Introduction
The Ewing sarcoma family of tumors (ESFTs) is a
group of rare malignancies arising from the migrating cells of the
neural crest, characteristically composed of small cells arranged
in cords and embedded in fibrous tissue. This group of malignancies
includes classic Ewing sarcoma of the bone, extraskeletal Ewing
sarcoma, peripheral primitive neuroectodermal tumors (PNETs) and
Askin tumors. The tumors that occur in the thoracopulmonary region
have been referred to as Askin tumors since the first report of a
PNET of the chest wall by Askin et al(1) in 1979. The generally accepted view is
that the neoplasms are essentially the same entity showing various
degrees of neuroectodermal differentiation, and that Ewing sarcoma
is considered to be the beginning of the Ewing sarcoma/PNET
spectrum, while PNET is the end.
An Askin tumor is a soft tissue sarcoma belonging to
the ESFTs that is localized in the thoracopulmonary region. This
type of tumor mainly occurs in children and adolescents. Although
rare, individual cases have been occasionally reported in older
patients (2) and newborns (3). The incidence of Askin tumors is more
frequent in males than in females (1.5:1) (4). This neoplasm usually arises from the
soft tissue of the chest wall, although certain tumors have been
reported to be localized in the lung (5). Pathologically, an Askin tumor is a
malignant small round cell tumor that is known to be derived from
neuroectodermal cells due to their cytogenetic appearance. The
tumors show a common and unique chromosomal translocation,
t(11;22)(q24;q12) (6). Based on the
results following histological analysis, there are numerous
overlapping clinical and pathological characteristics and
therapeutic approaches between Askin tumors/PNETs and Ewing
sarcoma. Written informed consent was obtained from the
patient.
Case report
A 30-year-old male was admitted to the People’s
Hospital of Tai’an City in May 2012 presenting with a cough and
intermittent vertigo. The patient was diagnosed with a pulmonary
infection of the left lung and the condition improved with
antibiotic treatment. Unfortunately, the same syndrome recurred 2
months later along with the presence of a swelling mass in the left
chest wall and pain at the site of the swelling of the affected
thoracopulmonary region. There was no history of fever, hemoptysis,
dyspnea or joint pain. The patient was subsequently referred to the
Shandong Cancer Hospital and Institute (Jinan, China) for further
examination. A physical examination indicated swelling over the
left chest wall and supraclavicular region, with decreased breath
sounds over the left lung and dull percussion notes in the left
hemithorax. No lymph nodes were palpable. Horner syndrome was also
observed, as characterized by the symptoms of vertigo and
left-sided anhidrosis and hypothermia without ptosis of the face
and body, as well as miosis and conjunctival congestion in the left
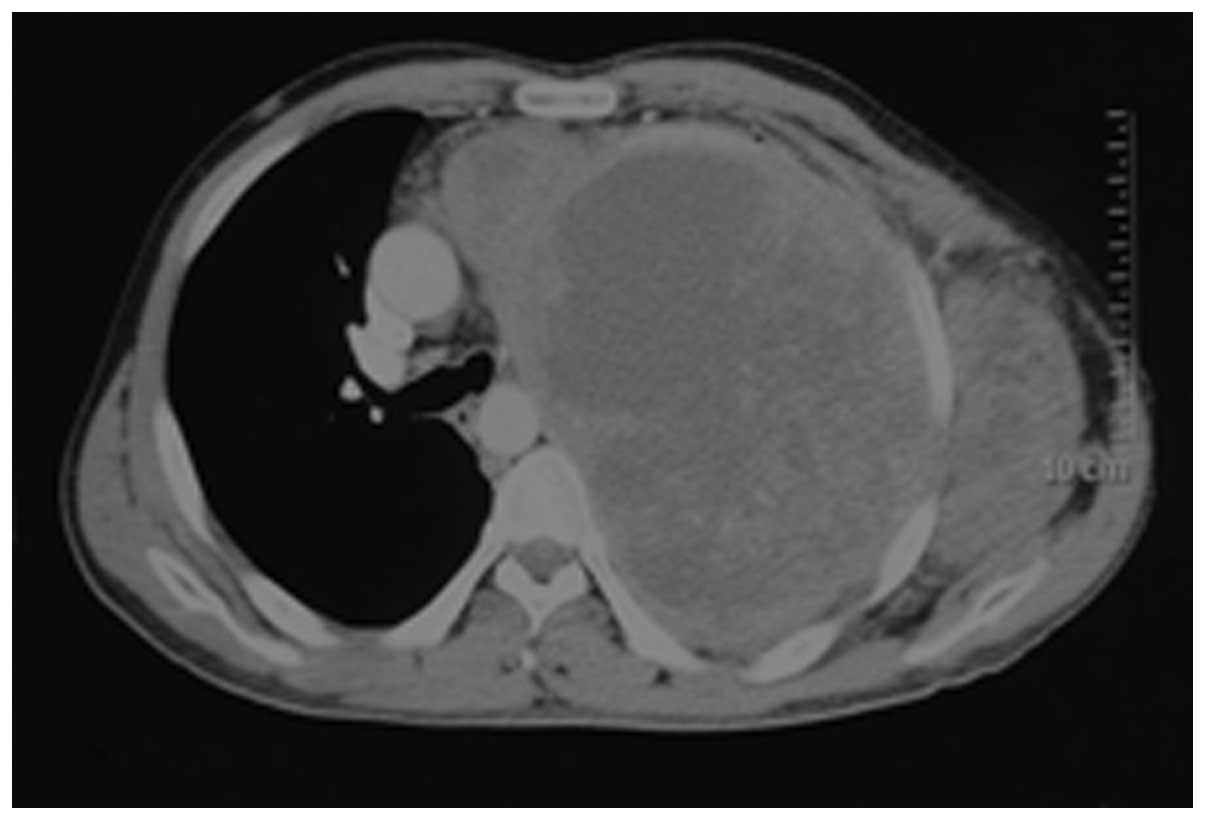
eye. Contrast-enhanced computed tomography (CT) showed a large,
heterogeneous density compatible with areas of non-enhancing
necrosis; the maximum cross-sectional area of the mass was 15×22.5
cm2. Compression of the mediastinum and right lung was
evident. The large mass extended into the lung, chest wall and
axilla, accompanied by destruction of the 2nd rib (Fig. 1). A needle biopsy was performed
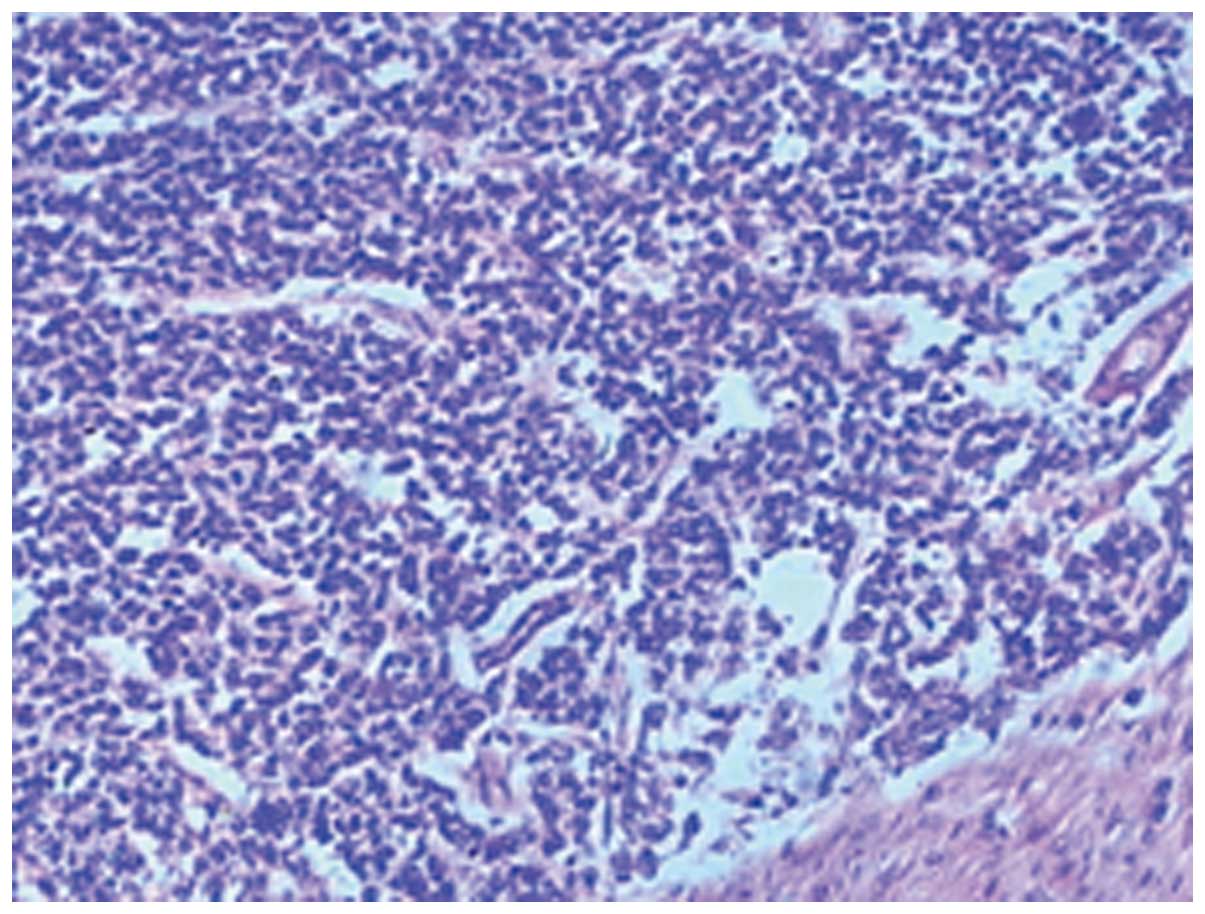
under radiological guidance, and a subsequent histological analysis
showed that the tumor was composed of small round cells with scant
cytoplasm and stained positive for neuron-specific enolase (NSE),
CD99 and vimentin (Fig. 2). The
diagnosis of an Askin tumor was confirmed following a morphological
and immunohistochemical analysis.
The patient underwent chemoradiotherapy instead of
surgery due to the anatomical complexities of the involved
structures. The patient was administered 4 cycles of combined
chemotherapy (1 cycle every 3 weeks), including 2 mg vincristine,
30 mg epirubicin and 60 mg cisplatin; the 4th cycle was
administered in combination with radiotherapy at a dose of 1.8 Gy
per fraction. The patient refused further treatment when 20
fractions of radiotherapy were completed. The patient response to
the aforementioned treatment was good and a clinical response was
achieved. A physical examination indicated disappearance of the
swelling in the chest wall and relief of the symptoms, although a
CT scan indicated that the limited reduction in tumor size was not
ideal. At the time of writing this manuscript, the patient was well
with no evidence of metastasis.
Discussion
Askin tumors/PNETs are highly aggressive, and local
recurrence and remote metastases are common. This type of neoplasm
typically involves the periosteum, soft tissues and extrapulmonary
tissue of the thoracic wall, but it may also involve peripheral
lung tissue by direct local extension or occur in the primary
peripheral lung cancer tissue (7).
PNETs of the lung have been suggested to be more aggressive
compared with PNETs in other locations (8). The metastatic sites include the lungs,
the mediastinal and retroperitoneal lymph nodes, the extrathoracic
skeleton, the liver, the adrenal glands and the sympathetic nerve
chain (9). Askin et
al(1) reported that 14/18
patients succumbed to the disease 4–44 months following the
diagnosis and that the mean survival period was 8 months. Contesso
et al(10) reported that the
2- and 6-year survival rates were 38 and 14%, respectively. After
failure of local control, the mean survival rate was reported to be
reduced to 11 months (11). The
initial tumor mass (>100 ml), the histopathological response to
initial chemotherapy and the presence of metastases at diagnosis
were identified as major prognostic factors (12,13).
An early diagnosis and multidisciplinary treatment modalities are
considered to be significant factors for improving treatment
outcomes.
The treatment of an Askin tumor should aim to
control local disease and distant metastasis. Thus, the prevailing
treatment of an Askin tumor is a combination of neoadjuvant
chemotherapy, radical surgical resection and adjuvant chemotherapy
and radiotherapy. Several studies have proved that this aggressive
therapy may lead to a longer relapse-free survival (14,15).
Consequently, in the present study, recent cases of Askin tumors
and their treatment were reviewed. It was identified that almost
all the patients underwent a multidisciplinary treatment including
surgery, radiotherapy and chemotherapy.
Surgery
The satisfactory outcomes that have been achieved to
date may be attributed to comprehensive treatment. Surgery, as an
option in the treatment of an Askin tumor, is crucial for local
control due to the removal of the tumor itself, while
chemotherapy/radiotherapy are administered as supplementary
treatment. Numerous researchers have reported satisfactory results
following surgery. According to a study on malignant chest wall
tumors in children and young adults by Dang et al(16), surgical resection with en bloc
removal of adjacent muscles or organs and chest wall reconstruction
provided excellent local control of malignant chest wall tumors.
According to a study conducted in the Memorial Sloan-Kettering
Cancer Center (MSKCC) (17),
complete remission was achieved in patients following surgery and
chemotherapy rather than chemotherapy alone, thus underlining the
importance of surgery. In this study (17), patients with large primary tumors
that underwent surgical resection within 3 months of diagnosis were
correlated with a significantly improved progression-free survival.
Improved survival following surgical treatment has also been
observed in other patient groups. Christiansen et
al(14) described the cases of
8 patients with Askin tumors who underwent integrated treatment.
Overall, 4/8 patients who underwent a complete resection were alive
after a median follow-up time of 30 months, while the remaining 4
patients with extended disease and marginal surgery succumbed to
the disease. Demir et al(18) showed that patients who underwent a
complete resection exhibited a higher 5-year survival rate compared
with patients who underwent an incomplete resection (56 vs. 25%;
P=0.13). With regard to repeated relapse cases, a surgical
resection combined with chemoradiotherapy resulted in satisfactory
survival rates. The case of a 16-year-old male with an Askin tumor
was described by Takanami et al(11). The patient underwent surgery for the
excision of the tumor and subsequently underwent 6 excisions for
local recurrence, with follow-up post-operative chemotherapy and
radiotherapy for 7 years. This was the first case to be reported
with long-term survival after repeated resections of local
recurrences of an Askin tumor.
However, there are contradictory opinions with
regard to the necessity and importance of surgery. Firstly,
although the most effective treatment is surgery, a macroscopically
complete resection does not guarantee absence of local recurrence.
This means that early local recurrence and distant metastases may
still be observed following complete resection and adjuvant
therapy. Secondly, surgery combined with chemoradiotherapy is
effective in the case of localized tumors. However, the role of
surgery remains controversial for some cases of metastatic disease.
Gunluoglu et al(19) showed
that pre-operative therapy reduces the tumor size and facilitates
surgery, while it does not decrease the probability of early local
recurrence and distant metastasis. Incomplete surgical excision is
not effective for the treatment of this type of tumor. Although
surgery appears to be an effective treatment strategy, no effect
has been observed on the disease-free period.
It is widely accepted that certain conditions are
required prior to surgery, including a small initial volume of the
tumor, a good response to systemic therapy, an operable location
and sufficient resection margins. Otherwise, surgery should not be
used as the only local therapy.
With regard to the implementation of the surgery, it
should be aggressive with the aim of achieving good margins of
resection, since a PNET has a high tendency to recur locally.
According to a retrospective analysis of 42 patients, 10/12
patients with relapsed disease underwent an intralesional or
marginal resection (20). Soyer
et al(21) suggested that
the general approach should be to perform a wide excision of all
the involved structures, regardless of the tumor size. Concerning
primary tumors of the ribs, all the involved ribs and their
adjacent muscles and underlying pleura should be excised. However,
not all tumors are suitable for complete resection since the tumor
is usually extensive and is located in a difficult anatomical site.
Therefore, adjuvant therapy is often administered to decrease the
size of the tumor prior to resection, as well as to treat potential
micrometastasis.
Radiotherapy
The main role of radiotherapy is to achieve a
satisfactory control of the primary disease; radiotherapy is also
administered as adjuvant therapy prior to or following resection.
Pre-operative radiotherapy is particularly suitable for patients
with a poor clinical response to the initial chemotherapy or for
patients in whom further tumor regression would allow the
possibility of function-preserving surgery. In Cooperative Ewing’s
Sarcoma Studies (CESS) 81 and 86 and European Intergroup
Cooperative Ewing’s Sarcoma Study (EICESS) 92, radiotherapy was
used in ~87% of patients as a pro- or post-operative adjuvant
therapy or as radical radiotherapy for unresectable tumors
(22,23). Based on these studies, Schuck et
al(22) concluded that
irradiation alone or post-operative irradiation as local therapy
had satisfactory outcomes in local control and patient survival.
Based on the protocol used in the CESS 81 study, radiotherapy was
demonstrated to be similarly efficient to surgery for small lesions
(lesion volume, <100 ml); however, a trend towards a better
prognosis in surgically treated patients with large lesions >100
ml in volume was observed (12).
The implementation of three dimensional conformal radiotherapy
(3D-CRT) and intensity-modulated radiotherapy (IMRT), including
meticulous delineation of planning target volumes (PTVs), treatment
planning and accurate execution, result in the local failure rate
reducing from an unacceptable 28.5% in the patients with PNET of
the chest wall in the CESS 81 study to 8.6% in the CESS 86 study.
The results from the Memorial Sloan-Kettering study demonstrated
that radiotherapy was an effective modality for local control,
particularly for patients without metastases (13). Although certain studies have
performed radical resections, in multiple metastatic settings,
chemotherapy combined with irradiation is the standard treatment in
patients with macroscopic lesions (14,24,25).
External-beam radiotherapy constitutes an alternative treatment
strategy in patients whose pathological complete response rates are
low, indicating a high risk of local relapse. Furthermore, certain
studies have also suggested intraoperative radiotherapy to be an
alternative treatment method in which satisfactory efficacy was
observed in patients with cancer recurrence (26).
The standard radiotherapy approach, where the
treatment program included exposure to 45–60 Gy plus intensive use
of cyclophosphamide/doxorubicin, was established by Miser et
al(27). According to this
study, patients with PNETs of the chest wall received 45 Gy to the
tumor with a surrounding 2 cm margin. Following treatment, no
recurrence was observed in the radiation field, while pleural
recurrence was observed in 3 patients. Schuck et al(22) suggested that in-field recurrence
should be expected when doses of >50 Gy are used. Although
appropriate radiation fields being provided, marginal relapses
cannot be completely prevented. Currently, the dose for adjuvant
radiotherapy is usually between 20 and 60 Gy. Radiotherapy is
rarely used as the preferred treatment of a primary tumor, unless
the tumor is anatomically unresectable, such as the case presented
in the current study.
However, in view of the radiation-related chest wall
deformities of growing bones and the neurodevelopmental
complications, such as pulmonary fibrosis, when large volumes of
the lung are in the radiation field, as well as the fear of the
late second malignancy, the radiotherapy should be individualized,
particularly in younger patients. Radiation to the heart also
increases the possibility of cardiomyopathy induced by doxorubicin
(28). However, the application of
IMRT solved the problem and provided superior dose coverage of the
PTV with the aim of keeping the dose to the organ at risk (OAR) as
low as possible (29). Adjuvant
radiotherapy is used when there is a high risk of recurrence, such
as in a incomplete tumor resection, residual microscopic disease or
tumor contamination during surgery. Currently, most patients
undergoing definitive surgery following initial chemotherapy have
an increased possibility of a successful complete resection of the
tumor, and as a result, radiation to the chest is avoided.
Chemotherapy
Chemotherapy is the first choice for the treatment
of Ewing sarcoma, and the subsequent combination of surgery and
radiation constitute the standard therapy (30). However, no standard therapy is
available for the treatment of Askin tumors due to the rarity of
this disease and the poor prognosis. Based on the grouping of these
two diseases into the same WHO classification in 2002, the
therapeutic guidelines for Ewing sarcoma may be useful in guiding
the treatment of Askin tumors. In certain cases, extended surgery
followed by post-operative chemotherapy and radiotherapy is the
first choice of treatment (31). A
number of researchers have reported the potential advantages of
pre-operative chemotherapy with reference to the treatment of Ewing
sarcoma (32,33). Veronesi et al(32) summarized the benefits of
pre-operative chemotherapy, including the reduction in the risk of
intraoperative tumor rupture and tumor cell dissemination, the
increase in the probability of R0 resection, the enhanced
probability of post-operative function preservation by a more
conservative surgical approach, the decrease in any occult distant
spread and the provision of a pathological and clinical evaluation
of the response that favors the choice of the best post-operative
regimen of chemotherapy and radiotherapy. Sawin et
al(33) demonstrated that
pre-operative chemotherapy resulted not only in a reduced tumor
volume (from 7,054 to 911 cm3), but also in prolonged
survival rates. Demir et al(18) reported that neoadjuvant chemotherapy
significantly increased the complete resection rate (P=0.027) and
that the 5-year survival rates of the patients with or without
neoadjuvant therapy were 77 and 37%, respectively (P=0.22).
Concerning older patients, radical surgery may be avoided when a
complete response is achieved following neoadjuvant chemotherapy
(34).
Askin tumors/PNETs are highly sensitive to
chemotherapy. Due to the characteristic high recurrence rates and
the high likelihood of metastases of this disease, systemic
chemotherapy should be prompt even though the disease is
organ-confined (8). Originally,
chemotherapy for Ewing sarcoma consisted of 12 cycles of
vincristine, actinomycin D, cyclophosphamide and adriamycin (ADM;
VACA) in low-risk tumors (extremity tumors <100 cm3).
In the case of patients with a high risk of recurrence (central
tumors ≥100 cm3), the chemotherapeutic regimen used
included vincristine, actinomycin D, ifosfamide (IFM) and ADM
(VAIA) (35). Subsequently, similar
to the EICESS 92 protocol for high-risk patients, the chemotherapy
regime included 14 cycles of etoposide, vincristine, actinomycin D,
IFM and ADM (EVAIA). The therapy was repeated every 3 weeks (1
cycle), while ADM was replaced with actinomycin D (36). Grier et al(37) reported that adding IFM and etoposide
to the standard therapy (VACA) significantly improved the outcomes
for patients with non-metastatic Ewing sarcoma, primitive
neuroectodermal tumor of the bone or primitive sarcoma of the bone.
However, according to Miser et al(38), the outcomes were disappointing in
patients with metastatic disease.
The improvement of the treatment strategy for Ewing
sarcoma and PNETs may be inspirational to the treatment of Askin
tumors. Currently, the regimen includes multiple agents, including
doxorubicin, vincristine and cyclophosphamide, alternating with IFM
and etoposide (39). Additional
effective chemotherapy regimens have also been reported. Askin
et al(1) and Contesso et
al(10) considered the commonly
used chemotherapy regimen to be VAC, although a combination of DDP
and 5-fluorouracil (5-FU) were also effective. We demonstrated that
a chemotherapy regimen of a combination of vincristine, epirubicin
and cisplatin was effective and well-tolerated by patients with
less side-effects. Recently, Japanese researchers (40) reported satisfactory results when an
adult with a PNET with multiple lung metastases was treated with
the ADM and IFM (AI) regimen. After 7 cycles of chemotherapy, the
size of the primary tumor and the multiple lung metastases
decreased. Therefore, the authors suggested that the AI regimen was
effective for PNET treatment.
The main side-effect of intensive chemotherapy is
bone marrow suppression. In the event of sufficient bone marrow
recovery, the next cycle of chemotherapy may be postponed, with the
addition of granulocyte-colony stimulating factor (G-CSF).
Autologous bone marrow transplantation has been suggested to be an
effective rescue treatment, but this remains controversial. It has
been demonstrated that the use of high-dose chemotherapy followed
by either autologous bone marrow transplantation or peripheral stem
cell rescue is valuable in the treatment of Ewing sarcoma (41). This potential benefit may be applied
to PNET, particularly to patients with metastasis whose prognosis
is usually poor. Young et al(42) reported the use of bone marrow
transplantation after intensive chemoradiotherapy in patients with
sarcomas of the chest wall. The preliminary results were
satisfactory with a significant complete response rate and
acceptable morbidity rates. In addition, cardiac toxicity with the
use of anthracyclines should attract considerable attention during
chemotherapy, particularly in children.
In conclusion, Askin tumors/PNETs develop in soft
tissues and the disease is detectable only when the tumor is large.
Thus, Askin tumors are mostly diagnosed at an advanced stage, which
is one of the most important reasons for a poor prognosis.
Moreover, due to the rarity of this disease, relevant studies are
mostly small-scale, single-institution clinical trials. The
diagnosis and treatment of Askin tumors/PNETs remains a challenge
for clinicians and surgeons due to the absence of standard
guidelines. Notably, a wide resection combined with
polychemotherapy has been shown to result in satisfactory local
control. The results of local control and decreased distant relapse
in patients administered neoadjuvant chemotherapy followed by a
complete resection are superior compared with the results in
patients who undergo primary surgery followed by chemotherapy
and/or radiotherapy. Consequently, large-scale randomized trials
are required for the development of effective treatment strategies
for Askin tumors.
References
|
1
|
Askin FB, Rosai J, Sibley RK, Dehner LP
and McAlister WH: Malignant small cell tumor of the
thoracopulmonary region in childhood: a distinctive
clinicopathologic entity of uncertain histogenesis. Cancer.
43:2438–2451. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kara Gedik G, Sari O, Altinok T, Tavli L,
Kaya B and Ozcan Kara P: Askin’s tumor in an adult: case report and
findings on 18F-FDG PET/CT. Case Rep Med. 2009:5173292009.
|
|
3
|
Crocoli A, Bagolan P, Boldrini R, Natali
GL, De Ioris MA and Morini F: Congenital Askin tumor with favorable
outcome: case report and review of the literature. J Pediatr Surg.
47:1440–1444. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shrestha B, Kapur BN, Karmacharya K,
Kakkar S and Ghuliani R: Askin’s tumor: a dual case study. Int J
Pediatr. 2011:2521962011.
|
|
5
|
Verfaillie G, Hoorens A and Lamote J:
Primary primitive neuro-ectodermal tumor of the lung. Acta Chir
Belg. 109:381–384. 2009.PubMed/NCBI
|
|
6
|
Delattre O, Zucman J, Melot T, et al: The
Ewing family of tumors - a subgroup small-round-cell tumurs defined
by specific chimeric transcripts. N Engl J Med. 331:294–299. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hicks MJ, Smith JD Jr, Carter AB, Flaitz
CM, Barrish JP and Hawkins EP: Recurrent intrapulmonary malignant
small cell tumor of the thoracopulmonary region with metastasis to
the oral cavity: review of literature and case report. Ultrastruct
Pathol. 19:297–303. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mikami Y, Nakajima M, Hashimoto H, Irei I,
Matsushima T, Kawabata S and Manabe T: Primary pulmonary primitive
neuroectodermal tumor (PNET). A case report. Pathol Res Pract.
197:113–119. 2001.PubMed/NCBI
|
|
9
|
Fink M, Salisbury J and Gishen P: Askin
tumor: three case histories and a review of the literature. Eur J
Radiol. 14:178–180. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Contesso G, Llombart-Bosch A, Terrier P,
et al: Does malignant small round cell tumor of the
thoracopulmonary region (Askin tumor) constitute a
clinicopathologic entity? An analysis of 30 cases with
immunohistochemical and electron-microscopic support treated at the
Institute Gustave Roussy. Cancer. 69:1012–1020. 1992. View Article : Google Scholar
|
|
11
|
Takanami I, Imamura T, Naruke M and
Kodaira S: Long-term survival after repeated resections of Askin
tumor recurrences. Eur J Cardiothorac Surg. 13:313–315. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sauer R, Jürgens H, Burgers JM, Dunst J,
Hawlicek R and Michaelis J: Prognostic factors in the treatment of
Ewing’s sarcoma. The Ewing’s Sarcoma Study Group of the German
Society of Paediatric Oncology CESS 81. Radiother Oncol.
10:101–110. 1987.
|
|
13
|
La TH, Meyers PA, Wexler LH, et al:
Radiation therapy for Ewing’s sarcoma: results from Memorial
Sloan-Kettering in the modern era. Int J Radiat Oncol Biol Phys.
64:544–550. 2006.
|
|
14
|
Christiansen S, Semik M,
Dockhorn-Dworniczak B, et al: Diagnosis, treatment and outcome of
patients with Askin-tumors. Thorac Cardiovasc Surg. 48:311–315.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nakajima Y, Koizumi K, Hirata T, et al:
Long-term survival of Askin tumor for 10 years with 2 relapses. Ann
Thorac Cardiovasc Surg. 12:137–140. 2006.PubMed/NCBI
|
|
16
|
Dang NC, Siegel SE and Phillips JD:
Malignant chest wall tumors in children and young adults. J Pediatr
Surg. 34:1773–1778. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kushner BH, Hajdu SI, Gulati SC, Erlandson
RA, Exelby PR and Lieberman PH: Extracranial primitive
neuroectodermal tumors. The Memorial Sloan-Kettering Cancer Center
experience. Cancer. 67:1825–1829. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Demir A, Gunluoglu MZ, Dagoglu N, et al:
Surgical treatment and prognosis of primitive neuroectodermal
tumors of the thorax. J Thorac Oncol. 4:185–192. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gunluoglu MZ, Kara HV, Demir A and Dincer
SI: Results of multimodal treatment of two patients with thoracic
primitive neuroectodermal tumor. Is surgery really helpful for
survival? Thorac Cardiovasc Surg. 55:460–461. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jürgens H, Bier V, Harms D, et al:
Malignant peripheral neuroectodermal tumors. A retrospective
analysis of 42 patients. Cancer. 61:349–357. 1988.PubMed/NCBI
|
|
21
|
Soyer T, Karnak I, Ciftci AO, Senocak ME,
Tanyel FC and Büyükpamukçu N: The results of surgical treatment of
chest wall tumors in childhood. Pediatr Surg Int. 22:135–139. 2006.
View Article : Google Scholar
|
|
22
|
Schuck A, Hofmann J, Rübe C, et al:
Radiotherapy in Ewing’s sarcoma and PNET of the chest wall: results
of the trials of CESS 81, CESS 86, and EICESS 92. Int J Radiat
Oncol Biol Phys. 42:1001–1006. 1998.
|
|
23
|
Schuck A, Ahrens S, Konarzewska A, et al:
Hemithorax irradiation for Ewing tumors of the chest wall. Int J
Radiat Oncol Biol Phys. 54:830–838. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Eralp Y, Bavbek S, Başaran M, et al:
Prognostic factors and survival in late adolescent and adult
patients with small round cell tumors. Am J Clin Oncol. 25:418–424.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cañizares MA, Arnau A and Cantó A: Askin’s
tumor of the chest wall with early metastasis. Arch Bronconeumol.
37:215–216. 2001.(In Spanish).
|
|
26
|
Yeste L, Sierra A, Cañon R, Aristu J and
Torre W: Successful use of intraoperative radiotherapy for local
control of an Askin’s tumor recurrence. J Cardiovasc Surg (Torino).
42:143–145. 2001.PubMed/NCBI
|
|
27
|
Miser JS, Kinsella TJ, Triche TJ, et al:
Treatment of peripheral neuroepithelioma in children and young
adults. J Clin Oncol. 5:1752–1758. 1987.PubMed/NCBI
|
|
28
|
Corn BW, Trock BJ and Goodman RL:
Irradiation-related ischemic heart disease. J Clin Oncol.
8:741–750. 1990.PubMed/NCBI
|
|
29
|
Jamema SV, Sharma PK, Laskar S, et al:
Treatment planning comparison of electron arc therapy and photon
intensity modulated radiotherapy for Askin’s tumor of chest wall.
Radiother Oncol. 84:257–265. 2007.PubMed/NCBI
|
|
30
|
Nesbit ME Jr, Gehan EA, Burgert EO Jr, et
al: Multimodal therapy for the management of primary, nonmetastatic
Ewing’s sarcoma of bone: a long-term follow-up of the First
Intergroup study. J Clin Oncol. 8:1664–1674. 1990.
|
|
31
|
Takanami I and Imamura T: The treatment of
Askin tumor: results of two cases. J Thorac Cardiovasc Surg.
123:391–392. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Veronesi G, Spaggiari L, De Pas T, et al:
Preoperative chemotherapy is essential for conservative surgery of
Askin tumors. J Thorac Cardiovasc Surg. 125:428–429. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sawin RS, Conrad EU III, Park JR and
Waldhausen JH: Preresection chemotherapy improves survival for
children with Askin tumors. Arch Surg. 131:877–880. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mulsow J, Jeffers M, McDermott R, Geraghty
J and Rothwell J: Complete clinical response to neoadjuvant
chemotherapy in a 54-year-old male with Askin tumor. Thorac
Cardiovasc Surg. 58:306–308. 2010.PubMed/NCBI
|
|
35
|
Paulussen M, Ahrens S, Dunst J, et al:
Localized Ewing tumor of bone: final results of the Cooperative
Ewing’s Sarcoma Study CESS 86. J Clin Oncol. 19:1818–1829.
2001.PubMed/NCBI
|
|
36
|
Schuck A, Ahrens S, Paulussen M, et al:
Local therapy in localized Ewing tumors: results of 1058 patients
treated in the CESS 81, CESS 86 and EICESS 92 trials. Int J Radiat
Oncol Biol Phys. 55:168–177. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Grier HE, Krailo MD, Tarbell NJ, et al:
Addition of ifosfamide and etoposide to standard chemotherapy for
Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl
J Med. 348:694–701. 2003.
|
|
38
|
Miser JS, Krailo MD, Tarbell NJ, et al:
Treatment of metastatic Ewing’s sarcoma or primitive
neuroectodermal tumor of bone: evaluation of combination ifosfamide
and etoposide - a Children’s Cancer Group and Pediatric Oncology
Group study. J Clin Oncol. 22:2873–2876. 2004.
|
|
39
|
Windfuhr JP: Primitive neuroectodermal
tumor of the head and neck: incidence, diagnosis, and management.
Ann Otol Rhinol Laryngol. 113:533–543. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ishiyama T, Jinguu A, Matsumoto H, Kikuchi
J, Suzuki T and Yamakawa M: A case of adult primitive
neuroectodermal tumor (PNET) with multiple lung metastases
effectively treated with ADM, IFM (AI) regimen. Gan To Kagaku
Ryoho. 39:1287–1289. 2012.(In Japanese).
|
|
41
|
Burdach S, Jürgens H, Peters C, et al:
Myeloablative radiochemotherapy and hematopoietic stem-cell rescue
in poor-prognosis Ewing’s sarcoma. J Clin Oncol. 11:1482–1488.
1993.
|
|
42
|
Young MM, Kinsella TJ, Miser JS, Triche
TJ, Glaubiger DL, Steinberg SM and Glatstein E: Treatment of
sarcomas of the chest wall using intensive combined modality
therapy. Int J Radiat Oncol Biol Phys. 16:49–57. 1989. View Article : Google Scholar : PubMed/NCBI
|