Introduction
Cells are constantly exposed to varieties of
genotoxic stress, including UV radiation, ionizing radiation (IR),
chemical agents and reactive oxygen species, which induce
potentially harmful DNA lesions (1). Human beings have evolved a highly
efficient and complex system, the DNA damage response (DDR)
pathway, to cope with damaged DNA (2). The DDR process includes cell cycle
checkpoint activation to stop the cell cycle progression in order
to allow time for DNA repair or apoptosis when the DNA damage is
irreparable (3). Failure to
properly sense and repair DNA may promote the accumulation of
chromosomal rearrangements, which in turn fuels malignant
transformation and finally leads to the occurrence of a tumor
(4). Of the various forms of DNA
damage, DNA double-strand breaks (DSBs) result in the most
deleterious damage (5). One single
DSB is sufficient to kill a mammalian cell.
In mammalian cells, DSBs are mainly repaired through
non-homologous end joining (NHEJ), which is susceptible to errors,
and homologous recombination (HR), which has a high fidelity
(6). HR repair occurs in the S and
G2 phases of the cell cycle due to its requirement of a
homologous chain as a template to complete the repair process,
whereas NHEJ repair joins the broken DNA together with no or simple
processing of the ends of the DNA (7). Thus, HR-mediated and NHEJ-mediated DSB
repair are essential for genome integrity.
The response to DSBs is initially detected by the
Mre11-Rad50-Nbs1 (MRN) complex (8).
In particular, cells activate the DDR protein kinases, ataxia
telangiectasia mutated gene (ATM), ataxia telangiectasia and
Rad3-related protein (ATR) and DNA-dependent protein kinase
(DNA-PK; also known as PRKDC) (1).
These then trigger histone H2AX phosphorylation and the
accumulation of proteins, including MDC1, 53BP1, BRCA1, CtIP, RNF8
and RNF168/RIDDLIN, into ionizing radiation-induced foci (IRIF)
that amplify DSB signaling and promote DSB repair (9). Following DSB formation, the attachment
of a small ubiquitin-related modifier (SUMO) of the target proteins
also accumulates at the DSB sites, which is a significant
modification in the DDR pathway (10).
Protein inhibitors of activated STAT (PIAS) proteins
are often identified to be associated with SUMO-modified
substrates, further emphasizing their role as potential SUMO
ligases (11). In this mode of
function, the PIAS proteins are believed to act as adapter proteins
that enhance the interactions between the SUMO conjugating enzyme,
Ubc9, and the substrate proteins (12). In previous studies, PIAS1 has been
established to recruit to damage sites and to promote DSB repair,
indicating a significant role in the DDR pathway (13). However, the function of other PIAS
members in DSB repair and which method of repair they are involved
in remains largely unknown. The present study investigated whether
another PIAS member, PIAS3 was involved in the components of the
DDR and its actions at the DSB sites, in the processes of NHEJ or
HR.
Materials and methods
Cell lines, plasmids and antibodies
The human 293T and HeLa cell lines were purchased
from the American Type Culture Collection (ATCC; Rockville, MD,
USA). The green fluorescent protein (GFP) reporter system for
HR-mediated DSB repair direct repeat (DR)-GFP 293T cells, the GFP
reporter system for NHEJ-mediated DSB repair EJ5-GFP 293T cells and
the I-SceI expression construct were obtained from the City of Hope
National Medical Center/Beckman Research Institute (Duarte, CA,
USA). All the cell lines were cultured in Dulbecco’s modified
Eagle’s medium (DMEM; Hyclone, Logan, UT, USA) with 10% fetal
bovine serum (FBS; Hyclone) at 37°C in the presence of 5%
CO2. The full-length coding sequences of PIAS1 and PIAS3
were amplified using PCR and cloned into a pXJ-40-myc vector.
Hemagglutinin-tagged BRCA1 (HA-BRCA1) was constructed as previously
described (14). The antibody for
ATM, the myc-horseradish peroxidase (HRP), the HA-HRP and the
HRP-conjugated secondary antibody were purchased from Sigma-Aldrich
(St. Louis, MO, USA).
HR- or NHEJ-mediated DSB repair GFP
reporter systems
The HR-mediated DSB repair assay was performed as
previously described (15).
Briefly, DR-GFP 293T cells were delivered with ATM-RNAi
(Lipofectamine; Invitrogen, Carlsbad, CA, USA) to the DR-GFP 293T
cells using lipid-mediated transfection (RNAiMAX, Lipofectamine;
Invitrogen) according to the specifications. Certain DR-GFP 293T
cells were transfected with HA-BRCA1 or myc-PIAS1/3 using
Lipofectamine 2000 (Invitrogen) according to the manufacturer’s
instructions. At 24 h post-transfection, the cells were transfected
with an I-SceI expression plasmid (pCBA Sce) using Jet Prime
(Polyplus, Illkirch, France). Two days later, the GFP+
cells were assayed by FACScan (BD Biosciences, San Jose, CA, USA).
The NHEJ-mediated DSB repair assays in the EJ5-GFP 293 cells were
performed as previously described (15). Briefly, the EJ5-GFP 293 cells with
the EJ5-GFP reporter stably integrated into their genome were
transfected with HA-BRCA1 or myc-PIAS1/3. A second transfection was
performed 24 h later with an empty vector or an I-SceI-expressing
construct. Following the second transfection, the cells were
harvested for 72 h and the fraction of the GFP+ cells
was determined using flow cytometry (BD Biosciences).
Immunoblotting
The total cell lysate was extracted with RIPA buffer
and a protease inhibitor mixture (Roche, Basel, Switzerland).
Precipitates or total cell lysates were resolved in 10% SDS-PAGE
and transferred onto a nitrocellulose membrane. The blots on the
nitrocellulose membrane were blocked using 5% skimmed milk in TBST
(PBS with 0.05% Tween-20) and sequentially incubated with primary
antibodies and HRP-conjugated secondary antibodies in 5% skimmed
milk in TBST. The blots were washed with TBST following a 1-h
incubation period. The immunoreactive bands were visualized using
Peroxide Solution and Luminol/Enhancer Solution (Amersham
Pharmacia, Amersham, UK).
IR survival assay
The HeLa cells were transfected with myc-PIAS and
empty vector and exposed to IR. The cells were left for 10–14 days
at 37°C to allow colony formation. The colonies were stained with
0.5% crystal violet/20% ethanol and counted. The results were
normalized to plating efficiencies.
Results
Establishment of the DR- and
NHEJ-mediated DSB repair system
The HR-and NHEJ-mediated DSB repair systems were
established to study the effect of PIAS3 in DSB repair. The
reporter system that was stably integrated in the DR-GFP 293T cells
was used to measure the HR-mediated DSB repair efficiency and the
GFP-based chromosomal reporter EJ5-GFP in the 293T cells was used
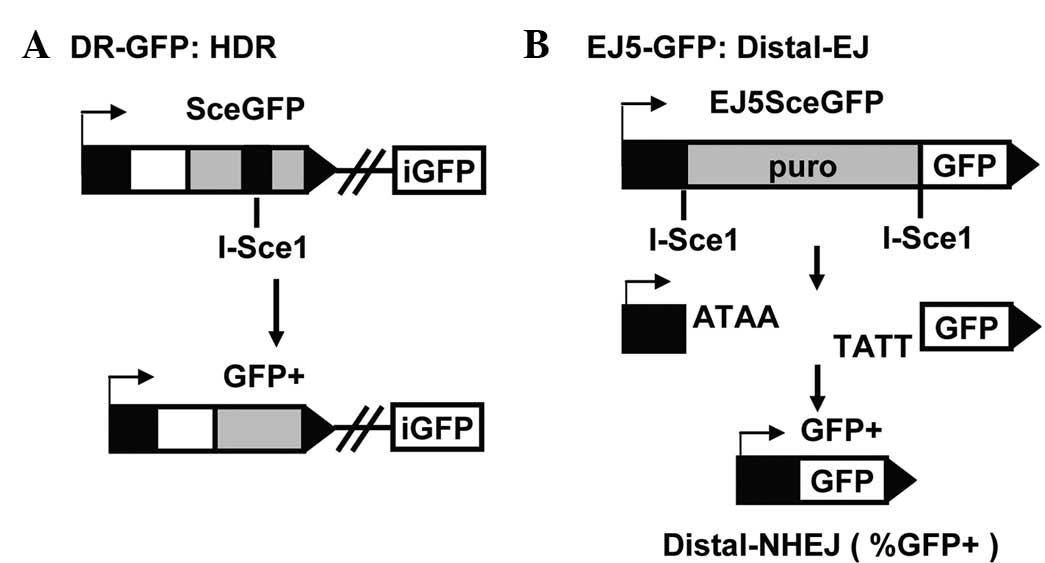
to measure the total NHEJ repair efficiency. DR-GFP was constructed
using the homology-directed repair (HDR) product that uses intense
GFP (iGFP) as the template for nascent DNA synthesis, which results
in the restoration of a GFP expression cassette (Fig. 1A). EJ5-GFP contains a promoter that
is separated from a GFP coding cassette by a puromycin gene flanked
by two I-SceI sites in the same orientation. Once the puromycin
gene is excised by the two I-SceI-induced DSBs, the promoter is
joined to the rest of the expression cassette by NHEJ repair,
leading to restoration of the GFP+ gene (Fig. 1B). Therefore, the number of
GFP+ cells is a measure of the NHEJ-mediated DSB repair.
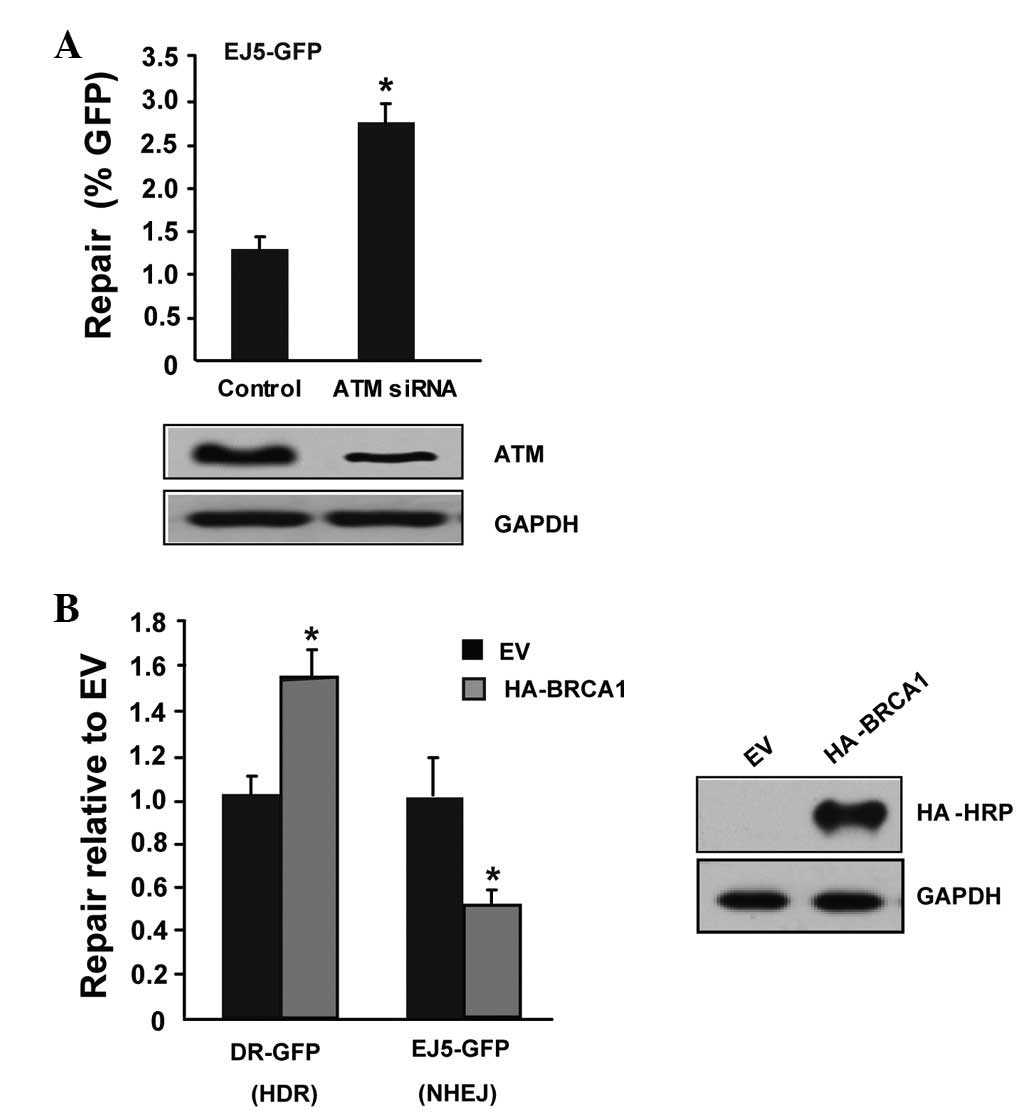
To test the accuracy of the two systems, the present study utilized
two factors with known functions that are involved in the DSB
pathway to the systems, ATM and BRCA1. As expected, knockdown of
ATM with specific ATM siRNA increased NHEJ-mediated DSB repair
(Fig. 2A). Transfection of HA-BRCA1
increased the level of HDR and reduced the level of NHEJ-mediated
DSB repair (Fig. 2B), which is
consistent with a previous study (16). Taken together, the results of the
tests of the two classical factors, ATM and BRCA1, indicated that
the HR- and NHEJ-mediated DSB repair systems were established
successfully.
PIAS3 promotes HDR and distal-NHEJ
Mammalian SUMO E3-ligase PIAS1 was reported to
promote the response to DNA DSBs (17). Therefore, other PIAS family members,
including PIAS3, may also be involved in DSB repair. A PIAS3
expression vector was transfected into the well-established HR- and
NHEJ-mediated DSB repair systems, respectively. PIAS1 was used as a
positive control. The overexpression of PIAS1 and PIAS3 resulted in
a 1.6-fold increase of GFP+ cells in comparison with the
empty vector cells (Fig. 3), and
the co-transfection of PIAS1 and PIAS3 did not synergistically
increase the GFP+ cells. This result indicates that
PIAS3 promotes HDR and distal-NHEJ, as does PIAS1. PIAS3 and PIAS1
do not have a synergistic effect on HDR and distal NHEJ.
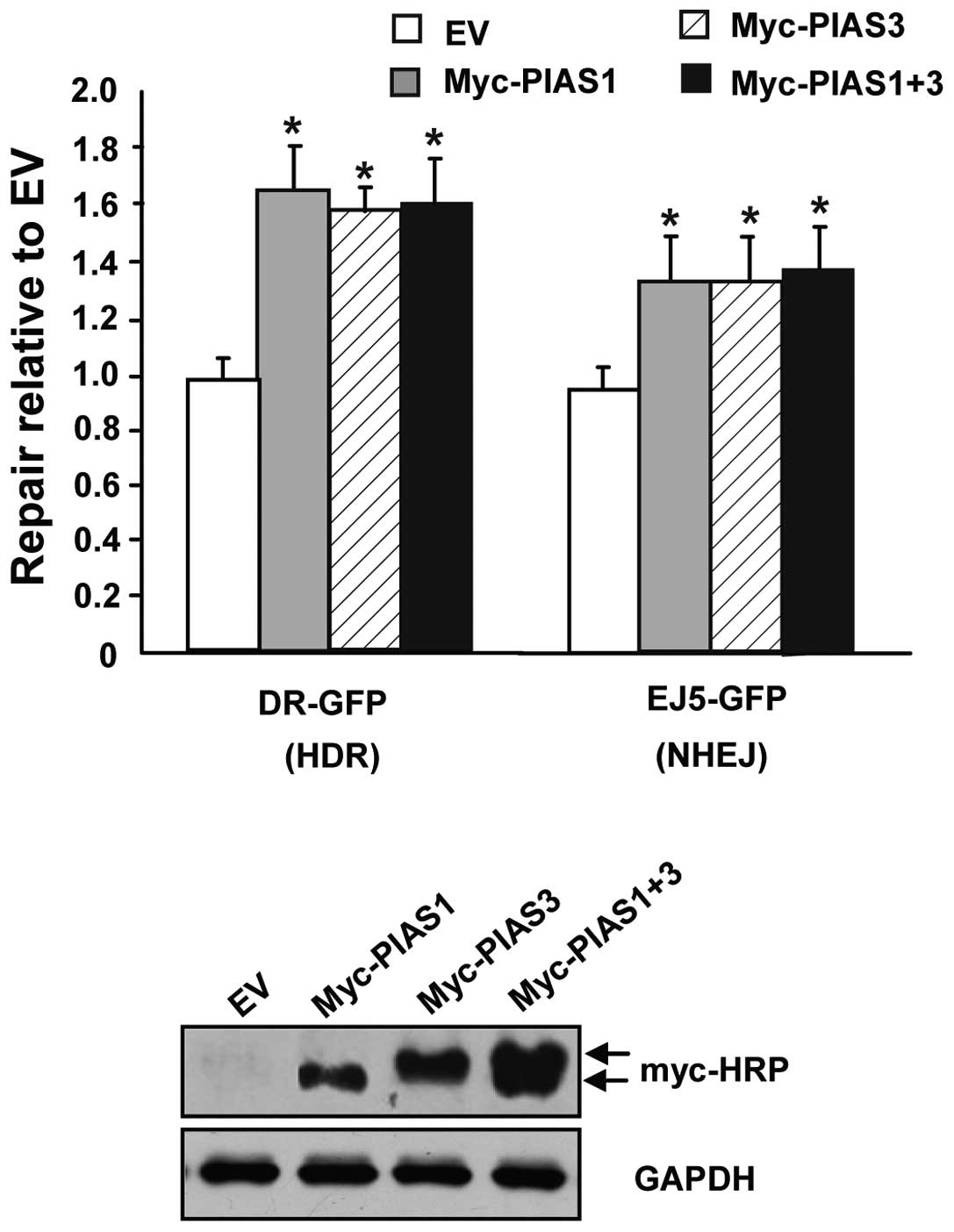 | Figure 3PIAS3 promotes HDR and distal NHEJ.
The human 293T cells were transfected with an expression vector for
I-SceI, along with a complementation vector for PIAS1, PIAS3, P1AS1
plus PIAS3 or the empty expression vector (EV). Repair is measured
as the percentage of GFP+ cells.
(*P<0.001, statistical differences between EV and
PIAS1, PIAS3 or P1AS1 plus PIAS3 treatments). Representative
western blots of PIAS1 and PIAS3 expression in human 293T cells are
shown in the lower panel, with PIAS1 and PIAS3 carrying Myc-HRP.
PIAS, protein inhibitor of activated STAT; HDR, homology-directed
repair; NHEJ, non-homolous end joining; GFP, green fluorescent
protein; HRP, horseradish peroxidase. |
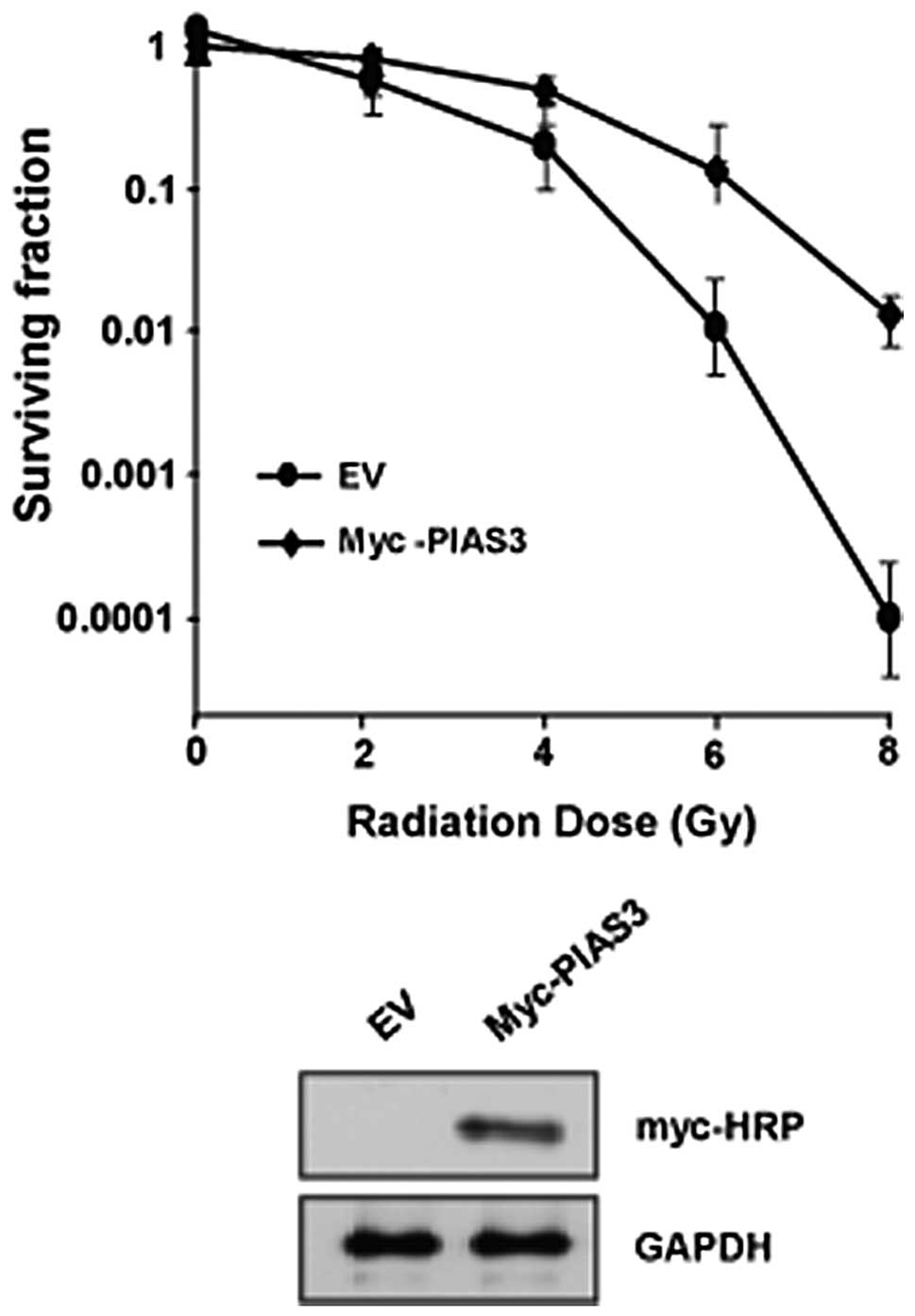
Overexpression of PIAS3 confers IR
resistance
The overexpression of PIAS3 resulted in an increase
of HR- and NHEJ-mediated DSB repair. PIAS3 was able to upregulate
IR resistance. The expression of PIAS3 in the HeLa cells (Fig. 4) increased the cell resistance to
IR. PIAS3 plays a significant role in promoting IR resistance,
therefore, PIAS3 may be a potentially promising therapeutic
approach for cancer treatment.
Discussion
The PIAS family of proteins was named based on the
identification of the founding member, PIAS3, as a repressor of the
activity of the STAT3 transcription factor (18). Since then, three additional family
members, PIAS1, PIAS2 and PIAS4, have been identified and are
characterized by a high degree of sequence conservation throughout
the proteins (19). The PIAS
proteins have been shown to impact on the function of a number of
proteins, but a major process on which all these proteins act is
the control of gene transcription. Thus, PIAS proteins may be
considered to be transcriptional coregulators. PIAS protein action
may be activated or repressed, although the mechanism of action
apparently differs depending on the target gene or interacting
transcriptional regulator. The other major functional part of PIAS
proteins is the SP-RING domain, which is associated with the
zinc-binding RING fingers and is most similar to the domains that
have been identified in a subclass of ubiquitin E3 ligases
(18,20). These somewhat functionally-redundant
proteins are structurally associated with ubiquitin and are
covalently attached to target proteins by a SUMO-conjugation system
consisting of an E1 activating enzyme (SAE1/SAE2), an E2 ligase
(Ubc9) and various E3 ligases with differing target-protein
specificities (20). The present
study identified that PIAS3 not only promotes HR repair, but that
it also promotes NHEJ repair. Given the fact that PIAS3 serves as
the ligase for protein sumoylation in DSB repair (21), PIAS3 may modulate the sumoylation
status of key DDR factors, including CtIP and DNA-PKcs/Ku70/Ku80.
However, the molecular mechanisms of PIAS3 in DSB repair require
further investigation.
A number of tumor-associated mutations, including
ATM, BRCA1, BRCA2, CHK2 and p53, have been identified to be
clustered in the HR pathway (19,22–24).
Therefore, promoting HR in human tumors may be a highly useful
strategy to combat cancer by enhancing the effect of radiation or
DSB-inducing chemotherapy. This may be of particular significance
to breast cancer therapy, as a significant percentage of hereditary
breast cancers carry the BRCA1 or BRCA2 mutations and thus are
deficient in HR (22,25,26).
Therefore, PIAS3 mimetics are promising candidates for the
development of sensitizers for the treatment of BRCA-deficient
breast cancers using DNA-damaging chemotherapeutic drugs and
radiation. This study serves as a proof-of-principle of targeting
SUMO-dependent functions in the development of novel therapeutics,
as well as in uncovering the role of SUMO modifications in various
cellular functions.
In conclusion, PIAS3 is an enhancer of HR- and
NHEJ-mediated DSB repair that increases cell resistance to IR.
Acknowledgements
The authors would like to thank the members of the
Ye Laboratory, Department of Medical Molecular Biology (Beijing,
China). This study was supported by the National Natural Science
Foundation of China (no. 31100604).
References
|
1
|
Yang J, Yu Y, Hamrick HE and
Duerksen-Hughes PJ: ATM, ATR and DNA-PK: initiators of the cellular
genotoxic stress responses. Carcinogenesis. 24:1571–1580. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
David R: DNA damage response: restricting
repair. Nat Rev Mol Cell Biol. 13:6012012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schwartz D and Rotter V: p53-dependent
cell cycle control: response to genotoxic stress. Semin Cancer
Biol. 8:325–336. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Surova O and Zhivotovsky B: Various modes
of cell death induced by DNA damage. Oncogene. Dec 3–2012.(Epub
ahead of print).
|
|
5
|
Helmink BA and Sleckman BP: The response
to and repair of RAG-mediated DNA double-strand breaks. Annu Rev
Immunol. 30:175–202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Takata M, Sasaki MS, Sonoda E, et al:
Homologous recombination and non-homologous end-joining pathways of
DNA double-strand break repair have overlapping roles in the
maintenance of chromosomal integrity in vertebrate cells. EMBO J.
17:5497–5508. 1998. View Article : Google Scholar
|
|
7
|
Bennardo N, Cheng A, Huang N and Stark JM:
Alternative-NHEJ is a mechanistically distinct pathway of mammalian
chromosome break repair. PLoS Genet. 4:e10001102008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Panier S and Durocher D: Regulatory
ubiquitylation in response to DNA double-strand breaks. DNA Repair
(Amst). 8:436–443. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Galanty Y, Belotserkovskaya R, Coates J,
Polo S, Miller KM and Jackson SP: Mammalian SUMO E3-ligases PIAS1
and PIAS4 promote responses to DNA double-strand breaks. Nature.
462:935–939. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Galanty Y, Belotserkovskaya R, Coates J
and Jackson SP: RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes
DNA double-strand break repair. Genes Dev. 26:1179–1195. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhu J, Zhu S, Guzzo CM, et al: Small
ubiquitin-related modifier (SUMO) binding determines substrate
recognition and paralog-selective SUMO modification. J Biol Chem.
283:29405–29415. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sharrocks AD: PIAS proteins and
transcriptional regulation - more than just SUMO E3 ligases. Genes
Dev. 20:754–758. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Polo SE and Jackson SP: Dynamics of DNA
damage response proteins at DNA breaks: a focus on protein
modifications. Genes Dev. 25:409–433. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yan J, Zhu J, Zhong H, Lu Q, Huang C and
Ye Q: BRCA1 interacts with FHL2 and enhances FHL2 transactivation
function. FEBS Lett. 553:183–189. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bennardo N, Gunn A, Cheng A, Hasty P and
Stark JM: Limiting the persistence of a chromosome break diminishes
its mutagenic potential. PLoS Genet. 5:e10006832009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Laulier C, Cheng A and Stark JM: The
relative efficiency of homology-directed repair has distinct
effects on proper anaphase chromosome separation. Nucleic Acids
Res. 39:5935–5944. 2011. View Article : Google Scholar
|
|
17
|
Yunus AA and Lima CD: Structure of the
Siz/PIAS SUMO E3 ligase Siz1 and determinants required for SUMO
modification of PCNA. Mol Cell. 35:669–682. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rosas-Acosta G, Langereis MA, Deyrieux A
and Wilson VG: Proteins of the PIAS family enhance the sumoylation
of the papillomavirus E1 protein. Virology. 331:190–203. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schmidt D and Müller S: Members of the
PIAS family act as SUMO ligases for c-Jun and p53 and repress p53
activity. Proc Natl Acad Sci USA. 99:2872–2877. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jackson PK: A new RING for SUMO: wrestling
transcriptional responses into nuclear bodies with PIAS family E3
SUMO ligases. Genes Dev. 15:3053–3058. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Duval D, Duval G, Kedinger C, Poch O and
Boeuf H: The ‘PINIT’ motif, of a newly identified conserved domain
of the PIAS protein family, is essential for nuclear retention of
PIAS3L. FEBS Lett. 554:111–118. 2003.
|
|
22
|
Weitzel JN, Clague J, Martir-Negron A, et
al: Prevalence and type of BRCA mutations in Hispanics undergoing
genetic cancer risk assessment in the southwestern United States: a
report from the Clinical Cancer Genetics Community Research
Network. J Clin Oncol. 31:210–216. 2013. View Article : Google Scholar
|
|
23
|
Gannon HS, Woda BA and Jones SN: ATM
phosphorylation of Mdm2 Ser394 regulates the amplitude and duration
of the DNA damage response in mice. Cancer Cell. 21:668–679. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Squatrito M, Brennan CW, Helmy K, Huse JT,
Petrini JH and Holland EC: Loss of ATM/Chk2/p53 pathway components
accelerates tumor development and contributes to radiation
resistance in gliomas. Cancer Cell. 18:619–629. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rodriguez-Wallberg KA and Oktay K:
Fertility preservation and pregnancy in women with and without BRCA
mutation-positive breast cancer. Oncologist. 17:1409–1417. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hartman AR, Kaldate RR, Sailer LM, et al:
Prevalence of BRCA mutations in an unselected population of
triple-negative breast cancer. Cancer. 118:2787–2795. 2012.
View Article : Google Scholar : PubMed/NCBI
|