Introduction
Despite advances in glioma treatments, including
neurosurgery, radiotherapy and chemotherapy, the prognosis of
patients diagnosed with malignant glioma has not improved in recent
years (1–4). The poor prognosis of glioblastoma
multiforme (GBM) is mainly due to tumor cell invasion of the brain
tissue beyond the resected areas (5–7).
Moreover, brain tumors are resistant to current therapies (6).
Glioma invasion is a complex cellular phenomenon
that involves changes in the intracellular and extracellular
biomechanical systems (4). Although
our understanding of glioma oncogenesis is steadily improving, the
molecular mechanisms that mediate glioma invasion remain poorly
understood (8). There have been
numerous studies describing the extracellular factors involved in
glioma cell invasion, including β-catenin, c-Myc and cyclin D1
(9–11). However, the intracellular and
molecular mechanisms that mediate glioma invasion require further
elucidation in order to identify new drug targets.
ID proteins (inhibitors of DNA
binding/differentiation) are helix-loop-helix transcription factors
(12,13). The reactivation of the ID proteins,
particularly ID1, promotes the development of several tumor types,
including high-grade astrocytoma, prostate and breast cancers and
non-small cell lung carcinoma (14–17).
ID1 controls the expression of a large number of genes that mediate
important cellular processes by inhibiting the activity of bHLH
proteins (18–20). The main role of ID1 is to inhibit
cell differentiation (18). In
addition, loss of differentiation, unrestricted proliferation and
increased cell invasion are hallmarks of malignancy. By maintaining
an immature phenotype, ID1 enhances cell proliferation and
invasion. Therefore, ID1 overexpression may induce invasion in
several cancer types (21–23). The importance of ID1 in promoting
tumor invasion and metastasis has emerged and a role as a possible
molecular marker of tumor aggressiveness has been proposed
(22). However, the role of ID1 in
GBM is poorly understood.
In the present study, ID1-small hairpin RNA
(shRNA)-expressing U87 cells and controls were constructed, and
Transwell invasion and scratch assays were performed to analyze the
effect of the knockdown of ID1 on cell invasion.
Materials and methods
Cell culture
The U87 human glioma cell line was obtained from the
American Type Culture Collection (Manassas, VA, USA). The cells
were grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco BRL,
Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum
(FBS) at 37°C in a humidified atmosphere containing 5%
CO2.
qPCR
The U87 cells were washed three times with ice-cold
phosphate-buffered saline (PBS) and total RNA was extracted using
TRIzol (Invitrogen, Carlsbad, CA, USA). An equal amount of total
RNA was used for first-strand cDNA synthesis using the oligo-dT
primer and M-myeloblastosis virus reverse transcriptase XL
(Promega, Madison, WI, USA) in a reaction volume of 25 μl,
according to the manufacturer’s instructions. Synthesized
first-strand cDNA (2 μl) was used for each PCR reaction.
qPCR experiments were performed using the SYBR Green
PCR Master Mix (Applied Biosystems, Foster City, CA, USA). The PCR
products were subjected to melting curve analysis to exclude the
synthesis of non-specific products. The Ct value was quantified
using a standard curve for the specific gene and relatively
quantified using GAPDH as an internal reference control. The Ct
value was then normalized to the average expression levels of
undifferentiated samples, calculated according to the
2-ΔΔCt method. All experiments were performed in
triplicate.
Western blotting
Cellular proteins (30 μg) were subjected to 12% SDS
polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto
Hybond ECL nitrocellulose membranes (Amersham, Piscataway, NJ,
USA). Subsequent to being washed with 0.1% TBS-T, the membranes
were blocked in 5% skimmed milk in TBS-T for 1 h at room
temperature, then incubated with the appropriate antibody [1/500
dilution, ID1 antibody sc-488 (Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA); 1/1,000 dilution β-actin antibody (Abcam,
London, England); 1/1,000 dilution, cyclin D1 antibody (Cell
Signaling Technology, Danvers, MA, USA); 1/1,000 dilution, c-Myc
antibody (Cell Signaling Technology); 1/1,000 dilution, β-catenin
antibody (Abcam); 1/500 dilution, E-cadherin antibody (Cell
Signaling Technology)] diluted in the same buffer overnight at 4°C.
Subsequent to being washed with 0.1% TBS-T, the membranes were
incubated with horseradish peroxidase-conjugated anti-rabbit or
anti-mouse antibody, as appropriate (1/10,000 dilution; Sigma, St.
Louis, MO, USA), for 2 h at room temperature. After washing with
0.1% TBS-T, specific protein bands were detected using western
blotting detection reagents (Odyssey; Licor, Lincoln, NE, USA).
Construction of U87 cells
The pGIPZ expression vector (Thermo Scientific,
Waltham, MA, USA) carrying the ID1-shRNA coding sequence (shRNA
sequence, TCGGAATCCGAAGTTGGAA) and the control empty vector were
transfected into the U87 cells using Lipofectamine 2000
(Invitrogen), according to the manufacturer’s instructions. Two
days after transfection, 800 μg/ml puromycin (Sigma) was added to
the growth medium for the selection of stable ID1-shRNA-expressing
cells. Colonies expressing marked green fluorescence were selected
for further studies.
BrdU proliferation of cells
Cell proliferation was assessed by
5-bromo-2′-deoxy-uridine (BrdU) incorporation using a Cell
Proliferation ELISA, BrdU kit (Roche Applied Science, Indianapolis,
IN, USA) according to the manufacturer’s instructions. The cells
were seeded onto a 96-well plate at a density of 1×105
cells/well in 100 μl culture medium and incubated at 37°C for 6,
12, 24 or 48 h. BrdU labeling solution was then added to a final
concentration of 10 μM and the cells were incubated for an
additional 2–4 h at 37°C. The medium was then removed and FixDenat
(200 μl/well) was added to the cells and incubated for 30 min at
25°C. The FixDenat solution was then completely removed and 100
μl/well anti-BrdU-POD working solution was added and incubated for
90 min at 25°C The antibody conjugate was then removed by flicking
and the wells were washed three times with 200 μl/well washing
solution. Substrate solution (100 μl/well) was added and the cells
were incubated at 25°C until color development was sufficient for
photometric detection (after 6, 12, 24 and 48 h). The absorbance
[optical density (OD)] at 450 nm was measured using a microplate
reader.
Cell adhesion assay
A 96-well plate was coated with Matrigel and
incubated at 37°C for 1 h. The U87 cells were then plated at
5×104 cells/well in serum-free MEM and the plate was
incubated for 30 min at 37°C, followed by a gentle rinse with PBS
to remove non-adherent cells. The cells were then fixed for 20 min
with 3.5% formalin, stained with 0.5% crystal violet for 1 h and
rinsed twice with PBS. The absorbance of the test samples and blank
controls was measured at 594 nm using a microplate reader. The OD
value of each test sample was designated the measured value and
that of the blank was designated the blank value. The final value =
measured value - blank value.
Transwell invasion assay
Matrigel (25 mg reconstituted basement membrane) was
added onto a polyvinylpyrrolidone-free polycarbonate filter
(Nuclepore; Whatman, Maidstone, UK) and dried. The cells were
harvested following brief exposure to 1 mM EDTA, then washed with
DMEM containing 0.1% bovine serum albumin and added to Boyden
chambers (2×105 cells/chamber). The chambers were
incubated for 24 h at 37°C. The cells that traversed the Matrigel
layer and became attached to the filter were stained using a
Diff-Quik kit (Dade Diagnostics, Aguada, PR, USA) and five
randomized fields were counted. The mean ± SE was calculated for
three independent experiments.
Scratch assay
The cells were cultured to 90% confluency in
six-well plates, then a thin scratch (wound) was made in the
central area using a 10-ml pipette tip. Detached and damaged cells
were carefully removed with PBS and the medium was replaced with
serum-free medium. Wound closure was observed by light microscopy
and images were captured at the indicated time points.
Immunofluorescence
The cells were cultured on glass coverslips in 35-mm
diameter dishes, washed with PBS and fixed with 4% p-formaldehyde
for 15 min. Subsequent to being washed with PBS, the cells were
incubated with 0.1% Triton X-100 for 25 min, washed three times
with PBS and blocked with 10% goat serum for 30 min at room
temperature. The cells were then incubated with an anti-actin
antibody (1/100; Abcam) in a humidified chamber either overnight at
4°C or for 2 h at room temperature, followed by incubation with
Alexa 488-conjugated goat anti-mouse antibody (1/200; Molecular
Probes, Eugene, OR, USA) for 1 h at room temperature in a
humidified chamber. DAPI staining was performed to identify the
cell nuclei, and the cells were observed using a confocal
Lasersharp 2000 version 5.1 microscope (Carl Zeiss, Jena, Germany)
equipped with a Plan-Apochromat 63x/1.40 oil objective lens.
Confocal images were acquired using LSM 5102.3 software (Alexa 488
emission was at 519 nm and Alexa 568 emission was at 603 nm).
Statistical analysis
Data are represented as the mean ± SE from at least
three independent experiments. All data were analyzed using an
independent samples t-test using GraphPad Prism 5 software (San
Diego, CA, USA). P<0.05 were considered to indicate a
statistically significant difference.
Results
ID1-knockdown inhibits the proliferation
of U87 cells
Our previous study showed that ID1 is highly
expressed in several GBM cell lines, including A172, T98g, U251 and
U87; of these, the U87 cells have the highest level of ID1
expression. Therefore, ID1 was knocked down in the U87 cells using
ID1-shRNA expression and the changes in cell proliferation and
invasion were measured between the ID1-silenced and control cells
transfected with empty vector. It appears that ID1 expression was
reduced in the U87 cells following shRNA-ID1 transfection (24).
Cell proliferation is necessary for the development
of all types of cancer, including gliomas. A previous study
demonstrated that ID1 promotes tumor progression, although there is
no direct evidence that ID1 is involved in GBM cell proliferation
(25). The BrdU cell proliferation
assay was therefore used to analyze the role of ID1 in GBM cell
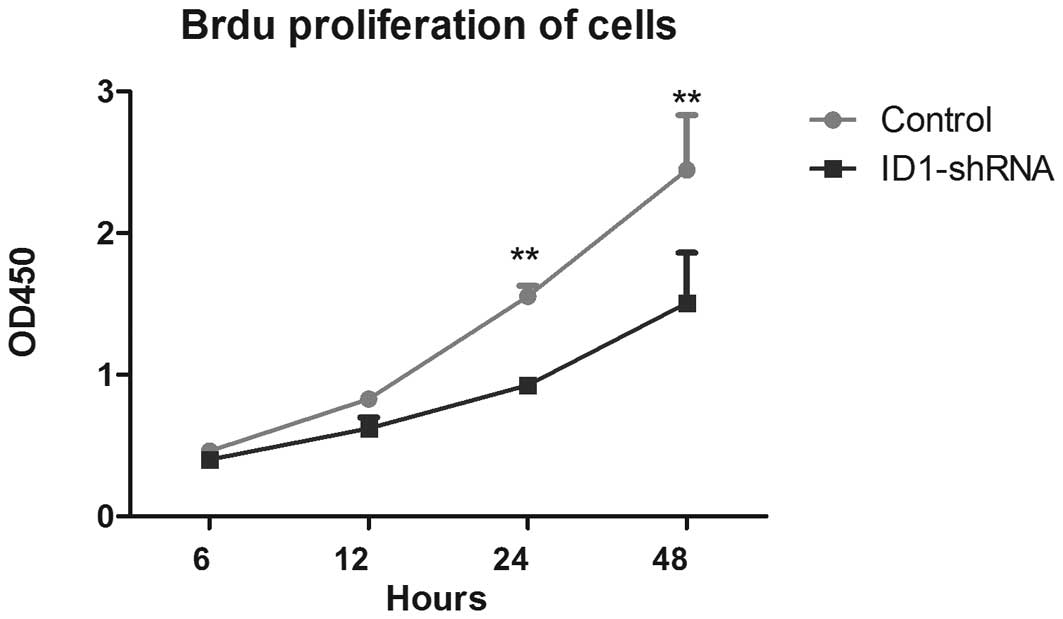
proliferation. As shown in Fig. 1,
there were significant differences in cell proliferation between
the ID1-silenced and control cells at the 24- and 48-h time points;
decreased ID1 expression correlated with a reduction in U87 cell
proliferation. These data suggest that ID1 has a role in blocking
aberrant U87 cell proliferation.
ID1-knockdown reduces the invasiveness of
U87 cells
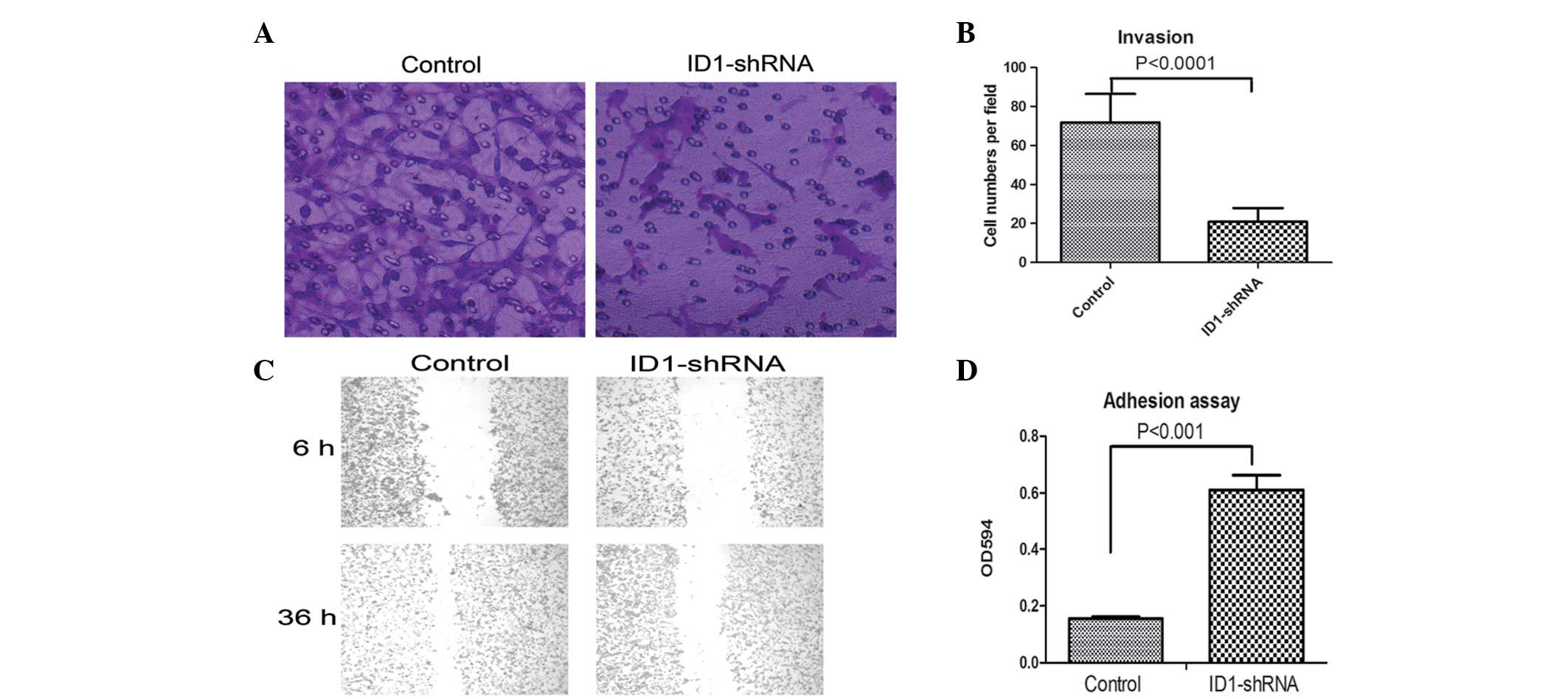
Transwell invasion and scratch assays were used to
measure the rate of invasion of the ID1-silenced and control U87
cells. The Transwell invasion assay showed that fewer
ID1-shRNA-expressing U87 cells passed through a polycarbonate
membrane compared with the controls (P<0.0001; Fig. 2A and B). The scratch assay showed
that the there was no significant difference in wound healing
between the two groups of cells after 6 h. However, after 36 h,
there was almost complete wound healing in the control cells, but
not in the U87 cells (Fig. 2C).
This result is consistent with the result of the Transwell invasion
assay, i.e., that wound healing and migration are inhibited by
reduced ID1 expression. These data suggest that ID1-knockdown
blocks U87 cell invasion.
ID1-knockdown enhances U87 cell
adhesion
Reduced adhesion is necessary for the increase in
cell mobility and invasion capacity. A cell adhesion assay was used
to compare the adhesive properties of ID1-silenced and control
cells. As shown in Fig. 2D, the U87
cells with ID1-knockdown were more adhesive compared with the
control cells, i.e., a change in ID1 expression led to a change in
cell adhesion. Compared with the controls, the ID1-silenced U87
cells showed more marked adhesive properties. This is consistent
with higher levels of cell invasion in normal U87 cells.
Effects of ID1-knockdown on U87
morphology and cytoskeleton
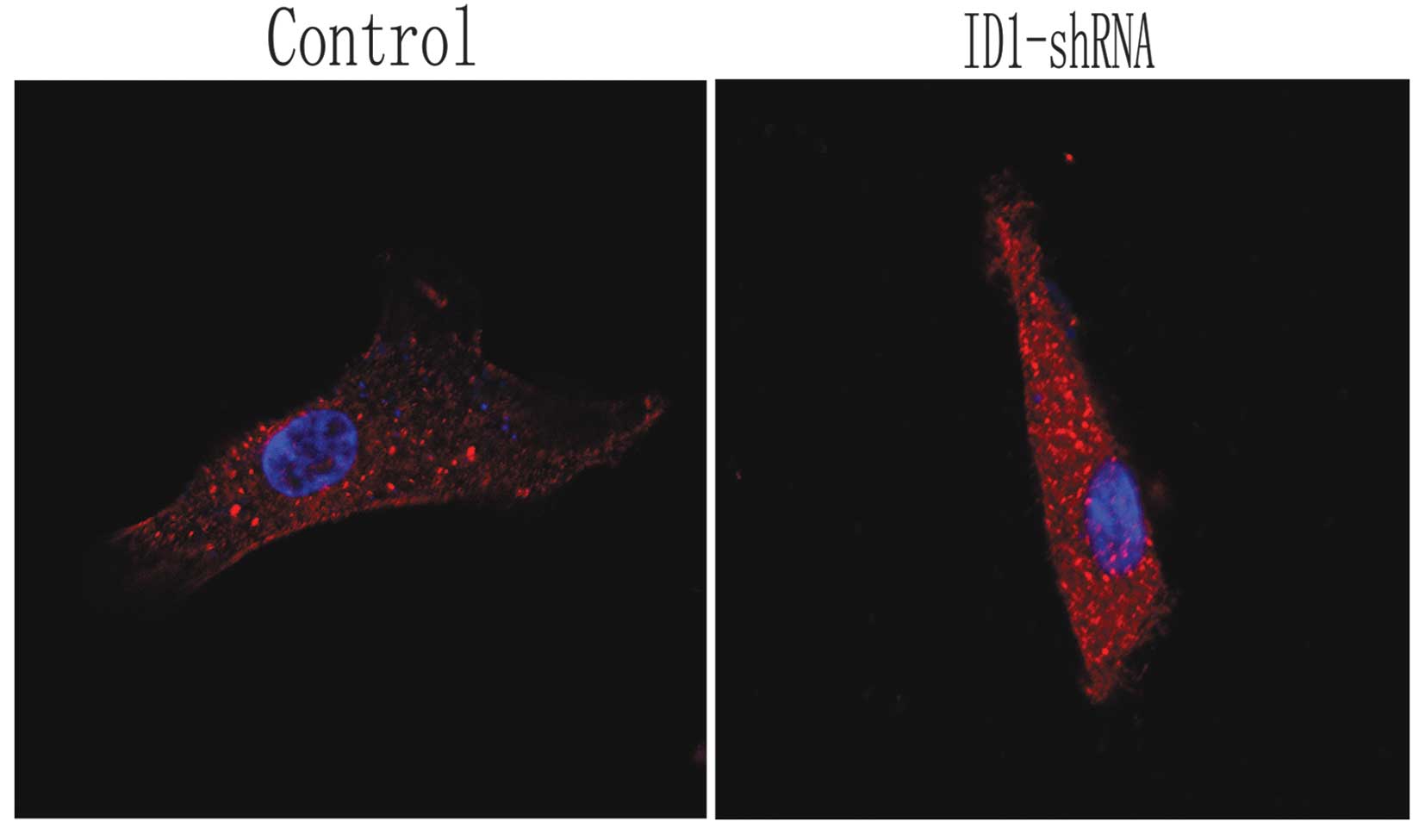
In order to observe the morphological and
cytoskeletal changes in the U87 cells following the silencing of
ID1, actin filaments were observed by immunofluorescence and
confocal scanning laser microscopy in the treated and control
cells. In the control cells, actin was localized and highly
expressed on pseudopodia around the nucleus and on stress fibers.
However, the U87 cells expressing ID1-shRNA became flatter and
smaller, with shorter lamellipodia compared with the controls
(Fig. 3). Thus, cytoskeletal
changes due to ID1 downregulation are associated with altered tumor
cell invasion properties. These data support the hypothesis that
U87 cell invasion is reduced following the knockdown of ID1.
ID1 regulates expression of β-catenin,
cyclin D1, c-Myc and E-cadherin
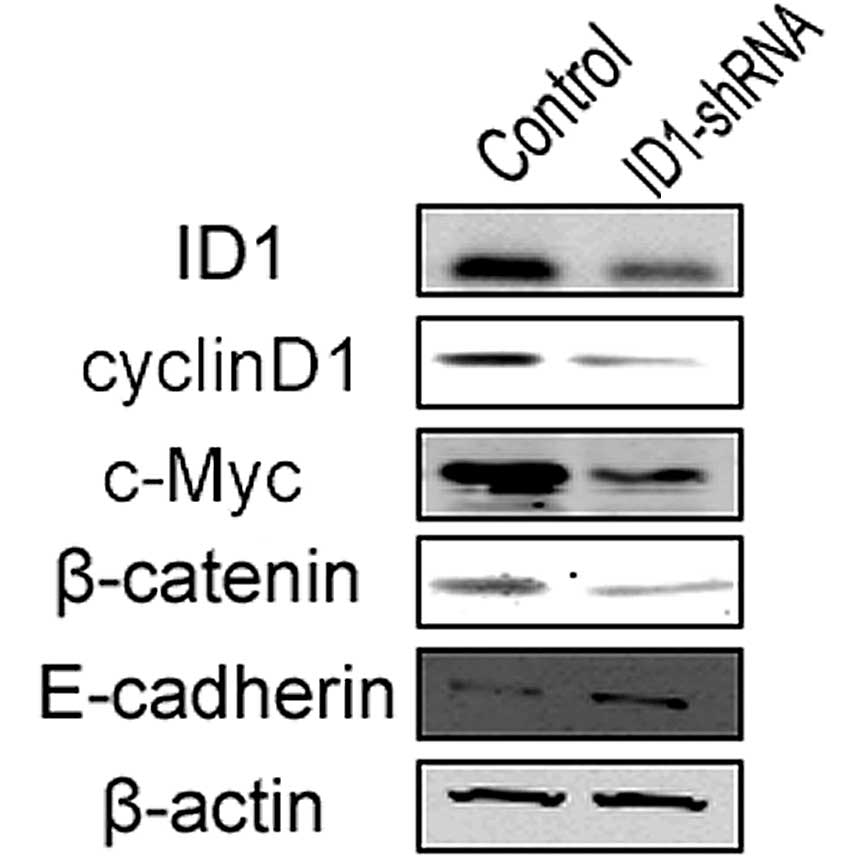
Several signaling molecules have been reported to
contribute to the increased invasion and proliferation properties
of certain types of cancer; these include c-Myc, cyclin D1 and
β-catenin. For example, the β-catenin pathway is critical in glioma
tumor invasion (10), while c-Myc
and cyclin D1 are also involved in several pathways that promote
GBM invasion. Therefore, the expression levels of these proteins
were measured in the ID1-silenced and control cells in order to
determine whether protein levels correlate with invasiveness. In
the U87 cells, ID1-knockdown led to the reduced expression of
c-Myc, cyclin D1 and β-catenin (Fig.
4). The epithelial-mesenchymal transition (EMT) increases the
invasive capacity of tumor cells (26). As previously reported, the level of
E-cadherin expression is a critical marker of EMT (27). The present data showed that
E-cadherin expression was increased following ID1-knockdown, i.e.,
a reduction in ID1 expression inhibits EMT and thus invasiveness in
U87 cells.
Discussion
GBM is the most common type of central nervous
system malignancy and the prognosis of patients with GBM is not
improved by standard treatments (1,3). The
majority of GBMs are poorly differentiated; this is linked to tumor
aggression and lethality (28).
Malignant glioma cell proliferation and invasion are key stages in
cancer progression that affect patient mortality (29). Clinically, there are limited
therapeutic interventions for malignant glioma. Therefore, more
research into the mechanisms of GBM invasion is essential for the
development of a curative therapy.
ID1, an inhibitor of basic helix-loop-helix
transcription factors, has been shown to be a key regulator of a
number of steps in cancer progression (12,13).
Moreover, ID1 inhibits cell differentiation and promotes invasion
in several types of malignant cancers, including breast and
prostate cancers and non-small cell lung carcinoma (14,16,23).
Generally, ID1 contributes to tumorigenesis by inhibiting cell
differentiation, stimulating proliferation, enhancing invasion and
facilitating tumor neoangiogenesis (30,31).
Perk et al, however, suggested that ID1 function may depend
on cell type (13). Meng et
al demonstrated that ID1 induces differentiation in mouse
embryonic carcinoma P19CL6 cells (32). Furthermore, Geng et al
reported that ID1 enhances docetaxel cytotoxicity in prostate
cancer cells through p21 inhibition and suggested that ID1 is a
novel prognostic marker and therapeutic target in prostate cancer
chemotherapy (33). There have been
several studies of ID1 in glioma. Vandeputte et al reported
that ID expression is lower in low-grade astrocytoma compared to
high-grade astrocytoma and therefore inferred that ID1 expression
levels are associated with the grade of glioma (17). Anido et al identified a cell
population enriched with glioma-initiating cells (GICs) that
express high levels of ID1 and suggested that high ID1 levels are
associated with a poor prognosis in GBM patients (34). By contrast, Barrett et al
reported that an improved prognosis is associated with higher ID1
expression in a preneural subgroup of GBM, even though ID1
overexpression correlates with increased self-renewal in GICs
(35). These contradictory studies
show that further investigations are required to determine the
function of ID1 in GBM.
Since ID1 promotes cell proliferation and invasion,
it has been proposed as an attractive target for cancer therapy.
Therefore, in the present study, an ID1-shRNA transfection system
was established for U87 cells to investigate the correlation
between ID1 expression and biological outcome. Using Transwell
invasion and scratch assays, it was observed that
ID1-shRNA-expressing U87 cells have a poorer invasion ability. In
addition, it was demonstrated that compared with ID1-silenced U87
cells, the controls had poorer adhesion properties. The effect of
ID1 silencing on cell proliferation was also investigated, as this
contributes to tumor invasion. The BrdU proliferation assay was
used to analyze cell proliferation in the U87 cells and
ID1-knockdown was observed to lead to decreased proliferation.
In general, cell movement requires morphological
changes that involve breaking down and reforming cytoskeletal
filaments (36). Therefore, to
determine whether changes in ID1-mediated cell invasion correlate
with cytoskeletal alterations, actin filaments were visualized by
immunofluorescence and changes in cell shape were determined
following ID1-knockdown. The ID1-silenced cells had fewer
lamellipodia and became rounder and smaller compared with the
control cells. Such changes in cell morphology are associated with
increased invasiveness and may be a consequence of the
dysregulation of ID1 signaling components mediating proliferation
and invasion (36). Factors such as
cyclin D1, c-Myc and β-catenin have been shown to promote GBM cell
invasion. Therefore, the expression of these proteins was examined
in the ID1-silenced and control U87 cells (37). These proteins were all observed to
be downregulated in the ID1-shRNA-expressing U87 cells. By
contrast, ID1-knockdown increased E-cadherin protein levels, which
are considered to be a marker of EMT. ID1 silencing may inhibit the
process of EMT, which is pivotal in cell invasion. The present
results therefore suggest that ID1-knockdown may inhibit U87 glioma
cell proliferation and invasion.
The present study demonstrated that the loss or
inhibition of ID1 expression may be important in GBM cell
proliferation and invasion. However, the specific mechanism through
which ID1 regulates these GBM cell properties requires further
research, which may lead to the identification of new strategies
and therapeutic targets for glioma treatment.
Acknowledgements
This study was supported by a project of building
hospital clinical key disciplines (no. RJ.4101307) and a grant from
the Shanghai government (no. 0952nm03900).
References
|
1
|
Ferguson SD: Malignant gliomas: diagnosis
and treatment. Dis Mon. 57:558–569. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Onishi M, Ichikawa T, Kurozumi K and Date
I: Angiogenesis and invasion in glioma. Brain Tumor Pathol.
28:13–24. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rampling R, James A and Papanastassiou V:
The present and future management of malignant brain tumours:
surgery, radiotherapy, chemotherapy. J Neurol Neurosurg Psychiatry.
75(Suppl 2): ii24–ii30. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Salhia B, Tran NL, Symons M, Winkles JA,
Rutka JT and Berens ME: Molecular pathways triggering glioma cell
invasion. Expert Rev Mol Diagn. 6:613–626. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Harpold HL, Alvord EC Jr and Swanson KR:
The evolution of mathematical modeling of glioma proliferation and
invasion. J Neuropathol Exp Neurol. 66:1–9. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Okita Y, Narita Y, Miyakita Y, et al:
Pathological findings and prognostic factors in recurrent
glioblastomas. Brain Tumor Pathol. 29:192–200. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Park SG, Jung S, Ryu HH, et al: Role of
14-3-3-beta in the migration and invasion in human malignant glioma
cell line U87MG. Neurol Res. 34:893–900. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
de Groot JF and Gilbert MR: New molecular
targets in malignant gliomas. Curr Opin Neurol. 20:712–718.
2007.PubMed/NCBI
|
|
9
|
Bredel M, Bredel C, Juric D, et al:
Functional network analysis reveals extended gliomagenesis pathway
maps and three novel MYC-interacting genes in human gliomas. Cancer
Res. 65:8679–8689. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nager M, Bhardwaj D, Cantí C, Medina L,
Nogués P and Herreros J: β-Catenin signalling in glioblastoma
multiforme and glioma-initiating cells. Chemother Res Pract.
2012:1923622012.
|
|
11
|
Yue X, Lan F, Yang W, et al: Interruption
of β-catenin suppresses the EGFR pathway by blocking multiple
oncogenic targets in human glioma cells. Brain Res. 1366:27–37.
2010.
|
|
12
|
Sikder HA, Devlin MK, Dunlap S, Ryu B and
Alani RM: Id proteins in cell growth and tumorigenesis. Cancer
Cell. 3:525–530. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Perk J, Iavarone A and Benezra R: Id
family of helix-loop-helix proteins in cancer. Nat Rev Cancer.
5:603–614. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Coppe JP, Itahana Y, Moore DH, Bennington
JL and Desprez PY: Id-1 and Id-2 proteins as molecular markers for
human prostate cancer progression. Clin Cancer Res. 10:2044–2051.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gupta GP, Perk J, Acharyya S, et al: ID
genes mediate tumor reinitiation during breast cancer lung
metastasis. Proc Natl Acad Sci USA. 104:19506–19511. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pillai S, Rizwani W, Li X, et al: ID1
facilitates the growth and metastasis of non-small cell lung cancer
in response to nicotinic acetylcholine receptor and epidermal
growth factor receptor signaling. Mol Cell Biol. 31:3052–3067.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vandeputte DA, Troost D, Leenstra S, et
al: Expression and distribution of id helix-loop-helix proteins in
human astrocytic tumors. Glia. 38:329–338. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Benezra R, Davis RL, Lockshon D, Turner DL
and Weintraub H: The protein Id: a negative regulator of
helix-loop-helix DNA binding proteins. Cell. 61:49–59. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kiewitz SD, Kruppa M, Riechers A, König B
and Cabrele C: Recognition of the helix-loop-helix domain of the Id
proteins by an artificial luminescent metal complex receptor. J Mol
Recognit. 21:79–88. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Langlands K, Yin X, Anand G and Prochownik
EV: Differential interactions of Id proteins with
basic-helix-loop-helix transcription factors. J Biol Chem.
272:19785–19793. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xie SQ, Zhang YH, Li Q, et al:
3-Nitro-naphthalimide and nitrogen mustard conjugate NNM-25 induces
hepatocellular carcinoma apoptosis via PARP-1/p53 pathway.
Apoptosis. 17:725–734. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ciarrocchi A, Piana S, Valcavi R, Gardini
G and Casali B: Inhibitor of DNA binding-1 induces mesenchymal
features and promotes invasiveness in thyroid tumour cells. Eur J
Cancer. 47:934–945. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lin CQ, Singh J, Murata K, et al: A role
for Id-1 in the aggressive phenotype and steroid hormone response
of human breast cancer cells. Cancer Res. 60:1332–1340.
2000.PubMed/NCBI
|
|
24
|
Guo Q, Guo P, Mao Q, et al: ID1 affects
the efficacy of radiotherapy in glioblastoma through inhibition of
DNA repair pathways. Med Oncol. 30:3252013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ling MT, Wang XH, Zhang XM and Wong YC:
The multiple roles of Id-1 in cancer progression. Differentiation.
74:481–487. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ahmad A, Aboukameel A, Kong D, et al:
Phosphoglucose isomerase/autocrine motility factor mediates
epithelial-mesenchymal transition regulated by miR-200 in breast
cancer cells. Cancer Res. 71:3400–3409. 2011. View Article : Google Scholar
|
|
27
|
Wells A, Yates C and Shepard CR:
E-cadherin as an indicator of mesenchymal to epithelial reverting
transitions during the metastatic seeding of disseminated
carcinomas. Clin Exp Metastasis. 25:621–628. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Borkar SA, Lakshmiprasad G, Subbarao KC,
et al: Giant cell glioblastoma in the pediatric age group: Report
of two cases. J Pediatr Neurosci. 8:38–42. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qi S, Song Y, Peng Y, et al: ZEB2 mediates
multiple pathways regulating cell proliferation, migration,
invasion, and apoptosis in glioma. PLoS One. 7:e388422012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cheng YJ, Tsai JW, Hsieh KC, et al: Id1
promotes lung cancer cell proliferation and tumor growth through
Akt-related pathway. Cancer Lett. 307:191–199. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tobin NP, Sims AH, Lundgren KL, Lehn S and
Landberg G: Cyclin D1, Id1 and EMT in breast cancer. BMC Cancer.
11:4172011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Meng Q, Jia Z, Wang W, Li B, Ma K and Zhou
C: Inhibitor of DNA binding 1 (Id1) induces differentiation and
proliferation of mouse embryonic carcinoma P19CL6 cells. Biochem
Biophys Res Commun. 412:253–259. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Geng H, Rademacher BL, Pittsenbarger J, et
al: ID1 enhances docetaxel cytotoxicity in prostate cancer cells
through inhibition of p21. Cancer Res. 70:3239–3248. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Anido J, Sáez-Borderías A, Gonzàlez-Juncà
A, et al: TGF-β Receptor Inhibitors Target the CD44(high)/Id1(high)
Glioma-Initiating Cell Population in Human Glioblastoma. Cancer
Cell. 18:655–668. 2010.
|
|
35
|
Barrett LE, Granot Z, Coker C, et al:
Self-renewal does not predict tumor growth potential in mouse
models of high-grade glioma. Cancer Cell. 21:11–24. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sarmiere PD and Bamburg JR: Regulation of
the neuronal actin cytoskeleton by ADF/cofilin. J Neurobiol.
58:103–117. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nager M, Bhardwaj D, Canti C, et al:
β-catenin signaling in glioblastoma multiforme and
glioma-initiating cells. Chemother Res Pract. 2012:1923622012.
|