Introduction
Esophageal cancer remains a major cause of cancer
mortality worldwide. Patients with this cancer generally have a
poor prognosis. Conducting detailed molecular and genetic
investigations is necessary to develop new strategies for
esophageal cancer.
Previous biological studies have shown that numerous
types of cancer are caused by the accumulation of multiple genetic
defects in dominant oncogenes and tumor suppressor genes involved
in the regulation of the cell cycle, induction of apoptosis and
modulation of checkpoints (1–4).
Checkpoint genes are well-known tumor suppressors.
Their main functions are to stop the cell cycle and repair errors
when cells are exposed to various stressors, thereby preventing DNA
damage (5). Failure of these
checkpoint functions results in genomic instability, a mutagenic
condition that predisposes cells to neoplastic transformation
(6,7).
Checkpoint with fork-head associated and ring finger
(CHFR), a checkpoint gene, was first identified by Scolnick and
Halazonetis in 2000 and described as a nuclear protein that plays
an important role in the early mitotic stress caused by microtubule
inhibitors (8). It has been
reported that CHFR expression levels are downregulated in various
digestive cancers, including esophageal cancer (9,10).
However, the regulatory mechanisms and clinical significance of the
CHFR gene remain unclear.
To date, a growing number of studies have reported
that the loss of tumor suppressor genes is often caused by
epigenetic alterations, including methylation of DNA (11,12). A
number of studies have described hypermethylation of the CpG island
in the promoter region as a key mechanism involved in gene
regulation (13–15). However, the role of CpG island
methylation in CHFR silencing in esophageal cancer has not been
fully investigated.
The objective of the present study was to evaluate
the correlation between CHFR expression and the methylation status
of the CpG island in the promoter region. The significance of CHFR
gene expression with respect to clinicopathological features and
survival was also examined.
Materials and methods
Sample collection and DNA/RNA
preparation
A total of 40 patients were enrolled in this study.
The patients had primary esophageal squamous cell carcinoma and
underwent primary resection at the Kanagawa Cancer Center
(Yokohama, Japan) between November 2001 and July 2003. There were
36 males and 4 females, with a mean age of 64.8 years (45–76
years). The study was approved by the institutional review board
committee of Kanagawa Cancer Center Hospital and written informed
consent for the study was obtained from all patients. The tumor
samples were obtained during surgical resection of esophageal
cancer and immediately stored at −80°C. Genomic DNA was extracted
from the tumor tissues using the DNA extraction kit, Sepa-Gene
(Sanko-Junyaku, Tokyo, Japan). RNA was extracted from the tumor
tissues using TRIzol reagent (Life Technologies, Tokyo, Japan).
Reverse transcription polymerase chain
reaction (RT-PCR)
Single-step RT-PCR was performed using CHFR
gene-specific oligonucleotide primers for reverse transcription and
PCR and fluorescent hybridization probes labeled with LC-Red 640 or
FITC for real-time detection, using the LightCycler RNA
Amplification kit HybriProbe and LightCycler (Roche Diagnostics,
Tokyo, Japan). The housekeeping gene, human porphobilinogen
deaminase (hPBGD), was simultaneously evaluated using to normalize
the amount of RNA used in the reaction. The reaction was designed
according to the manufacturer’s instructions without modifications
and consisted of the following settings: 50°C for 10 min for
reverse transcription and 95°C for 2 min for denaturation, followed
by 45 cycles of 95°C for 1 sec, 55°C for 15 sec and 72°C for 10
sec. The same conditions were used for CHFR and hPBGD. The
nucleotide sequences of the primers and hybriprobes are shown
below. For CHFR: forward 5′-gcctggccccgttttgtgagc-3′, reverse
5′-gacgggatgttacggccactg-3′, hybriprobe LC-Red 640
5′-ggccgtaacatcccgtcctga-3′ and hybriprobe FITC
5′-attcctgcttccgagttgccag-3′ For hPBGD: forward
5′-accctgccagagaagagtgt-3′, reverse 5′-ccacagcatacatgcattcc-3′,
hybriprobe LC-Red 640 5′-ctgaactccagatgcgggaactt-3′ and hybriprobe
FITC 5′-ggtgttgaggtttccccgaatact-3′.
Bisulfite modification
Genomic DNA obtained from the tumors was subjected
to bisulfite modification. A total of 2 μg genomic DNA was
incubated in 50 μl water and denatured in 0.2 M NaOH for 10 min at
37°C. The denatured DNA was then diluted in 550 μl of a solution
containing 30 μl hydroquinone (10 mM) and 520 μl sodium bisulfite
(3 M). The DNA solution was incubated for 16 h at 50°C. Following
incubation, the DNA sample was desalted using the Wizard DNA
Clean-Up System (Promega Corporation, Madison, WI, USA) and treated
with 0.3 M NaOH for 5 min at room temperature. Finally, the DNA
sample was precipitated with ethanol, dissolved in 32 μl TE buffer
and stored at −20°C.
Methylation-specific PCR (MSP)
MSP exploits the effects of sodium bisulfite on DNA,
which efficiently converts unmethylated cytosine to uracil while
leaving methylated cytosine intact (16). Consequently, following modification,
the unmethylated and methylated alleles have different sequences
that can be used to design allele-specific primers for PCR. The
primer sequences for hCHFR have been described previously (16,17).
The primers used for the unmethylated reaction were as follows:
5′-ata taa tat ggt gtt gat t-3′ (sense) and 5′-tca act aat cca caa
aac a-3′ (antisense). The primers used for the methylated reaction
were as follows: 5′-ata taa tat ggc gtc gat g-3′ (sense) and 5′-tca
act aat ccg cga aac g-3′ (antisense). PCR amplification was
performed in 20 μl reaction volumes containing 2.0 μl 10X PCR
buffer, 0.5 μl Taq DNA Polymerase, 0.8 μl primer mixture (10
μM each) and 2.0 μl modified DNA. The annealing temperature was
50°C for the unmethylated samples and 58°C for the methylated
samples. The PCR amplification conditions were as follows: 94°C for
2 min, 42 cycles of 94°C for 30 sec, the specific annealing
temperature for 30 sec and 68°C for 1 min. The PCR products were
206 bp in size for each sample. The PCR products were subjected to
gel electrophoresis through a 2% agarose gel, stained with ethidium
bromide and then visualized under UV illumination.
Statistical analyses
The statistical correlation between CHFR expression
and MSP status was analyzed using the Mann-Whitney U test. The CHFR
expression levels and patient characteristics, including age,
gender, histological type, depth of invasion, lymph node
metastasis, lymphatic invasion, venous invasion, pathological stage
and intraluminal metastasis were compared using the χ2
test.
The post-operative survival rate was analyzed
according to the Kaplan-Meier method, and differences in survival
rates were assessed using the log-rank test.
All statistical analyses were conducted using the
Dr. SPSS II software program, version 11.0.1J for Windows (SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
RT-PCR
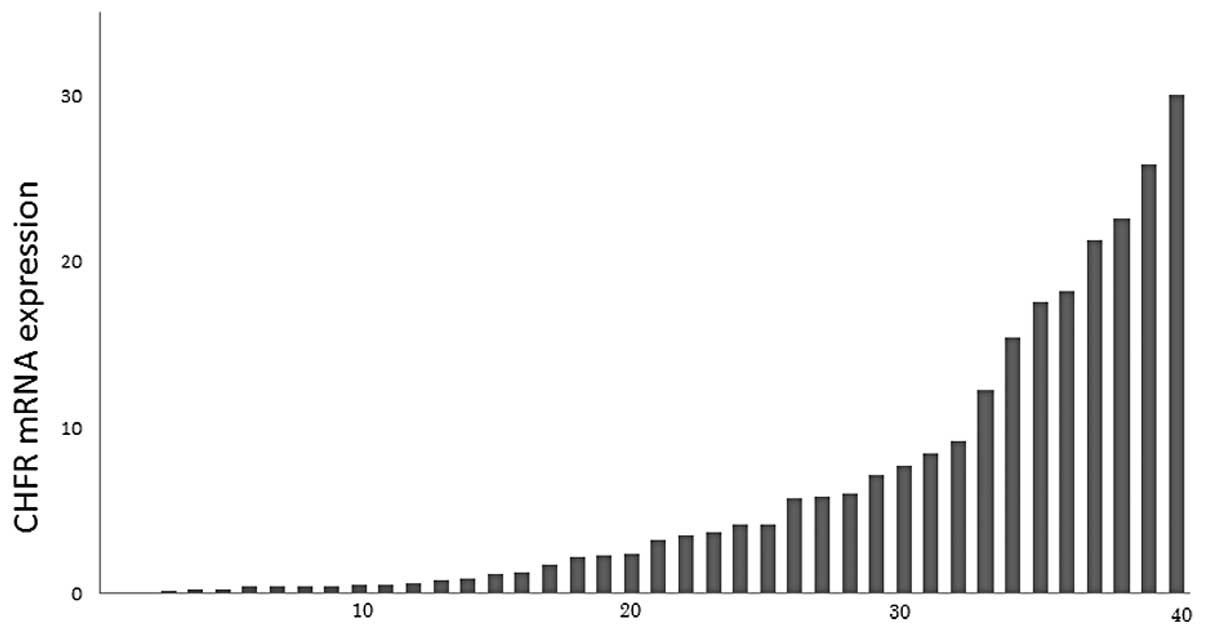
Using RT-PCR, CHFR gene expression in 40 primary
esophageal squamous cell carcinomas was quantified. The relative
levels of CHFR mRNA expression are shown as a ratio of hPBGD
expression. Esophageal cancers exhibited a variety of levels of
CHFR gene expression (Fig. 1).
MSP
MSP was subsequently successfully performed in 29
cases. Therefore, the methylation status in 29 of 40 primary
esophageal cancers was investigated using the MSP technique.
Amplification of the methylated DNA-specific PCR primers was
observed in 13 of 29 primary esophageal cancers (44.8%), while that
of the unmethylated primers was observed in 16 patients (55.2%).
Concurrent amplification of a methylated and unmethylated status
was defined as a methylated status (Fig. 2).
Correlation between the MSP status and
CHFR gene expression levels
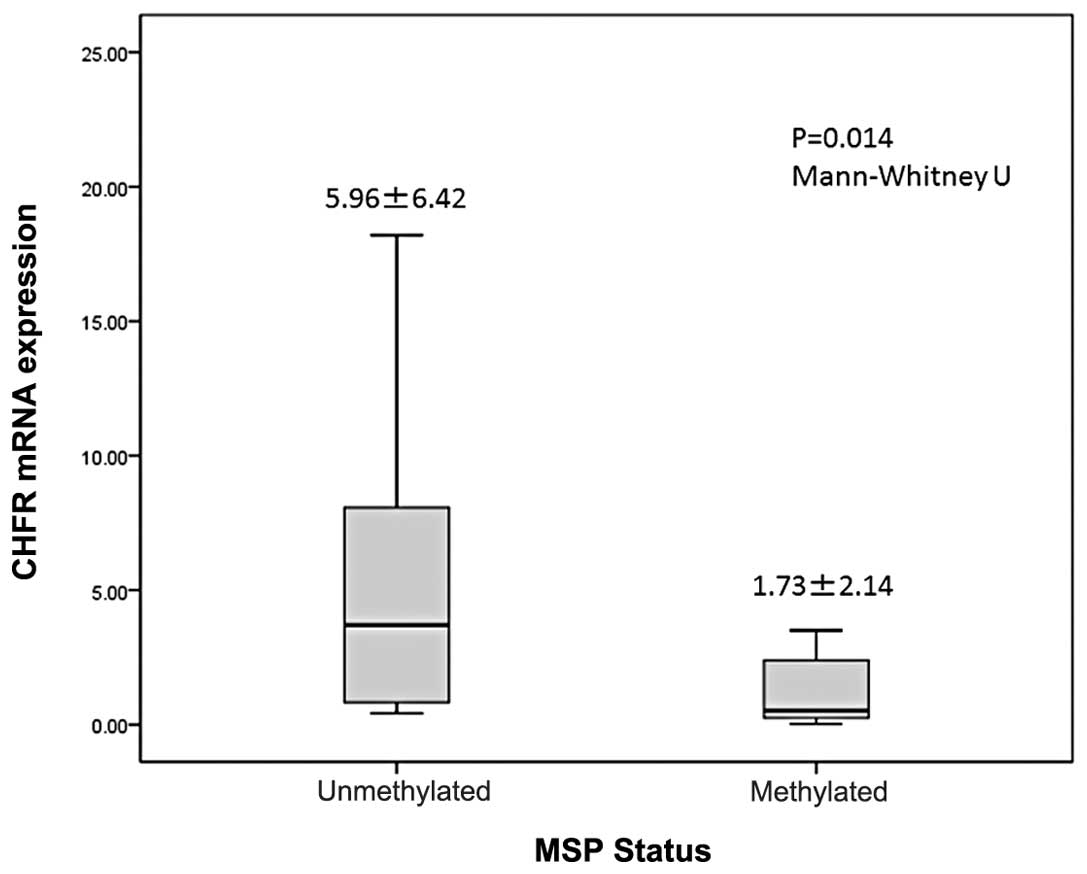
CHFR expression levels of the methylated status
samples were significantly lower than that of the unmethylated
status samples (1.735±2.149 vs. 5.966±6.429; P=0.014; Mann-Whitney
U test; Fig. 3).
Correlation between CHFR gene expression
levels and clinicopathological features
The expression levels of the CHFR gene were
categorized as low or high according to the median value. The
correlation between the expression levels of this gene and
clinicopathological features was then examined. None of the
clinicopathological features were found to correlate with CHFR
expression levels (Table I).
 | Table ICorrelation between expression of the
CHFR gene and clinicopathological features. |
Table I
Correlation between expression of the
CHFR gene and clinicopathological features.
| CHFR expression | |
|---|
|
| |
|---|
|
Variables/Categories | Low (n=20) | High (n=20) | P-value |
|---|
| Age | 67.0±5.26 | 62.7±6.86 | 0.156 |
| Gender |
| Male | 18 | 18 | 0.698 |
| Female | 2 | 2 | |
| Histological
type |
|
Well-differentiated | 4 | 3 | 0.264 |
|
Moderately-differentiated | 8 | 6 | |
|
Poorly-differentiated | 8 | 11 | |
| Depth of
invasion |
| T1 | 1 | 2 | 0.493 |
| T2 | 4 | 12 | |
| T3 | 15 | 15 | |
| T4 | 0 | 1 | |
| Lymph node
metastasis |
| Absent | 3 | 5 | 0.376 |
| Present | 17 | 15 | |
| Number of lymph node
metastasis |
| 0–3 | 15 | 11 | 0.347 |
| ≥4 | 5 | 9 | |
| Lymphatic
invasion |
| Absent | 9 | 11 | 0.376 |
| Present | 11 | 9 | |
| Venous invasion |
| Absent | 2 | 3 | 0.500 |
| Present | 18 | 17 | |
| Stage |
| I | 0 | 1 | 0.599 |
| II | 5 | 4 | |
| III | 14 | 13 | |
| IV | 1 | 2 | |
| Intraluminal
metastasis |
| Absent | 18 | 18 | 0.698 |
| Present | 2 | 2 | |
Correlation between CHFR gene expression
levels and survival
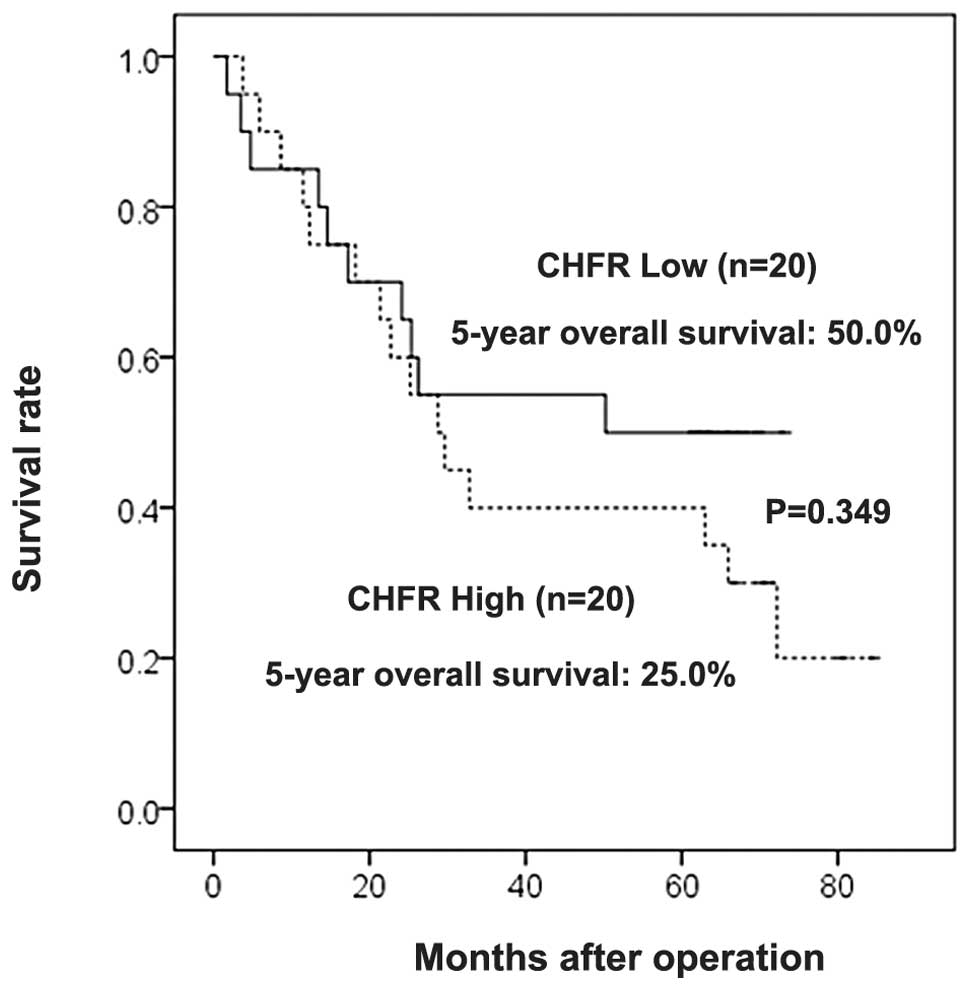
In the current study, the median follow-up period
was 50.3 months. The overall survival rates were not significantly
correlated with the CHFR expression levels (CHFR-high, 25.0%;
CHFR-low, 50.0%; P=0.349, log-rank test) (Fig. 4).
Discussion
Checkpoint genes, one of the surveillance mechanisms
of cells, act to maintain genomic stability against various types
of damage to the genome. The G1 checkpoint prevents replication of
damaged DNA, while genomic integrity prior to mitosis is monitored
by the G2 checkpoint, which promotes G2 arrest on detection of DNA
damage. As such, checkpoints are important for preventing the
propagation of cells with corrupted genomes that could potentially
cause tumor formation.
CHFR, a mitotic checkpoint gene, has been previously
cloned and is localized to chromosome 12q24. It has been reported
that the CHFR protein delays entry into mitosis at the G2 to M
entry site (8,18–20).
In fact, CHFR knockout mice are cancer-prone (21), indicating that the CHFR gene
functions as a tumor suppressor. Previous studies have revealed
that CHFR expression levels are frequently downregulated in
patients with digestive cancer, including esophageal cancer
(22,23).
The epigenetically-mediated loss-of-gene function is
a well-known mechanism involved in carcinogenesis (24). Several tumor suppressor genes
containing CpG islands can be silenced via methylation of the CpG
island. Previously, aberrant methylation of the CHFR gene
associated with gene silencing has been demonstrated in several
studies (11,25), although it has not been fully
clarified how the CHFR gene is regulated in esophageal cancer.
In the present study, mRNA expression of CHFR was
assessed in 40 primary tumor samples using RT-PCR, which showed a
variety of CHFR expression levels. Then, MSP was carried out to
evaluate the methylation status of CpG islands in the promoter
region of the CHFR gene in 29 cases. The results indicate that
aberrant methylation of the CHFR gene is frequently (44.8%)
observed in esophageal cancer. This frequency is higher than that
reported in previous studies, which have described a frequency of
16.3–24.0% (11,26,27).
The differences in this rate may be due to disparities in the
primer design, resulting in different degrees of CpG
amplification.
In the present study, downregulation mechanisms were
analyzed, revealing that aberrant methylation of the promoter
region correlated with a loss of CHFR mRNA expression. Shibata
et al(27) also confirmed
the presence of a significant correlation between CHFR methylation
and downregulation, which supports our previous results. Other data
have shown that the interplay between epigenetic and genetic
mechanisms underlies the loss of CHFR function in esophageal
adenocarcinoma (10), indicating
that there are various steps involved in suppressing mRNA
expression of the CHFR gene.
Subsequently, the effect of CHFR gene expression
levels on clinicopathological features was evaluated, and no
correlation was found. Our results also revealed that CHFR
expression levels do not have prognostic significance in patients
with esophageal cancer. The clinical importance of CHFR expression
in esophageal cancer is not well studied. To date, a single study
has been performed to investigate the correlation between CHFR
expression and patient characteristics in esophageal cancer, and
indicated no relative clinical factors (27). These results indicate the
possibility that CHFR silencing is associated with carcinogenesis,
without tumor progression.
Taken together, results of the present study
indicate that aberrant hypermethylation of CpG islands is the key
mechanism associated with transcriptional inactivation of the CHFR
gene in patients with esophageal cancer. It has been previously
shown that it is possible to reverse epigenetic changes and restore
gene function using treatment with DNA methylation inhibitors
(28). Further investigations are
likely to provide new insights into establishing novel strategies
for treating esophageal cancer.
Acknowledgements
The present study was supported by the Kanagawa
Cancer Center (Kanagawa, Japan).
References
|
1
|
Ding Y, Shimada Y, Kano M, et al:
PTEN/MMAC1 expression in esophageal squamous cell carcinomas. Int J
Oncol. 17:695–699. 2000.PubMed/NCBI
|
|
2
|
Roncalli M, Bosari S, Marchetti A, et al:
Cell cycle-related gene abnormalities and product expression in
esophageal carcinoma. Lab Invest. 78:1049–1057. 1998.PubMed/NCBI
|
|
3
|
Shimada Y, Sato F, Watanabe G, et al: Loss
of fragile histidine triad gene expression is associated with
progression of esophageal squamous cell carcinoma, but not with the
patient’s prognosis and smoking history. Cancer. 89:5–11. 2000.
|
|
4
|
Tanaka H, Shimada Y, Harada H, et al:
Methylation of the 5′ CpG island of the FHIT gene is closely
associated with transcriptional inactivation in esophageal squamous
cell carcinomas. Cancer Res. 58:3429–3434. 1998.
|
|
5
|
Privette LM and Petty EM: CHFR: A novel
mitotic checkpoint protein and regulator of tumorigenesis. Transl
Oncol. 1:57–64. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hartwell LH and Weinert TA: Checkpoints:
controls that ensure the order of cell cycle events. Science.
246:629–634. 1989. View Article : Google Scholar
|
|
7
|
Hartwell LH and Kastan MB: Cell cycle
control and cancer. Science. 266:1821–1828. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Scolnick DM and Halazonetis TD: Chfr
defines a mitotic stress checkpoint that delays entry into
metaphase. Nature. 406:430–435. 2000. View
Article : Google Scholar
|
|
9
|
Bertholon J, Wang Q, Falette N, et al:
Chfr inactivation is not associated to chromosomal instability in
colon cancers. Oncogene. 22:8956–8960. 2003. View Article : Google Scholar
|
|
10
|
Soutto M, Peng D, Razvi M, et al:
Epigenetic and genetic silencing of CHFR in esophageal
adenocarcinomas. Cancer. 116:4033–4042. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Morioka Y, Hibi K, Sakai M, et al:
Aberrant methylation of the CHFR gene in digestive tract cancer.
Anticancer Res. 26:1791–1795. 2006.PubMed/NCBI
|
|
12
|
Sharma S, Kelly TK and Jones PA:
Epigenetics in cancer. Carcinogenesis. 31:27–36. 2010. View Article : Google Scholar
|
|
13
|
Eads CA, Lord RV, Wickramasinghe K, et al:
Epigenetic patterns in the progression of esophageal
adenocarcinoma. Cancer Res. 61:3410–3418. 2001.PubMed/NCBI
|
|
14
|
Nie Y, Yang G, Song Y, et al: DNA
hypermethylation is a mechanism for loss of expression of the HLA
class I genes in human esophageal squamous cell carcinomas.
Carcinogenesis. 22:1615–1623. 2001. View Article : Google Scholar
|
|
15
|
Momparler RL and Bovenzi V: DNA
methylation and cancer. J Cell Physiol. 183:145–154. 2000.
View Article : Google Scholar
|
|
16
|
Herman JG, Graff JR, Myöhänen S, Nelkin BD
and Baylin SB: Methylation-specific PCR: a novel PCR assay for
methylation status of CpG islands. Proc Natl Acad Sci USA.
93:9821–9826. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Esteller M, Sanchez-Cespedes M, Rosell R,
Sidransky D, Baylin SB and Herman JG: Detection of aberrant
promoter hypermethylation of tumor suppressor genes in serum DNA
from non-small cell lung cancer patients. Cancer Res. 59:67–70.
1999.
|
|
18
|
Chaturvedi P, Sudakin V, Bobiak ML, et al:
Chfr regulates a mitotic stress pathway through its RING-finger
domain with ubiquitin ligase activity. Cancer Res. 62:1797–1801.
2002.
|
|
19
|
Ahel I, Ahel D, Matsusaka T, et al:
Poly(ADP-ribose)-binding zinc finger motifs in DNA
repair/checkpoint proteins. Nature. 451:81–85. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Oh YM, Kwon YE, Kim JM, et al: Chfr is
linked to tumour metastasis through the downregulation of HDAC1.
Nat Cell Biol. 11:295–302. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu X, Minter-Dykhouse K, Malureanu L, et
al: Chfr is required for tumor suppression and Aurora A regulation.
Nat Genet. 37:401–406. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kashima L, Toyota M, Mita H, et al: CHFR,
a potential tumor suppressor, downregulates interleukin-8 through
the inhibition of NF-kappaB. Oncogene. 28:2643–2653. 2009.
View Article : Google Scholar
|
|
23
|
Sanbhnani S and Yeong FM: CHFR: a key
checkpoint component implicated in a wide range of cancers. Cell
Mol Life Sci. 69:1669–1687. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Toyota M, Itoh F and Imai K: DNA
methylation and gastrointestinal malignancies: functional
consequences and clinical implications. J Gastroenterol.
35:727–734. 2000. View Article : Google Scholar
|
|
25
|
Jones PA and Laird PW: Cancer epigenetics
comes of age. Nat Genet. 21:163–167. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mizuno K, Osada H, Konishi H, et al:
Aberrant hypermethylation of the CHFR prophase checkpoint gene in
human lung cancers. Oncogene. 21:2328–2333. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shibata Y, Haruki N, Kuwabara Y, et al:
Chfr expression is downregulated by CpG island hypermethylation in
esophageal cancer. Carcinogenesis. 23:1695–1699. 2002. View Article : Google Scholar
|
|
28
|
Fang MZ, Jin Z, Wang Y, et al: Promoter
hypermethylation and inactivation of O6-methylguanine-DNA
methyltransferase in esophageal squamous cell carcinomas and its
reactivation in cell lines. Int J Oncol. 26:615–622.
2005.PubMed/NCBI
|