecular Probes) is highly
selective for O2•− among all the ROS. In
brief, 1×106 cells were incubated in a 60-mm culture
dish (Nunc) with 50 μM H2O2, with or
without 2 mM NAC, 400 μM PG or 10 μM MAPK inhibitors
for 24 h. The cells were then incubated with 20 μM
H2DCFDA or dihydroethidium (DHE) at 37°C for 30 min. The
fluorescence of DCF and DHE was detected using a FACStar flow
cytometer (Becton Dickinson). The ROS and
O2•− levels were expressed as the mean
fluorescence intensity (MFI), which was calculated by CellQuest
software (Becton Dickinson).
Detection of the intracellular GSH
The cellular GSH levels were analyzed using a
5-chloromethylfluorescein diacetate dye (CMFDA, Ex/Em = 522 nm/595
nm; Invitrogen Molecular Probes). In brief, 1×106 cells
were incubated in a 60-mm culture dish (Nunc) with 50 μM
H2O2, with or without 2 mM NAC, 400 μM
PG or 10 μM MAPK inhibitors for 24 h. The cells were then
incubated with 5 μM CMFDA at 37°C for 30 min. The CMF
fluorescence intensity was determined using a FACStar flow
cytometer (Becton Dickinson). CMF-negative (GSH-depleted) cells
were expressed as the percent of CMF− cells.
Statistical analysis
The results represent the mean of at least two
independent experiments (mean ± SD). The data were analyzed using
Instat software (GraphPad Prism4; GraphPad Software, San Diego, CA,
USA). The Student’s t-test and a one-way analysis of variance
(ANOVA) with post hoc analysis, using Tukey’s multiple comparison,
were applied to the parametric data. P<0.05 was considered to
indicate a statistically significant difference.
Results
Effects of NAC and PG on cell growth and
death and MMP (Δψm) levels in
H2O2-treated HPFs
The effects of NAC and PG on cell growth and death
and MMP (Δψm) levels were investigated in
H2O2-treated HPFs at 24 h using MTT assays. A
concentration of 50 μM H2O2 was used
as an optimal dose in this experiment; this inhibited the growth of
the HPFs by ∼45% in 24 h (Fig. 1A).
NAC and PG significantly reduced the growth inhibition caused by
H2O2, with PG showing a more marked effect
(Fig. 1A).
H2O2 increased the percentage of annexin
V-FITC stained cells among the HPFs, indirectly indicating that the
HPF cell death caused by H2O2 occurred via
apoptosis (Fig. 1B). NAC and PG
significantly reduced the number of annexin V-FITC-positive cells
in the H2O2-treated HPFs, while PG completely
prevented the HPF cell death caused by H2O2
(Fig. 1B). Notably, PG alone
increased the number of annexin V-FITC-positive cells among the
control HPFs (Fig. 1B). Since cell
death is closely associated with the collapse of MMP
(Δψm) (16), the effect
of H2O2 on MMP (Δψm) in the HPFs
was assessed using a rhodamine 123 dye. Treatment with 50 μM
H2O2 significantly induced the loss of MMP
(Δψm) in the HPFs (Fig.
1C). NAC and PG attenuated the loss of MMP (Δψm)
caused by H2O2, while PG totally prevented
this loss (Fig. 1C). Similar to the
number of annexin V-FITC-positive cells, PG also increased the
number of cells that lost MMP (Δψm) among the control
HPFs (Fig. 1C).
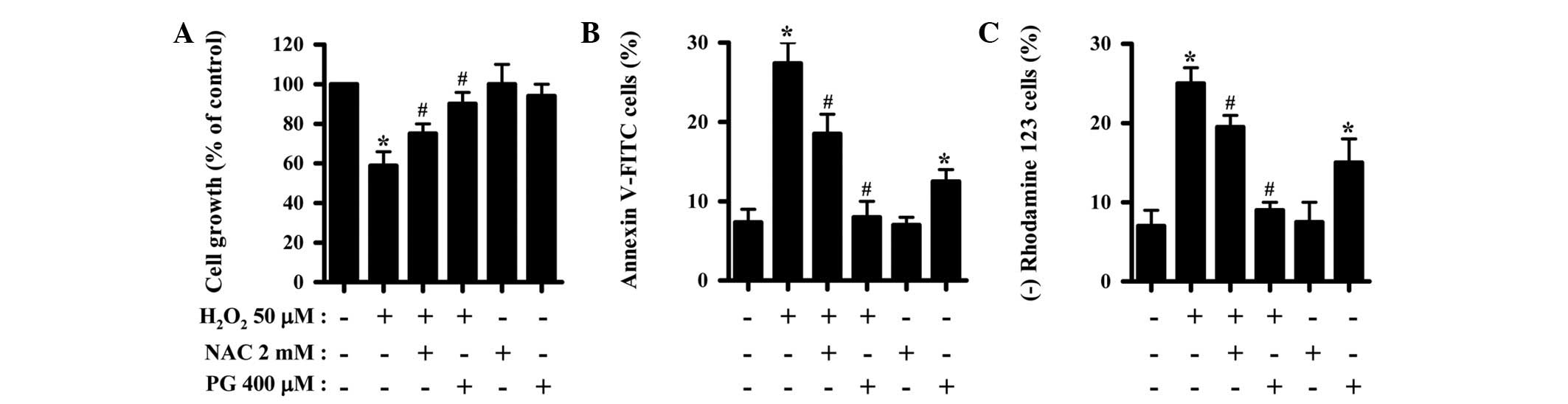 | Figure 1Effects of NAC and PG on cell growth
and death and MMP (Δψm) levels in the
H2O2-treated HPFs. Exponentially-growing HPFs
were treated with 50 μM H2O2 for 24 h
following a 1 h pre-incubation with 2 mM NAC or 400 μM PG.
(A) Cellular growth changes in HPFs, as assessed by MTT assays. (B)
Percentages of annexin V-FITC-positive cells, as measured by flow
cytometry. (C) Percentages of rhodamine 123-negative [MMP
(Δψm) loss] cells. *P<0.05 compared with
the control group. #P<0.05 compared with cells
treated with H2O2 only. NAC, N-acetyl
cysteine; PG, propyl gallate; MMP, mitochondrial membrane
potential; HPF, human pulmonary fibroblast; MTT,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide;
V-FITC, Annexin V-fluorescein isothiocyanate. |
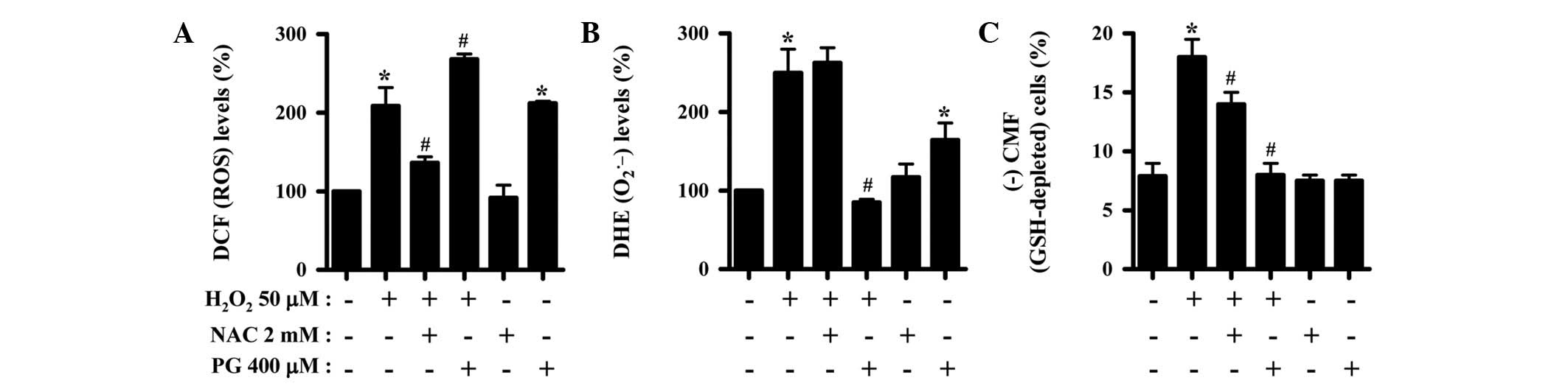
Effects of NAC and PG on intracellular
ROS and GSH levels in H2O2-treated HPFs
H2DCFDA and DHE dyes were used to assess
intracellular ROS levels in the H2O2-treated
HPFs. As shown in Fig. 2A, the
levels of ROS (DCF), such as H2O2, were
increased in the HPFs treated with 50 μM at 24 h. NAC
significantly suppressed the increased ROS levels in the
H2O2-treated HPFs, whereas PG enhanced the
increase in ROS levels caused by H2O2
(Fig. 2A). Moreover, PG alone
markedly increased the ROS (DCF) levels in the control HPFs
(Fig. 2A). When the intracellular
O2•− levels were assessed in the
H2O2-treated HPFs, the level of DHE
fluorescence dye, which specifically indicates
O2•− accumulation in cells, was increased at
24 h (Fig. 2B). While NAC did not
alter the O2•− level in the
H2O2-treated HPFs, PG entirely attenuated the
increase in these cells (Fig. 2B).
However, PG alone increased the O2•− level in
the control HPFs (Fig. 2B). When
the intracellular GSH levels were measured in the
H2O2-treated HPFs using a CMFDA dye, 50
μM H2O2 was shown to increase the
number of GSH-depleted cells in the HPFs at 24 h (Fig. 2C). NAC and PG significantly reduced
the number of GSH-depleted cells in the
H2O2-treated HPFs, while PG completely
prevented the GSH depletion (Fig.
2C).
Effects of MAPK inhibitors on cell growth
and death and MMP (Δψm) levels in
H2O2-treated HPFs
The effect of the MAPK inhibitors on cell growth and
death and MMP (Δψm) levels in the
H2O2-treated HPFs was evaluated. Based on
previous studies (17,18), 10 μM of each MAPK inhibitor
was used as an optimal dose in the present study. None of the MAPK
inhibitors affected the growth inhibition caused by
H2O2 (Fig.
3A). p38 inhibitor alone increased the growth of control HPFs
(Fig. 3A). Additionally, none of
the MAPK inhibitors affected the number of annexin V-stained cells
among the H2O2-treated or -untreated HPFs
(Fig. 3B). All the MAPK inhibitors
appeared to enhance the loss of MMP (Δψm) in the
H2O2-treated HPFs (Fig. 3C).
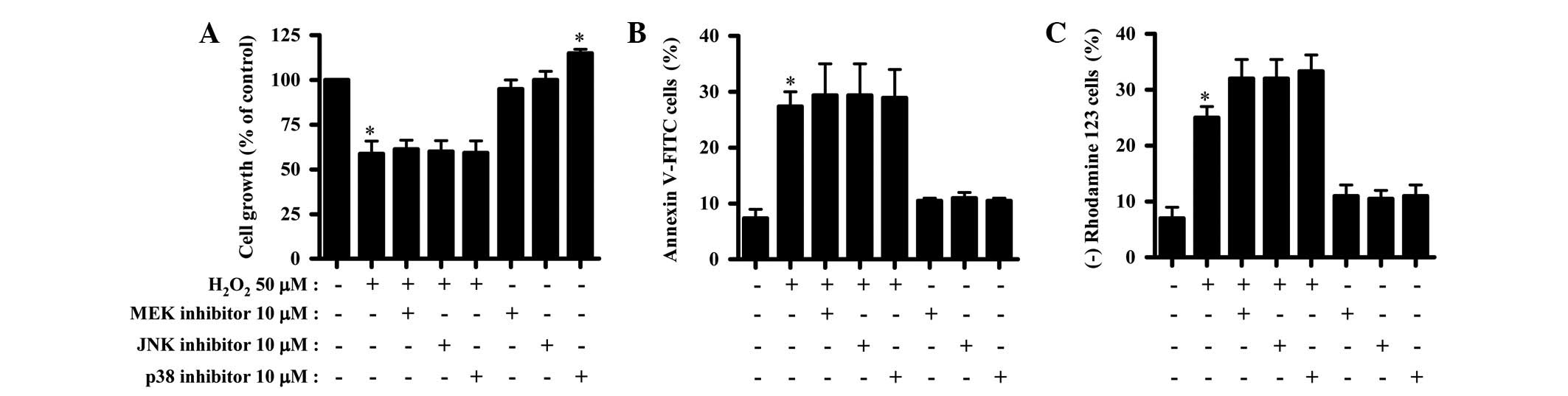 | Figure 3Effects of MAPK inhibitors on cell
growth and death and MMP (Δψm) levels in
H2O2-treated HPFs. Exponentially-growing HPFs
were treated with 50 μM H2O2 for 24 h
following a 1 h pre-incubation with each MAPK inhibitor. (A)
Cellular growth changes in the HPFs, as assessed by MTT assays. (B)
Percentages of annexin V-FITC-positive cells, as measured by
FACStar flow cytometry. (C) Percentages of rhodamine 123-negative
[MMP (Δψm) loss] cells. *P<0.05 compared
with the control group. #P<0.05 compared with cells
treated with H2O2 only. MAPK,
mitogen-activated protein kinase; MEK, MAP/ERK kinase; JNK, c-Jun
N-terminal kinase; MMP, mitochondrial membrane potential; HPF,
human pulmonary fibroblast; MTT,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide;
V-FITC, Annexin V-fluorescein isothiocyanate. |
Effects of MAPK inhibitors on
intracellular ROS and GSH levels in
H2O2-treated HPFs
The changes in intracellular ROS levels in the
H2O2 and/or each MAPK inhibitor-treated HPF
were assessed. As shown in Fig. 4A,
the MEK and JNK inhibitors only marginally enhanced the ROS levels
in the H2O2-treated HPFs, whereas the p38
inhibitor appeared to decrease the ROS levels (Fig. 4A). In addition, all the MAPK
inhibitors marginally intensified the O2•−
level increase caused by H2O2 (Fig. 4B). The p38 inhibitor alone increased
the ROS levels, including the O2•− level, in
the HPF control cells (Fig. 4A and
B). Moreover, none of the MAPK inhibitors affected the number
of GSH-depleted cells in the H2O2-treated or
untreated HPFs (Fig. 4C).
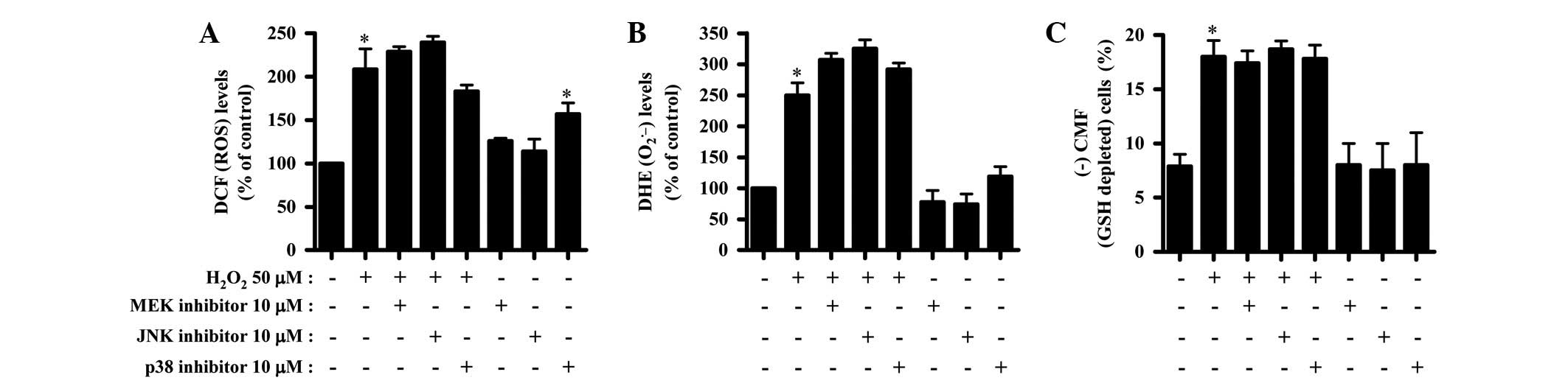 | Figure 4Effects of MAPK inhibitors on
intracellular ROS and GSH levels in
H2O2-treated HPFs. Exponentially-growing HPFs
were treated with 50 μM H2O2 for 24 h
following a 1 h pre-incubation with each MAPK inhibitor. ROS levels
in the HPFs were measured using a FACStar flow cytometer. (A) DCF
(ROS) levels (%) in the HPFs compared with the control cell group.
(B) DHE (O2•−) levels (%) in the
HPFs compared with the control cell group. (C) CMF−
(GSH-depleted) cells (%) in the HPFs. *P<0.05
compared with the control group. #P<0.05 compared
with cells treated with H2O2 only. MAPK,
mitogen-activated protein kinase; MEK, MAP/ERK kinase; JNK, c-Jun
N-terminal kinase; ROS, reactive oxygen species; HPF, human
pulmonary fibroblast; GSH, glutathione; DCF,
2′,7′-dichlorodihydrofluorescein diacetate; DHE, dihydroethidium;
CMF, 5-chloromethylfluorescein diacetate. |
Discussion
HPFs are pathophysiologically involved in lung
inflammation, fibrosis and cancer since these cells synthesize
extracellular matrix and collagen to maintain the structural and
functional integrity of the lung. The present study focused on
elucidating the cytotoxic effect of exogenous
H2O2 on cell growth and death in normal HPFs,
in relation to ROS and MAPKs. Treatment with 50 μM
H2O2 inhibited the growth of HPFs by ∼45% in
24 h. H2O2 increased the number of annexin
V-FITC-positive cells among the HPFs, indicating that
H2O2-induced HPF cell death occurred via
apoptosis. An increase in caspase-3 activity was also observed in
the H2O2-treated HPFs (data not shown). In
addition, H2O2 triggered the loss of MMP
(Δψm) in the HPFs. The level cells with MMP
(Δψm) loss appeared to be similar to that of the annexin
V stained cells, suggesting that cell death caused by
H2O2 was markedly correlated with the
collapse of MMP (Δψm).
ROS toxicity in cells is generally mediated by
•OH (19).
H2O2 and O2•− are the
main ROS involved in the cell signaling pathways. According to the
present results, the ROS levels, including those of
O2•−, were significantly increased in the
HPFs treated with H2O2. Since 50 μM
H2O2 induced cell death and MMP
(Δψm) loss in the HPFs, it is possible that exogenous
H2O2 generates O2•− by
damaging the mitochondria and that H2O2 and
O2•− may be efficiently converted into toxic
•OH via the Fenton reaction, resulting in the death of
the HPFs. It is also possible that H2O2
activates oxidases, such as NADPH oxidase and xanthine oxidase, in
HPFs to generate O2•−. In th epresent study,
NAC attenuated the growth inhibition and cell death of the
H2O2-treated HPFs and also significantly
attenuated the MMP (Δψm) loss in these cells. NAC
markedly decreased the ROS (DCF) levels in the
H2O2-treated HPFs. However, NAC did not
reduce the increased O2•− (DHE) level caused
by H2O2, suggesting that NAC did not block
the O2•− generation pathway induced by
exogenous H2O2. In addition, the
O2•− level in the HPFs co-treated with
H2O2 and NAC did not appear to be correlated
with HPF cell death.
PG, as a synthetic antioxidant, exerts a variety of
effects on tissues and cells. For example, PG is an efficient
protector of liver cells against lipid peroxidation by oxygen
radicals (20). By contrast, PG has
pro-oxidant properties (21,22).
The anti-oxidative and cytoprotective properties of PG may change
to pro-oxidative, cytotoxic and genotoxic properties in the
presence of Cu(II) (23). The
present results demonstrated that PG alone marginally inhibited the
growth of the HPFs and induced cell death accompanied by the loss
of MMP (Δψm). In addition, PG increased the ROS levels,
including those of O2•−, in the HPFs. Thus,
it is possible that PG, as a pro-oxidant, is able to directly
generate mitochondrial O2•− in the HPFs by
impairing mitochondrial function, consequently leading to HPF cell
death via oxidative stress. Similarly, it has been reported that PG
causes cytotoxic effects in isolated rat hepatocytes by causing
mitochondrial damage (24) and that
it also increases mitochondrial
O2•− levels in HeLa cells
(25). Notably, PG markedly
attenuated the growth inhibition and cell death in the
H2O2-treated HPFs and also prevented MMP
(Δψm) loss in these cells. Moreover, PG completely
abrogated the O2•− (DHE) level increase
caused by H2O2. Therefore, PG appeared to
protect the HPFs against exogenous H2O2 by
protecting the mitochondria. However, PG increased the ROS (DCF)
levels in the H2O2-treated HPFs. Numerous
studies, including the present study, support the hypothesis that
PG has a role as an antioxidant (20,26,27) or
as a pro-oxidant (21,22), depending on various conditions, such
as the cell culture media, the co-treated drugs and the cell types.
PG is likely to have differing effects on the levels of the
different ROS in the cells. Further studies are required to
elucidate the exact roles of the types of ROS in PG-treated
HPFs.
In the present study, the MEK inhibitor, which is
likely to inactivate ERK, did not affect growth inhibition or cell
death in the H2O2-treated HPFs. Thus,
H2O2 did not directly regulate the signaling
associated with ERK in the HPFs to induce their growth inhibition
and death. In addition, the JNK and p38 MAPKs, which are generally
associated with cell death (12,13),
were not likely to be affected by H2O2 in the
HPFs since none of the inhibitors affected the growth inhibition
and cell death caused by H2O2. The p38
inhibitor alone increased the growth of the control HPFs,
suggesting that p38 signaling is involved in the basal level of HPF
growth. With regard to MMP (Δψm), all the MAPK
inhibitors marginally increased the loss of MMP (Δψm) in
the H2O2-treated HPFs, indicating that the
dysregulation of these MAPK signalings enhanced the loss in these
cells. Moreover, the MAPK inhibitors marginally, but not
significantly, affected the ROS levels, including that of
O2•−, in the
H2O2-treated HPFs. The p38 inhibitor
increased the ROS levels in the HPF control cells regardless of the
level of cell death, while the other inhibitors mildly affected the
ROS levels, including that of O2•−. Thus, the
MAPK signalling in the H2O2-treated and
-untreated HPFs did not meaningfully change the redox state to
affect HPF death.
GSH is a key cellular non-protein antioxidant,
which reduces H2O2 to H2O using
GSH peroxidase (28). The
intra-cellular GSH content has a significant effect on anticancer
drug-induced apoptosis, indicating that apoptotic effects are
inversely proportional to the GSH content (29,30).
Similarly, in the present study, H2O2
increased the number of GSH-depleted cells in the HPFs. NAC and PG
demonstrated anti-apoptotic effects on the
H2O2-treated HPFs, significantly suppressing
the GSH depletion in these cells. In addition, none of the MAPK
inhibitors affected the GSH depletion in the
H2O2-treated HPFs. Therefore, the
intracellular GSH content appears to be a decisive factor in HPF
cell death. However, PG alone induced cell death in the HPF control
cells but it did not significantly induce GSH depletion,,
suggesting that PG-induced HPF cell death is not highly associated
with changes in the GSH level.
In conclusion, H2O2 induced
growth inhibition and death in the HPFs via GSH depletion. NAC and
PG attenuated H2O2-induced HPF cell growth
inhibition and death, but each antioxidant affected the ROS levels,
including that of O2•−, differently in the
H2O2-treated and -untreated HPFs. Treatment
with MAPK inhibitors did not affect cell death or the ROS levels in
the H2O2-treated HPFs. The present data
provide useful information concerning the toxicological effect of
exogenous H2O2 on normal HPFs with regard to
ROS and MAPKs.
Abbreviations:
|
HPF
|
human pulmonary fibroblast;
|
|
ROS
|
reactive oxygen species;
|
|
MAPK
|
mitogen-activated protein kinase;
|
|
MEK
|
MAP/ERK kinase;
|
|
ERK
|
extracellular signal-regulated
kinase;
|
|
JNK
|
c-Jun N-terminal kinase;
|
|
MMP (Δψm)
|
mitochondrial membrane potential;
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide;
|
|
FITC
|
fluorescein isothiocyanate;
|
|
H2DCFDA
|
2′,7′-dichloro dihydrofluorescein
diacetate;
|
|
DHE
|
dihydroethidium;
|
|
GSH
|
glutathione;
|
|
CMFDA
|
5-chloromethylfluorescein
diacetate;
|
|
NAC
|
N-acetyl cysteine;
|
|
PG
|
propyl gallate
|
Acknowledgements
The present study was supported by a
grant from the Ministry of Science and Technology (MoST)/Korea
Science and Engineering Foundation (KOSEF) through the Diabetes
Research Center at Chonbuk National University (2012-0009323).
References
|
1
|
Gonzalez C, Sanz-Alfayate G, Agapito MT,
Gomez-Niño A, Rocher A and Obeso A: Significance of ROS in oxygen
sensing in cell systems with sensitivity to physiological hypoxia.
Respir Physiol Neurobiol. 132:17–41. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baran CP, Zeigler MM, Tridandapani S and
Marsh CB: The role of ROS and RNS in regulating life and death of
blood monocytes. Curr Pharm Des. 10:855–866. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial ROS-induced ROS release: an update and review.
Biochim Biophys Acta. 1757:509–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zelko IN, Mariani TJ and Folz RJ:
Superoxide dismutase multigene family: a comparison of the CuZn-SOD
(SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures,
evolution, and expression. Free Radic Biol Med. 33:337–349. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wilcox CS: Reactive oxygen species: roles
in blood pressure and kidney function. Curr Hypertens Rep.
4:160–166. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen TJ, Jeng JY, Lin CW, Wu CY and Chen
YC: Quercetin inhibition of ROS-dependent and -independent
apoptosis in rat glioma C6 cells. Toxicology. 223:113–126. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dasmahapatra G, Rahmani M, Dent P and
Grant S: The tyrphostin adaphostin interacts synergistically with
proteasome inhibitors to induce apoptosis in human leukemia cells
through a reactive oxygen species (ROS)-dependent mechanism. Blood.
107:232–240. 2006. View Article : Google Scholar
|
|
8
|
Wallach-Dayan SB, Izbicki G, Cohen PY,
Gerstl-Golan R, Fine A and Breuer R: Bleomycin initiates apoptosis
of lung epithelial cells by ROS but not by Fas/FasL pathway. Am J
Physiol Lung Cell Mol Physiol. 290:L790–L796. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Genestra M: Oxyl radicals, redox-sensitive
signalling cascades and antioxidants. Cell Signal. 19:1807–1819.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Blenis J: Signal transduction via the MAP
kinases: proceed at your own RSK. Proc Natl Acad Sci USA.
90:5889–5892. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kusuhara M, Takahashi E, Peterson TE, Abe
J, Ishida M, Han J, Ulevitch R and Berk BC: p38 Kinase is a
negative regulator of angiotensin II signal transduction in
vascular smooth muscle cells: effects on
Na+/H+ exchange and ERK1/2. Circ Res.
83:824–831. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hsin YH, Chen CF, Huang S, Shih TS, Lai PS
and Chueh PJ: The apoptotic effect of nanosilver is mediated by a
ROS- and JNK-dependent mechanism involving the mitochondrial
pathway in NIH3T3 cells. Toxicol Lett. 179:130–139. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mao X, Yu CR, Li WH and Li WX: Induction
of apoptosis by shikonin through a ROS/JNK-mediated process in
Bcr/Abl-positive chronic myelogenous leukemia (CML) cells. Cell
Res. 18:879–888. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guyton KZ, Liu Y, Gorospe M, Xu Q and
Holbrook NJ: Activation of mitogen-activated protein kinase by
H2O2Role in cell survival following oxidant
injury. J Biol Chem. 271:4138–4142. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Henson ES and Gibson SB: Surviving cell
death through epidermal growth factor (EGF) signal transduction
pathways: implications for cancer therapy. Cell Signal.
18:2089–2097. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Park WH: Mitogen-activated protein kinase
inhibitors differently affect the growth inhibition and death of a
proteasome inhibitor, MG132-treated human pulmonary fibroblast
cells. Hum Exp Toxicol. 30:1945–1954. 2011. View Article : Google Scholar
|
|
18
|
Park WH: MAPK inhibitors and siRNAs
differentially affect cell death and ROS levels in arsenic
trioxide-treated human pulmonary fibroblast cells. Oncol Rep.
27:1611–1618. 2012.PubMed/NCBI
|
|
19
|
Perez-Vizcaino F, Cogolludo A and Moreno
L: Reactive oxygen species signaling in pulmonary vascular smooth
muscle. Respir Physiol Neurobiol. 174:212–220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu TW, Fung KP, Zeng LH, Wu J and Nakamura
H: Propyl gallate as a hepatoprotector in vitro and in vivo.
Biochem Pharmacol. 48:419–422. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kobayashi H, Oikawa S, Hirakawa K and
Kawanishi S: Metal-mediated oxidative damage to cellular and
isolated DNA by gallic acid, a metabolite of antioxidant propyl
gallate. Mutat Res. 558:111–120. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kawanishi S, Oikawa S and Murata M:
Evaluation for safety of antioxidant chemopreventive agents.
Antioxid Redox Signal. 7:1728–1739. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jacobi H, Eicke B and Witte I: DNA strand
break induction and enhanced cytotoxicity of propyl gallate in the
presence of copper(II). Free Radic Biol Med. 24:972–978. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nakagawa Y, Nakajima K, Tayama S and
Moldéus P: Metabolism and cytotoxicity of propyl gallate in
isolated rat hepatocytes: effects of a thiol reductant and an
esterase inhibitor. Mol Pharmacol. 47:1021–1027. 1995.PubMed/NCBI
|
|
25
|
Han YH and Park WH: Propyl gallate
inhibits the growth of HeLa cells via regulating intracellular GSH
level. Food Chem Toxicol. 47:2531–2538. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Reddan JR, Giblin FJ, Sevilla M,
Padgaonkar V, Dziedzic DC, Leverenz VR, Misra IC, Chang JS and Pena
JT: Propyl gallate is a superoxide dismutase mimic and protects
cultured lens epithelial cells from H2O2
insult. Exp Eye Res. 76:49–59. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen CH, Liu TZ, Chen CH, Wong CH, Chen
CH, Lu FJ and Chen SC: The efficacy of protective effects of tannic
acid, gallic acid, ellagic acid, and propyl gallate against
hydrogen peroxide-induced oxidative stress and DNA damages in
IMR-90 cells. Mol Nutr Food Res. 51:962–968. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rhee SG, Yang KS, Kang SW, Woo HA and
Chang TS: Controlled elimination of intracellular H(2)O(2):
regulation of peroxiredoxin, catalase, and glutathione peroxidase
via post-translational modification. Antioxid Redox Signal.
7:619–626. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Estrela JM, Ortega A and Obrador E:
Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci.
43:143–181. 2006. View Article : Google Scholar
|
|
30
|
Higuchi Y: Glutathione depletion-induced
chromosomal DNA fragmentation associated with apoptosis and
necrosis. J Cell Mol Med. 8:455–464. 2004. View Article : Google Scholar : PubMed/NCBI
|