Introduction
Lung cancer is the most commonly diagnosed type of
cancer in males and the leading cause of cancer mortality in each
gender in economically developed and developing countries (1). Non-small cell lung carcinoma (NSCLC)
accounted for ∼85% of the all lung cancer cases (2). Standard lung cancer treatment
modalities include surgery, chemotherapy, targeted therapy and
radiation therapy; however, not all patients benefit from routine
therapy. The overall 5-year survival rate of lung cancer patients
remains relatively low at ∼15% (2).
Therefore, the identification of useful biomarkers and exploration
of novel therapeutic targets are necessary and demanding tasks.
The protein kinase cAMP-dependent catalytic β
(PRKACB) gene is located at chromosome site 1p31.1 and encodes
cAMP-dependent protein kinase A (PKA) catalytic subunit β. The
PRKACB protein is a member of the Ser/Thr protein kinase family and
a key effector of the cAMP/PKA-induced signal pathway that is
involved in numerous cellular processes, including cell
proliferation, apoptosis, gene transcription, metabolism and
differentiation (3). Typically, PKA
is an inactive holoenzyme consisting of two catalytic (C) subunits
bound to a regulatory (R) subunit dimer. When four cAMP molecules
bind the R subunits, the C subunits are released (4) and free active catalytic subunits
phosphorylate serine and threonine residues on specific substrate
proteins, which include C-Raf, RhoA, Src and CUTL1, that are
involved in cellular proliferation, apoptosis, differentiation and
invasion (5–8). In the human enzyme, four different R
subunits (RIα, RIβ, RIIα and RIIβ) and four different C subunits
(Cα, Cβ, Cγ and PrKX) have been identified (3). In total, ten different splice variants
encoded by the PRKACB gene have been found and a certain number of
these were revealed to be expressed in human brain, lymphoid and
neuronal tissues (9–11). Multiple PRKACB subunits have also
been observed in human prostate specimens and it appears that the
PRKACB variants play varying roles in proliferation and
differentiation of prostate cancer progression (12). It has been demonstrated that
transcription of PRKACB may be directly activated by c-MYC, which
is associated with tumorigenesis by the promotion of cell
proliferation (13). It has also
been shown that a variant of PRKACB phosphorylates the p75
neutrophin receptor (p75NTR) and regulates its localization to
lipid rafts (14). PRKACB was
identified as a candidate gene that is directly or indirectly
involved in apoptosis in human mantle cell lymphoma (MCL) tumors
(15). In addition, a novel
interaction between PRKACB, the cell cycle and apoptosis regulatory
protein-1 (CARP-1) was identified and confirmed by
glutathione-S-transferase (GST) pull-down experiments in brain
tissue (16). However, limited
information is known with regard to its expression and role in
human NSCLC.
The present study aimed to assess the role of PRKACB
in the development and progress of human NSCLC. The mRNA and
protein expression patterns of PRKACB were first examined in the
NSCLC and corresponding normal tissues. Moreover, plasmid vectors
containing full-length PRKACB and transfected human adenocarcinoma
LTEP-A2 cells were constructed to increase the PRKACB expression.
The effects of PRKACB upregulation on cell proliferation,
clonogenicity, apoptosis and invasion were then investigated in the
LTEP-A2 cells.
Materials and methods
Tissue samples and patients
NSCLC tissues (12 cases of lung squamous cell
carcinoma tissues, 18 cases of lung adenocarcinoma tissues; 22 of
these 30 cases presented with lymph node metastasis) and their
corresponding normal tissues (30 cases) were collected from 30
patients who underwent surgery at the Department of Thoracic
Surgery, The Fourth Affiliated Hospital of China Medical
University, Shenyang, Liaoning, China, between 2008 and 2012. All
tumor tissues were diagnosed histopathologically by at least two
trained pathologists. Written informed consent was obtained from
all patients prior to surgery and the study protocol was approved
by the Institutional Review Board for the use of Human Subjects at
China Medical University (Shenyang, China). None of the patients
received pre-operative chemotherapy or radiation therapy.
Surgically-removed tumors and matched normal tissues were
immediately frozen in liquid nitrogen and kept at −80°C until the
extraction of the RNA and protein.
RNA extraction and real-time RT-PCR
Total RNA from the frozen tissues was isolated using
TRIzol reagent (Takara Bio Inc., Dalian, Liaoning, China).
Quantitative real-time polymerase chain reaction (QPCR) was
conducted using SYBR Premix Ex Taq (Takara Bio Inc.) in a total
volume of 20 μl using a 7300 Real-Time PCR System (Applied
Biosystems, Foster City, CA, USA). The PCR conditions were;
denaturation at 95°C for 30 sec, followed by a further 40 cycles of
denaturation at 95°C for 5 sec, and finally annealing at 60°C for
31 sec. The sequences of the primer pairs are as follows: PRKACB
forward, 5′-AGTGGTTTGCCACGACAGATTG-3′; and reverse,
5′-TTGCTGGTACCAGAGCCTCTAA-3′; GAPDH forward
5′-GCACCGTCAAGGCTGAGAAC-3′; and reverse, 5′-TGGTGAAGACGCCAGTGGA-3′.
GAPDH was used as the reference gene. The relative levels of gene
expression were calculated using the 2−ΔCt method (ΔCt =
Ct of PRKACB − Ct of GAPDH) and the fold change of gene expression
was calculated by the 2−ΔΔCt method. All experiments
were repeated in triplicate.
Western blot analysis
The total protein from the frozen tissues was
extracted in a lysis buffer (Beyotime Biotechnology, Haimen,
Jiangsu, China) and the protein content was determined using the
bicinchoninic acid (BCA) assay (Beyotime Biotechnology). A total of
80 μg total protein was separated by sodium dodecyl sulfate
polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred
onto polyvinylidene fluoride (PVDF) membranes. Subsequent to
blocking with 5% bovine serum albumin (BSA), PRKACB antibody
(1:500; Santa Cruz) and GAPDH antibody (1:500; Santa Cruz) were
incubated on membranes for PRKACB and GAPDH protein overnight at
4°C. The membranes were then incubated for 2 h at 37°C with goat
anti-rabbit IgG (1:4000; Beijing Biosynthesis Biotechnology Co.,
Ltd., Beijing, China). Immunoreactive strips were identified using
the enhanced chemiluminescence (ECL) system (Beyotime
Biotechnology) following the manufacturer’s instructions. The DNR
Imaging System (DNR Bio-Imaging Systems, Israel) was used to
identify the specific bands, and the optical density of each band
was measured using Image J software (NIH, Bethesda, MD, USA). The
ratio between the integrated optical density (IOD) of PRKACB and
GAPDH of the same sample was calculated as the relative content and
expressed graphically.
Cell culture and transfection
Lung adenocarcinoma LTEP-A2 cells were obtained from
the Shanghai Cell Bank (Shanghai, China). The cells were grown in
RPMI-1640, supplemented with 10% fetal bovine serum (FBS; Hyclone,
USA) and placed in an incubator with 5% CO2 at 37°C. To
increase the PRKACB expression for subsequent experiments, the
LTEP-A2 cells (60–70% confluence) were transfected with a plasmid
containing full-length PRKACB (pEGFP-C1-PRKACB) and the vector
control (pEGFP-C1; Takara Bio Inc.) for 48 h using Lipofectamine
LTX with PLUS reagent (Invitrogen, Carlsbad, CA, USA), according to
the manufacturer’s instructions. The experiments were repeated at
least three times. The efficiency of the transfection in the
experiments was >50%. Following 36–48 h of transfection, the
cells with high PRKACB expression were confirmed by real-time
RT-PCR and western blot analysis.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
The MTT assay was used to evaluate the proliferation
of the transfected cells. The cells were detached and seeded into
five 96-well plates (5×103 cells/100μl/well) in
parallel and transfected with PRKACB and the vector control. During
the following 4 days, the absorbance of one indicated plate was
examined each day, and the cells in the other plates were cultured
continuously. A total of 20 μl MTT (5 mg/ml) was added to
each well of the indicated plate, and 4 h later the liquids were
removed and 150 μl dimethyl sulphoxide (DMSO) was added.
Following 10 min of agitation, the absorbance was measured using a
microplate reader (TECAN, Männedorf, Switzerland) at 492 nm. The
results were plotted as the mean ± SD of five determinations.
Colony formation assay
The cells were transfected with PRKACB and the
vector for 24 h. Thereafter, 200 cells were planted into 6-cm cell
culture dishes and incubated for 14 days. The plates were stained
with Giemsa, and colonies with >50 cells were counted.
Cell apoptosis assay
Cell apoptosis was examined by flow cytometry using
an Annexin V-PE/7-aminoactinomycin D (7-AAD) apoptosis detection
kit (KeyGEN Biotech., Nanjing, China), following the manufacturer’s
instructions. At 24 h post-transfection, the cells were washed
twice in ice-cold PBS. The cells (100 μl; 1×105)
were gently mixed with 50 μl binding buffer and 5 μl
7-AAD and then incubated for 15 min at room temperature in the
dark. Subsequent to supplementation with another 450 μl
binding buffer, 1 μl Annexin V-PE was added to the cells,
which were then incubated for another 15 min at room temperature in
the dark. Cell apoptosis was detected using a flow cytometer. The
results are representative of three individual experiments.
Cell invasion assay
The cell invasion assay was performed using a
24-well Transwell chamber (Costar, Cambridge, MA, USA). At 24 h
post-transfection, the cells (4×104) were seeded in the
upper chamber of a 8-μm pore size insert pre-coated with
Matrigel (BD Biosciences-Pharmingen, San Diego, CA, USA), and
cultured in RPMI-1640 without FBS for a further 24 h. The cells
were allowed to migrate towards the medium containing 10% FBS in
the bottom chamber. The non-migratory cells on the upper membrane
surface were removed with a cotton tip, and the migratory cells
attached to the lower membrane surface were fixed with 4%
paraformaldehyde and stained with crystal violet (Sigma, St. Louis,
MO, USA). The number of invaded cells were counted in 10 randomly
selected power fields under a microscope (magnification, ×200)
(Olympus CK30; Olympus, Tokyo, Japan). The experiments were
performed in triplicate.
Statistical analysis
The SPSS for Windows version 17.0 statistical
analysis software (SPSS, Inc., Chicago, IL, USA) was applied to
complete the data processing. A paired-samples t-test was used to
compare the differences between the PRKACB expression in the NSCLC
and corresponding normal tissues. One-way ANOVA was used to compare
the differences in PRKACB expression in the transfected LTEP-A2
cells or controls. All data are represented as the mean ± SD.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Expression of PRKACB mRNA and protein in
human NSCLC tissues and their corresponding normal tissues
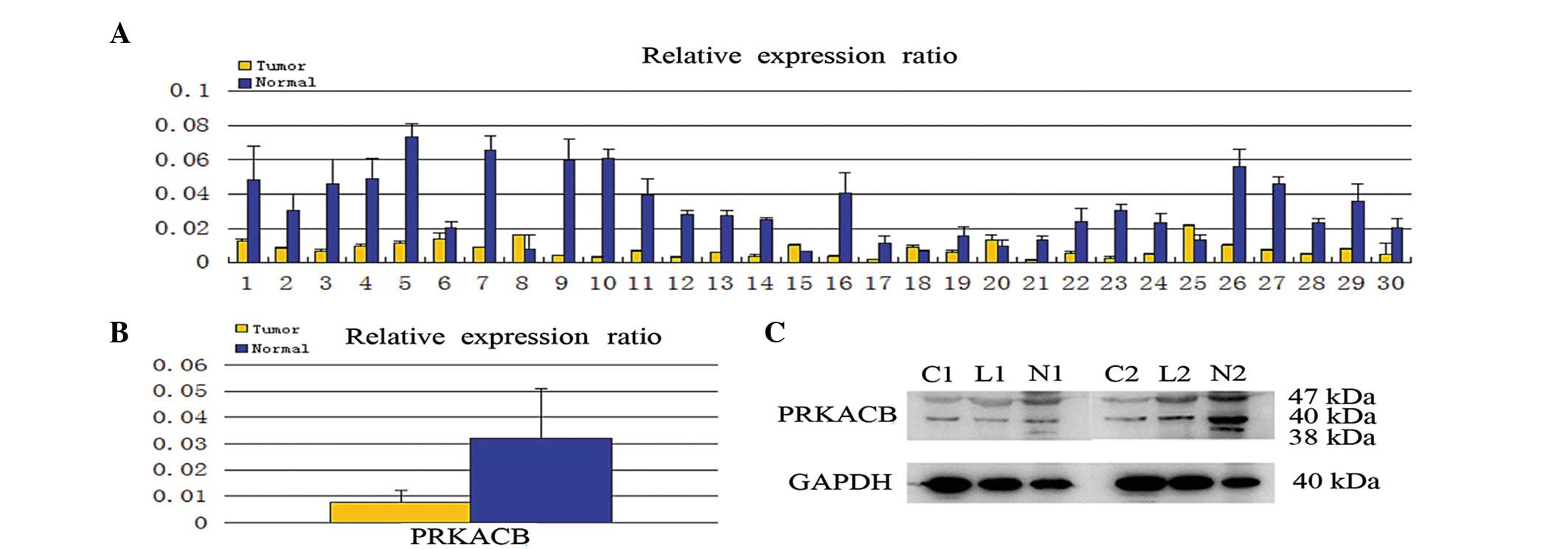
The PRKACB mRNA expression was first quantitatively
determined in the clinical samples using real-time RT-PCR. Of the
30 patients, 25 (83.3%) demonstrated a lower expression level of
PRKACB mRNA in the NSCLC tissues compared with the corresponding
normal tissues (Fig. 1A). In
addition, the mean expression value of the PRKACB mRNA in NSCLC
tissues (relative ratio of PRKABC/GAPDH; 0.007677±0.004608) was
significantly weaker than the value in the normal tissues
(0.031936±0.018996; P<0.05; Fig.
1B). Consistent with the mRNA level, the protein levels of
PRKACB were downregulated in the NSCLC tissues compared with the
normal tissues (0.350±0.124 vs. 0.964±0.245, respectively;
P<0.05; Fig. 1C). The study also
demonstrated that PRKACB protein expression was downregulated in
lymph node metastasis tissues (data not shown).
PRKACB upregulation inhibits
proliferation and clonogenicity in NSCLC cells
To elucidate the biological role of PRKACB during
carcinogenesis, the physiological effects of PRKACB upregulation on
cell proliferation and clonogenicity were examined using the
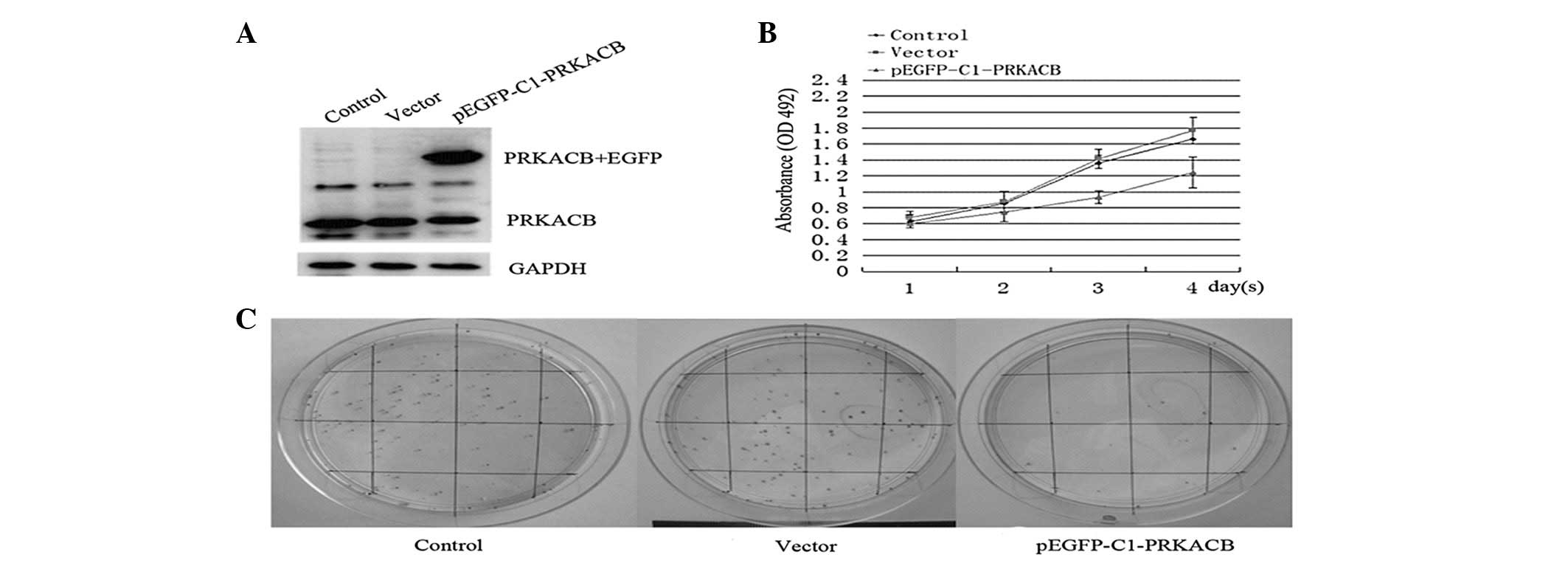
LTEP-A2 cells. Fig. 2A shows the
overexpression of PRKACB in the transfected cells. The study showed
that 3 days after PRKACB transfection, the absorbance values in the
PRKACB, vector and control groups were 0.93±0.08, 1.41±0.12 and
1.36±0.09, respectively (one-way ANOVA, P<0.05). The growth
curve shows that the cells transfected with pEGFP-C1-PRKACB grew
more slowly than the empty vector-transfected cells and control
group cells, indicating that PRKACB inhibits proliferation in NSCLC
cells (Fig. 2B).
The colony formation efficiencies of the LTEP-A2
cells transfected with PRKACB and the vector control for 24 h were
compared next. In total, 200 cells were planted on 6-cm cell
culture dishes. At two weeks post-transfection, the plates were
stained with Giemsa and colonies with >50 cells were counted.
The numbers of cell colonies in the PRKACB, vector and control
groups were 23.42±5.38, 89.28±7.15 and 86.85±6.86, respectively
(one-way ANOVA, P<0.05; Fig.
2C). These results showed that the increased expression of
PRKACB significantly inhibited the colony formation efficiencies of
the LTEP-A2 cells. Collectively, these data suggest that PRKACB may
act as a negative regulator of cell growth and that its
downregulation plays a significant role in NSCLC
carcinogenesis.
Elevated apoptotic rate in PRKACB
transfected cells
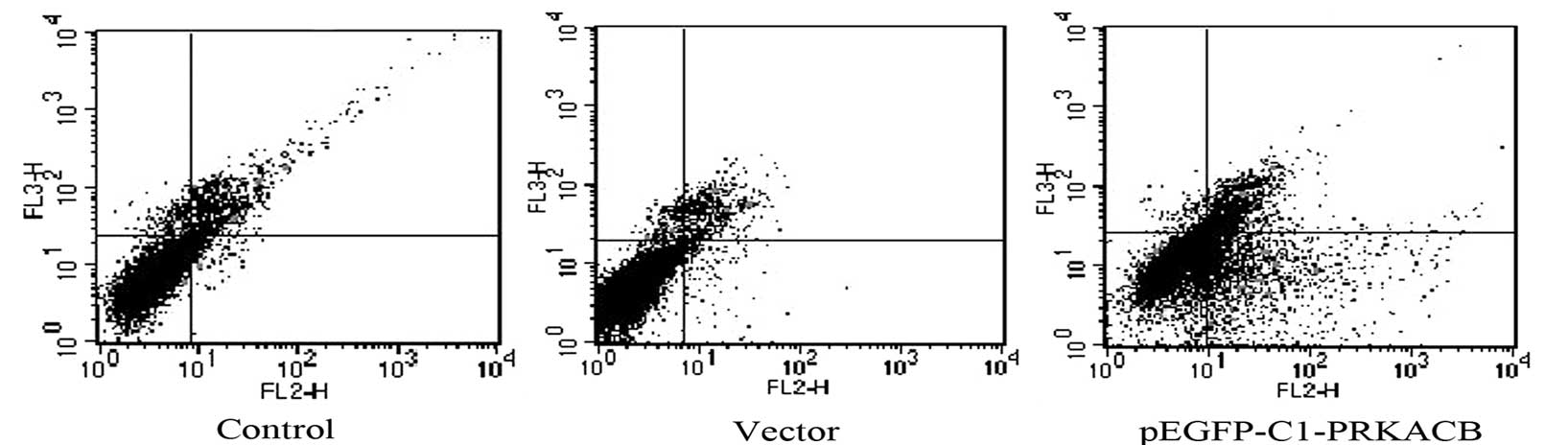
PRKACB has been considered to prevent the overgrowth
of cells by inducing cell apoptosis (15,16).
Therefore, apoptosis was examined following PRKACB transfection
using Annexin V-PE/7-AAD assay and flow cytometry. It was confirmed
that PRKACB was upregulated in the transfected cells. The apoptotic
rates of the LTEP-A2 cells in the PRKACB, vector and control groups
were 24.43±3.42, 4.39±1.63 and 3.48±1.44%, respectively (one-way
ANOVA, P<0.05; Fig. 3). The
results showed that apoptosis was significantly induced in the
PRKACB overexpressed cells.
Effect of PRKACB upregulation on the
invasive potential of transfected cells
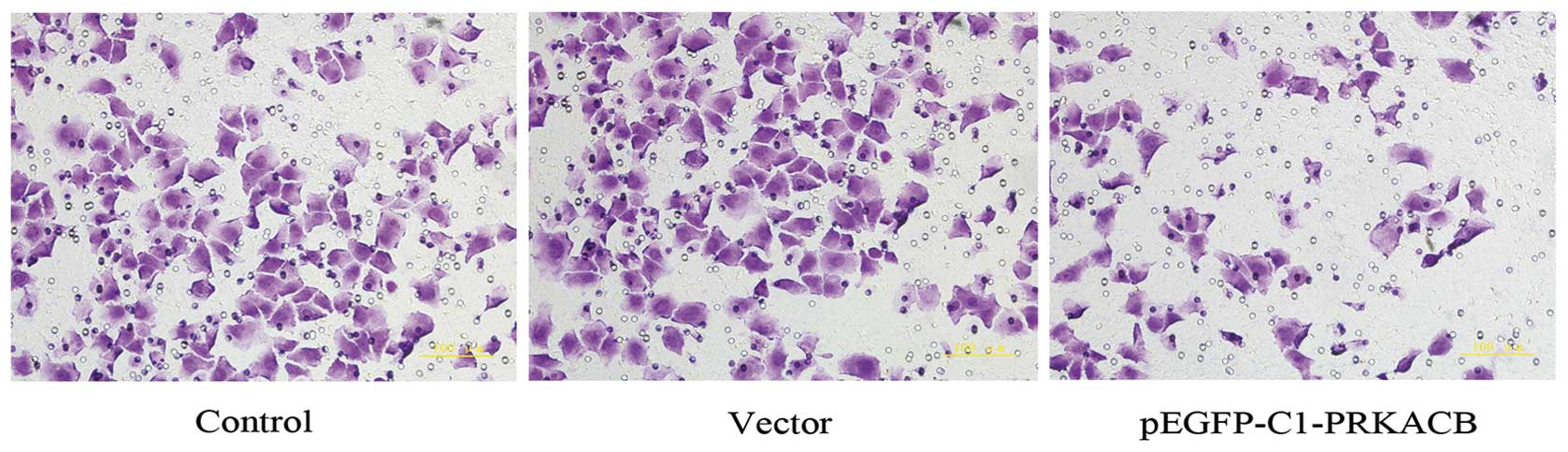
It has been acknowledged that PKA may inhibit RhoA
signaling, which has been implicated in the process of tumor cell
invasion and metastasis (6). To
determine whether PRKACB expression further affects the invasion of
LTEP-A2, the present study compared the invasive ability of the
three cell groups. The number of invasive cells in the PRKACB,
vector and control groups were 83.6±9.5, 156.9±13.7 and 154.2±12.9,
respectively (one-way ANOVA, P<0.05; Fig. 4). These results show that the
increased expression of PRKACB significantly inhibited the invasion
of the LTEP-A2 cells, as demonstrated by the Matrigel invasion
assay.
Discussion
The PRKACB gene is located at the 1p31.1 chromosome
site and encodes PKA catalytic subunit β, which is a member of the
Ser/Thr protein kinase family. As a key effector of the
cAMP/PKA-induced signaling pathway, the free C subunits
phosphorylate serine and threonine residues on specific substrate
proteins and regulate a wide range of cellular processes. Previous
studies have identified the loss of 1p31.1 in MCL patients and the
MCL cell line. PRKACB has been identified as an apoptotic candidate
gene and it appears that decreased expression of PRKACB is
implicated in human MCL (15).
PRKACB tissue-specific expression has also been found in human
brain, neuronal, lymphoid and prostate cancer tissues, and has been
reported to be correlated with cellular proliferative or
differentiation processes (9–12).
However, there are no studies investigating the role of PRKACB in
lung cancer. In the present study, the mRNA and protein levels of
PRKACB were downregulated in the human NSCLC tissues compared with
their corresponding normal tissues. These results suggest that
PRKACB has a critical effect in the tumorigenesis and aggression of
NSCLC.
A recent study discovered a novel interaction
between PRKACB, the cell cycle and CARP-1; this was confirmed by
GST pull-down experiments in brain tissue (16). A study has also demonstrated that
PRKACB interacts with p75NTR, which phosphorylates p75NTR at Ser304
(14). In the majority of cases,
the most prominent biological function of p75NTR is that it induces
cell death and induces the activity of the JNK-p53-Bax apoptosis
pathway and other proteins that regulate cell death, such as NRIF
(17). PKA-mediated phosphorylation
at Ser304 has been shown to promote the translocation of p75NTR to
lipid rafts and to regulate the downstream signals of p75NTR,
including the inactivation of RhoA, which has been implicated in
the process of tumor cell invasion and metastasis. In addition, PKA
may also directly inhibit RhoA signaling; when Ser188 is
phosphorylated, RhoA becomes inactive and thereby induces
characteristic morphological changes, causing cell rounding
(6). These data suggest that
decreased PRKACB is associated with cellular apoptosis, invasion
and metastasis. With the aim of assessing the role of PRKACB in the
development and progress of human NSCLC, the present study examined
the effects of exogenously-transfected PRKACB on the apoptosis and
invasion of LTEP-A2 cells. Consistent with the aforementioned
findings, the present study concluded that the upregulation of
PRKACB increased the number of apoptotic cells and decreased the
number of invasive cells. The results demonstrate the potential
role of PRKACB in the development and progression of human
NSCLC.
As previously described, PKA was able to induce the
signal pathway that is involved in numerous cellular process,
including cell proliferation, apoptosis and gene transcription
(3). cAMP-mediated PKA activation
has been shown to have anti-proliferative effects in a number of
cell types, including thyroid papillary carcinoma, ovarian
epithelial cancer, breast cancer and malignant glioma cells
(18–26). These anti-proliferative effects are
mainly associated with the negative regulation of the
Ras-Raf-MEK-ERK signaling pathway by interfering with the
activation of Raf-1 directly or via Ras in the Raf-1 pathway
(5,24,27).
Several other mechanisms have been proposed to explain the
anti-proliferative effects of activated PKA on various other cells
and tissues, including a decrease in the expression level of cyclin
D3 and an upregulation of the amount of p27kip1 (26). PKA is able to inhibit CUTL1-mediated
proliferation and migration (8), as
well as the LPA stimulation of SRF by promoting the dissolution of
F-actin (19). In this study, we
further examined the effects of exogenously transfected PRKACB on
the proliferation of LTEP-A2 cells. The observation that the
upregulation of PRKACB induces decreased proliferation of the
LTEP-A2 cells is consistent with a negative role for PKA in the
proliferation of these cells. Exogenously expressed PRKACB may
effectively inhibit the progression of lung cancer. However, the
fact that the excess of free PRKACB subunits may generate signals
different from those generated by the cAMP/PKA-induced signal
pathway cannot be excluded. It has also been previously shown that
the activation of PKA has either proliferative or anti-apoptotic
effects in cultured cells, and that these opposite responses may be
due to the existence of cell type-specific targets of this
signaling pathway (12,13).
The present study demonstrated that PRKACB was
down-regulated in human NSCLC tissues. Decreased PRKACB appears to
be associated with cellular apoptosis, invasion and proliferation.
However, the molecular mechanisms for these processes remain
primarily unknown. Increased PRKACB expression is possibly an
effective inhibitor of lung cancer. The upregulation of PRKACB may
provide a useful strategy for future NSCLC inhibitory
therapies.
Acknowledgements
This study was supported by the
National Nature Science Foundation of China (30973502).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Molina JR, Yang P, Cassivi SD, Schild SE
and Adjei AA: Non-small cell lung cancer: epidemiology, risk
factors, treatment, and survivorship. Mayo Clin Proc. 83:584–594.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Skalhegg BS and Tasken K: Specificity in
the cAMP/PKA signaling pathway: Differential expression,
regulation, and subcellular localization of subunits of PKA. Front
Biosci. 5:D678–D693. 2000. View
Article : Google Scholar
|
|
4
|
Corbin JD, Sugden PH, West L, Flockhart
DA, Lincoln TM and McCarthy D: Studies on the properties and mode
of action of the purified regulatory subunit of bovine heart
adenosine 3′:5′-monophosphate-dependent protein kinase. J Biol
Chem. 253:3997–4003. 1978.
|
|
5
|
Cook SJ and McCormick F: Inhibition by
cAMP of Ras-dependent activation of Raf. Science. 262:1069–1072.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lang P, Gesbert F, Delespine-Carmagnat M,
et al: Protein kinase A phosphorylation of RhoA mediates the
morphological and functional effects of cyclic AMP in cytotoxic
lymphocytes. EMBO J. 15:510–519. 1996.PubMed/NCBI
|
|
7
|
Schmitt JM and Stork PJ: PKA
phosphorylation of Src mediates cAMP’s inhibition of cell growth
via Rap1. Mol Cell. 9:85–94. 2002.
|
|
8
|
Michl P, Knobel B and Downward J: CUTL1 is
phosphorylated by protein kinase A, modulating its effects on cell
proliferation and motility. J Biol Chem. 281:15138–15144. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Larsen AC, Kvissel AK, Hafte TT, Avellan
CI, Eikvar S, et al: Inactive forms of the catalytic subunit of
protein kinase A are expressed in the brain of higher primates.
FEBS J. 275:250–262. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ørstavik S, Reinton N, Frengen E,
Langeland BT, Jahnsen T and Skålhegg BS: Identification of novel
splice variants of the human catalytic subunit Cbeta of
cAMP-dependent protein kinase. Eur J Biochem. 268:5066–5073.
2001.PubMed/NCBI
|
|
11
|
Kvissel AK, Ørstavik S, Øistad P, Rootwelt
T, Jahnsen T and Skålhegg BS: Induction of Cbeta splice variants
and formation of novel forms of protein kinase A type II
holoenzymes during retinoic acid-induced differentiation of human
NT2 cells. Cell Signal. 16:577–587. 2004. View Article : Google Scholar
|
|
12
|
Kvissel AK, Ramberg H, Eide T, Svindland
A, et al: Androgen dependent regulation of protein kinase A
subunits in prostate cancer cells. Cell Signal. 19:401–409. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu KJ, Mattioli M, Morse HC III and
Dalla-Favera R: c-MYC activates protein kinase A (PKA) by direct
transcriptional activation of the PKA catalytic subunit beta
(PKA-Cbeta) gene. Oncogene. 21:7872–7882. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Higuchi H, Yamashita T, Yoshikawa H and
Tohyama M: PKA phosphorylates the p75 receptor and regulates its
localization to lipid rafts. EMBO J. 22:1790–1800. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schraders M, Jares P, Bea S, Schoenmakers
EF, et al: Integrated genomic and expression profiling in mantle
cell lymphoma: identification of gene-dosage regulated candidate
genes. Br J Haematol. 143:210–221. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Erlbruch A, Hung CW, Seidler J, Borrmann
K, et al: Uncoupling of bait-protein expression from the prey
protein environment adds versatility for cell and tissue
interaction proteomics and reveals a complex of CARP-1 and the PKA
Cbeta1 subunit. Proteomics. 10:2890–2900. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kaplan DR and Miller FD: Neurotrophin
signal transduction in the nervous system. Curr Opin Neurobiol.
10:381–391. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Matsumoto H, Sakamoto A, Fujiwara M, et
al: Cyclic AMP-mediated growth suppression and MAPK phosphorylation
in thyroid papillary carcinoma cells. Mol Med Rep. 1:245–249.
2008.PubMed/NCBI
|
|
19
|
Nguyen GH, French R and Radhakrishna H:
Protein kinase A inhibits lysophosphatidic acid induction of serum
response factor via alterations in the actin cytoskeleton. Cell
Signal. 16:1141–1151. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen TC, Hinton DR, Zidovetzki R and
Hofman FM: Up-regulation of the cAMP/PKA pathway inhibits
proliferation, induces differentiation, and leads to apoptosis in
malignant gliomas. Lab Invest. 78:165–174. 1998.PubMed/NCBI
|
|
21
|
Cassoni P, Sapino A, Fortunati N, Munaron
L, Chini B and Bussolati G: Oxytocin inhibits the proliferation of
MDA-MB231 human breast-cancer cells via cyclic adenosine
monophosphate and protein kinase A. Int J Cancer. 72:340–344. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hewer RC, Sala-Newby GB, Wu YJ, Newby AC
and Bond M: PKA and Epac synergistically inhibit smooth muscle cell
proliferation. J Mol Cell Cardiol. 50:87–98. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu J, Li XD, Ora A, Heikkilä P, Vaheri A
and Voutilainen R: cAMP-dependent protein kinase activation
inhibits proliferation and enhances apoptotic effect of tumor
necrosis factor-alpha in NCI-H295R adrenocortical cells. J Mol
Endocrinol. 33:511–522. 2004. View Article : Google Scholar
|
|
24
|
D’Angelo G, Lee H and Weiner RI:
cAMP-dependent protein kinase inhibits the mitogenic action of
vascular endothelial growth factor and fibroblast growth factor in
capillary endothelial cells by blocking Raf activation. J Cell
Biochem. 67:353–366. 1997.
|
|
25
|
Hordijk PL, Verlaan I, Jalink K, van
Corven EJ and Moolenaar WH: cAMP abrogates the
p21ras-mitogen-activated protein kinase pathway in fibroblasts. J
Biol Chem. 269:3534–3538. 1994.PubMed/NCBI
|
|
26
|
van Oirschot BA, Stahl M, Lens SM and
Medema RH: Protein kinase A regulates expression of p27(kip1) and
cyclin D3 to suppress proliferation of leukemic T cell lines. J
Biol Chem. 276:33854–33860. 2001.PubMed/NCBI
|
|
27
|
Al-Wadei HA and Schuller HM: Cyclic
adenosine monophosphate-dependent cell type-specific modulation of
mitogenic signaling by retinoids in normal and neoplastic lung
cells. Cancer Detect Prev. 30:403–411. 2006. View Article : Google Scholar : PubMed/NCBI
|