Introduction
Hepatocellular carcinoma (HCC) is the third most
common lethal malignancy and the fifth most common type of cancer
worldwide. In 2008, there were an estimated 748,300 new liver
cancer cases and 695,900 cancer mortalities worldwide (1,2). Half
of these cancer cases and cancer-associated mortalities were
estimated to occur in China (3).
Liver transplantation or surgical resection remain the first-line
treatments for HCC (4). However,
patients often present symptoms at the advanced stages of HCC and,
therefore, the first-line treatments are not effective for the
majority of HCC cases. Due to high recurrence rates, even following
surgical resection, the long-term prognosis of HCC remains
unsatisfactory. For patients with HCC at the advanced stages,
chemotherapy via either transarterial chemoembolization or a
systemic route is the second-line treatment. Unfortunately, the
efficacy of the regimens is limited due to the highly
chemoresistant nature of the tumor and the toxicity of traditional
chemotherapy (5–7).
The results of previous studies support the theory
that tumors may be initiated and maintained by a small population
of cells that have stem-like features, and this highly tumorigenic
cell subpopulation within the tumor bulk has been considered as
cancer stem cells (CSCs) (8).
CD133+ liver CSCs were first reported to mark a CSC
subpopulation in HCC by Suetsugu et al (9). Subsequently, Yin et al found
that CD133+ cells isolated from HCC SMMC-7721 cells
demonstrated an enhanced clonogenicity in vitro and
tumorigenicity in vivo (10). Several studies on HCC cell lines
have reported that CD133+ liver cancer stem cells
(LCSCs) had an enhanced ability to differentiate and self-renew,
were highly proliferative, demonstrated epithelial-mesenchymal
transition (EMT), formed sphere clusters and had greater
tumorigenicity and chemoresistance (11,12).
EMT is a crucial and dynamic process that
facilitates the invasion of cancer cells. The induction of EMT was
found to generate CSCs in breast cancer (13,14).
Several studies have reported a conjunction between CSCs and EMT.
EMT refers to morphological and molecular changes that occur when
epithelial cells lose their characteristics and acquire mesenchymal
properties, such as increased invasive and motility features
(15). The expression of Twist,
along with the expression of mesenchymal markers, such as
N-cadherin, and the loss of E-cadherin, are key molecular markers
of EMT (16). Transcription
factors, including Twist and Snail, bind to consensus E-box
sequences in the E-cadherin gene promoter and downregulate
E-cadherin transcription. Twist, a transcription factor of the
basic helix-loop-helix class that represses E-cadherin, has been
reported to regulate HCC metastasis through the induction of EMT
(17). The strategies aimed at
targeting EMT of CSCs represent rational approaches for cancer
prevention and treatment (18).
This may prompt us to discover more preventive strategies for
cancer management by reducing cancer resistance and recurrence, and
improving patient survival. Several studies have reported that
sulforaphane has the capability to suppress pancreatic tumor
initiating cells and breast CSCs (19,20).
These studies provide a basis for preclinical and clinical
evaluation of dietary compounds for chemoprevention of CSCs.
Casticin is one of the main active components from
Fructus Viticis (Chinese name, Manjingzi), which has been shown to
inhibit the proliferation of breast cancer (21), lung cancer (22), colon cancer (23) and human myelogenous leukemia cells
(24) in vitro, as well as
exerting an anti-inflammatory effect in vivo (25). Casticin was also reported to induce
apoptosis (26), growth suppression
and cell cycle arrest through the activation of FOXO3a in HCC cell
lines (27). However, to the best
of our knowledge, to date there are no studies regarding the
inhibition of casticin on LCSCs.
In the present study, we aimed to investigate the
effects of casticin on the EMT of LCSCs derived from the SMMC-7721
cell line.
Materials and methods
Cell culture and reagents
The human HCC SMMC-7721 cell line was obtained from
the Chinese Academy of Sciences (Shanghai, China). Cells were
maintained in Dulbecco’s modified Eagle’s medium (DMEM)
supplemented with 10% fetal bovine serum (FBS; Hangzhou Sijiqing
Biological Engineering Materials Co., Ltd., Hangzhou, China), 100
U/ml penicillin and 100 μg/ml streptomycin (Invitrogen Life
Technologies, Carlsbad, CA, USA) in an incubator containing 5%
CO2 at 37°C. Casticin was purchased from Chengdu
Biopurify Phytochemicals Ltd. (purity, ≥98%; Chengdu, China) and
dissolved in dimethyl sulfoxide (DMSO) as a 10 mmol/l stock
solution, and diluted in a medium to the indicated concentration
before use. Trypsin and DMSO were purchased from Amersco Inc.
(Solon, OH, USA).
Cell sorting and sphere culture
Cells were labeled with primary CD133/1 antibody
[mouse immunoglobulin 1 (IgG1), 1 μl per million cells; Miltenyi
Biotec, Bergisch Gladbach, Germany], subsequently magnetically
labeled with rat anti-mouse IgG1 microbeads (20 μl per 10 million
cells; Miltenyi Biotec) and separated on MACS LS column (Miltenyi
Biotec). All procedures were performed according to the
manufacturer’s instructions. The purity of sorted cells was
evaluated by flow cytometry (FCM) and western blot analysis. The
FCM was performed with Epics Altra Cell Sorter (Beckman Coulter,
Inc., Pasadena, CA, USA) using CD133/1 primary antibody (Miltenyi
Biotec) and phycoerythrin-conjugated secondary antibody
(Sigma-Aldrich, St. Louis, MO, USA). The CD133+ cells
and the parental cells were collected and washed to remove serum,
and then suspended in serum-free DMEM/F12 supplemented with 20
ng/ml human recombinant epidermal growth factor (Invitrogen Life
Technologies), 20 ng/ml human recombinant basic fibroblast growth
factor (Invitrogen Life Technologies), 2% B27 supplement without
vitamin A (Invitrogen Life Technologies), 0.4% bull serum albumin
(Invitrogen Life Technologies), 4 ng/ml insulin (Invitrogen Life
Technologies), 100 IU/ml penicillin and 100 μg/ml streptomycin. The
cells were subsequently cultured in ultra low attachment six-well
plates (Corning Inc., Corning, NY, USA) at a density of no more
than 2,000 cells/well.
Spheroid passage and sphere formation
assay
The CD133+ sphere-forming cells (SFCs) of
the SMMC-7721 cell line were collected by gentle centrifugation,
then dissociated with trypsin-EDTA and mechanically disrupted with
a pipette. The resulting single cells were centrifuged to remove
the enzyme, resuspended in stem cell-conditioned culture medium and
allowed to reform spheres. The tumor spheres were passaged every 6
days before they reached a diameter of 50 mm. The volume of HCC
spheres was calculated by the following: V = (4/3)πr3,
where r is the radius of the sphere. The number and volume of the
spheres was observed and compared. To examine the effects of
casticin on the sphere formation, the resulting single cells with a
density of 2×103 cells/ml were plated to ultra low
attachment six-well plates and supplemented with serum-free medium
containing various concentrations of casticin.
In vivo tumorigenicity assay
Pathogen-free male Balb/c-nu mice (age, 4–5 weeks)
were purchased from the Animal Institute of the Chinese Academy of
Medical Science (Beijing, China). All animal studies were performed
in accordance with the standard protocols approved by the ethics
committee of the Hunan Normal University (Changsha, China) and the
committee of experimental animal feeding and management. Male
Balb/c-nu mice (age, 4–5 weeks) were randomly divided into five
groups (four mice per group) and maintained under standard
conditions, according to the standard protocols. Cells were
suspended in serum free-DMEM/Matrigel (BD Biosciences, Franklin
Lakes, NJ, USA) mixture (1:1 volume). Each Balb/c-nu mouse was
inoculated subcutaneously with different numbers of
CD133+ SFCs (5×102, 1×103,
5×103, 1×104 and 5×104 cells) in
one flank and parental cells (5×104, 1×105,
2×105, 5×105 and 1×106 cells) in
the other, respectively. Tumorigenicity experiments were terminated
2 months after cell inoculation. Tumor size was measured with a
caliper and the volume was calculated using the following: V
(mm3) = L×W2×0.5; where L is the length and W
is the width. Harvested tumors were examined and weighed
immediately. Moreover, specimens from tumor tissue samples were
fixed in 10% neutral-buffered formalin, processed in paraffin
blocks and sectioned. The tissue sections were stained with
hematoxylin and eosin and examined under light microscopy (IX-70,
Olympus Corporation, Tokyo, Japan) for identification of the
histopathological changes.
pcDNA3-Twist1 transfection
pcDNA3-Twist1 was purchased from GenePharma
(Shanghai, China). To generate Twist1-expressing stable
transfectants, the parental cells and SFCs of the SMMC-7721 cell
line were transfected with pcDNA3-Twist1, and stable clones were
selected with 1,000 μg/ml of G418 (EMD Millipore, San Diego, CA,
USA) for 4 weeks.
Western blot analysis
Protein extracts were resolved by 12% SDS-PAGE and
transferred onto nitrocellulose membranes (Amersham Bioscience,
Shanghai, China) blocked in 5% skimmed milk. Primary antibodies
were used as indicated by the manufacturer’s instructions and are
as follows: Mouse anti-CD133 (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA), Twist (Cell Signaling Technology, Inc., Beverly,
MA, USA), N-cadherin (EMD-Millipore) and E-cadherin (BD
Biosciences). After incubation with horseradish
peroxidase-conjugated anti-mouse or anti-rabbit secondary
antibodies (Amersham Biosciences, Cardiff, UK), specific protein
bands were visualized by enhanced chemiluminescence (Amersham
Biosciences). β-actin (Sigma-Aldrich) was used as a loading
control.
Statistical analysis
All values in the figures and text are expressed as
the means ± SD. Statistical analyses were performed using SPSS
software, version 16.0 (SPSS, Inc., Chicago, IL, USA). Any
significant differences among the mean values were evaluated by the
Student’s t-test. A two-sided P<0.05 was considered to indicate
a statistically significant difference.
Results
Sorting and identification of LCSCs
derived from the HCC SMMC-7721 cell line
In order to isolate LCSCs, SMMC-7721 cells were
enzymatically dispersed into single-cell suspensions and incubated
using an anti-CD133 antibody (Miltenyi Biotec) and then analyzed by
FCM. Following separation on MACS LS column for purity, FCM
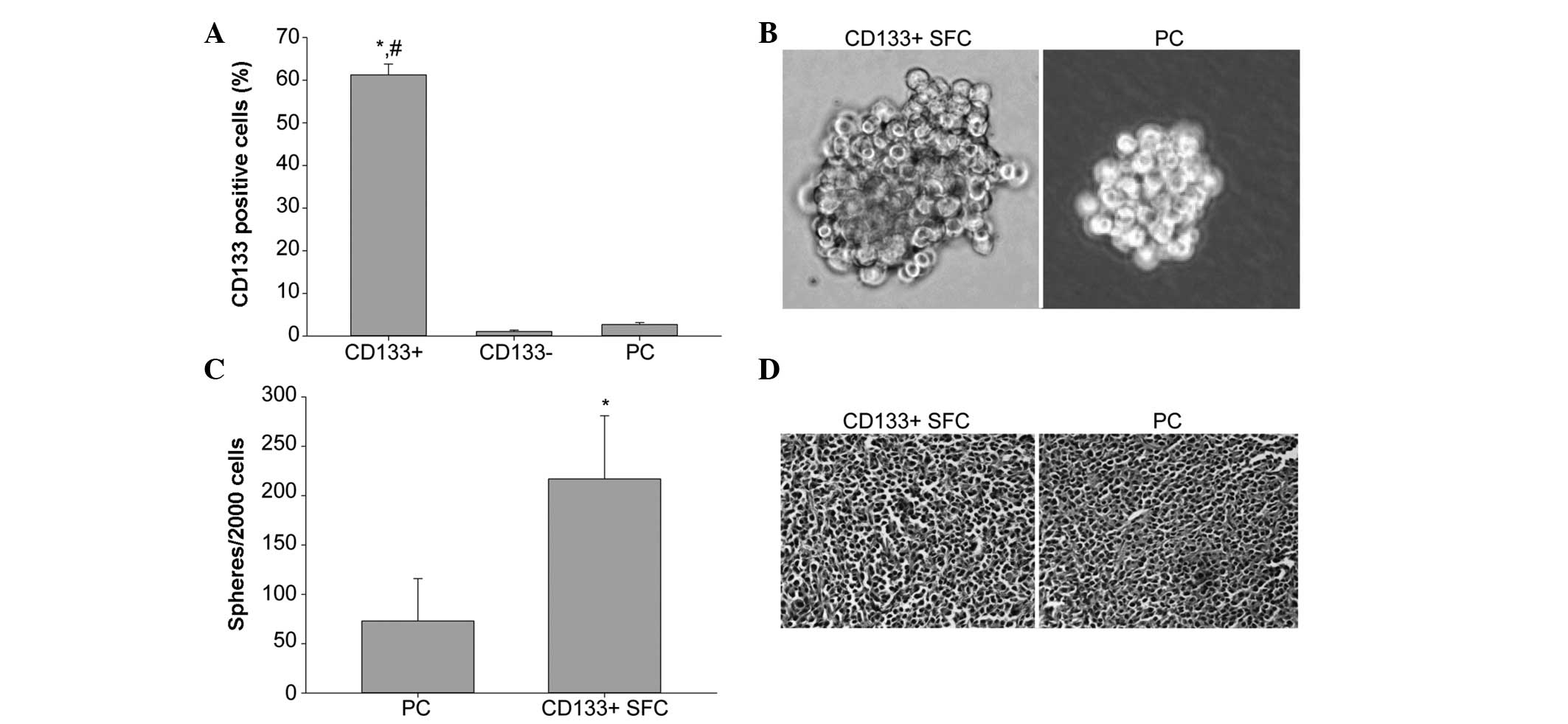
analysis showed that the CD133-positive cell percentage of
CD133+ subpopulation was 61.24±2.54%, which was
obviously higher than that of CD133− population
(1.07±0.34%) and the parental cells (2.72±0.46%), indicating our
immunomagnetic separation system was efficient (Fig. 1A).
We performed the tumor sphere formation assay with
stem cell-conditioned medium to enrich and expand LCSCs from sorted
CD133+ cells of the SMMC-7721 cell line. In the case of
inoculation of 2,000 cells per well, an increased number of spheres
formed in the group of CD133+ cells, while at the same
time as sphere formation, the sphere volume of CD133+
cells was greater than that of the parental cells (Fig. 1B and C).
To examine the tumor initiating capability, the male
Balb/c-nu mice were transplanted with various amounts of
CD133+ SFCs of the SMMC-7721 cell line and parental
cells. Our results demonstrated that as few as 1,000
CD133+ SFCs were sufficient to induce tumor development,
whereas, at least 2×105 SMMC-7721 parental cells were
necessary to consistently generate a tumor in the same model and
required a longer time period (Table
I). Hematoxylin and eosin staining indicated that
CD133+ SFCs formed similar histological features as the
tumor xenografts in the parental cells (Fig. 1D). These results demonstrated that
CD133+ SFCs sorted from the SMMC-7721 cell line were
highly tumorigenic and possessed LCSC properties.
 | Table ITumorigenicity experiments of
CD133+ SFCs and the parental cells in Balb/c-nu
mice. |
Table I
Tumorigenicity experiments of
CD133+ SFCs and the parental cells in Balb/c-nu
mice.
| Cell type | Cell nos.
injected | Tumor
incidencea | Latency
(days)b |
|---|
| SMMC-7721 parental
cells |
5×104 | 0/4 | - |
|
1×105 | 0/4 | - |
|
2×105 | 3/4 | 41 |
|
5×105 | 4/4 | 35 |
|
1×106 | 4/4 | 12 |
| CD133+
cells |
5×102 | 0/4 | - |
|
1×103 | 4/4 | 39 |
|
5×103 | 4/4 | 23 |
|
1×104 | 4/4 | 9 |
|
5×104 | 4/4 | 6 |
Casticin inhibits EMT of LCSCs derived
from the HCC SMMC-7721 cell line
EMT is a key process during the metastasis of LCSCs.
Therefore, we sought to examine whether morphological changes
existed between LCSCs and parental SMMC-7721 cells cultured
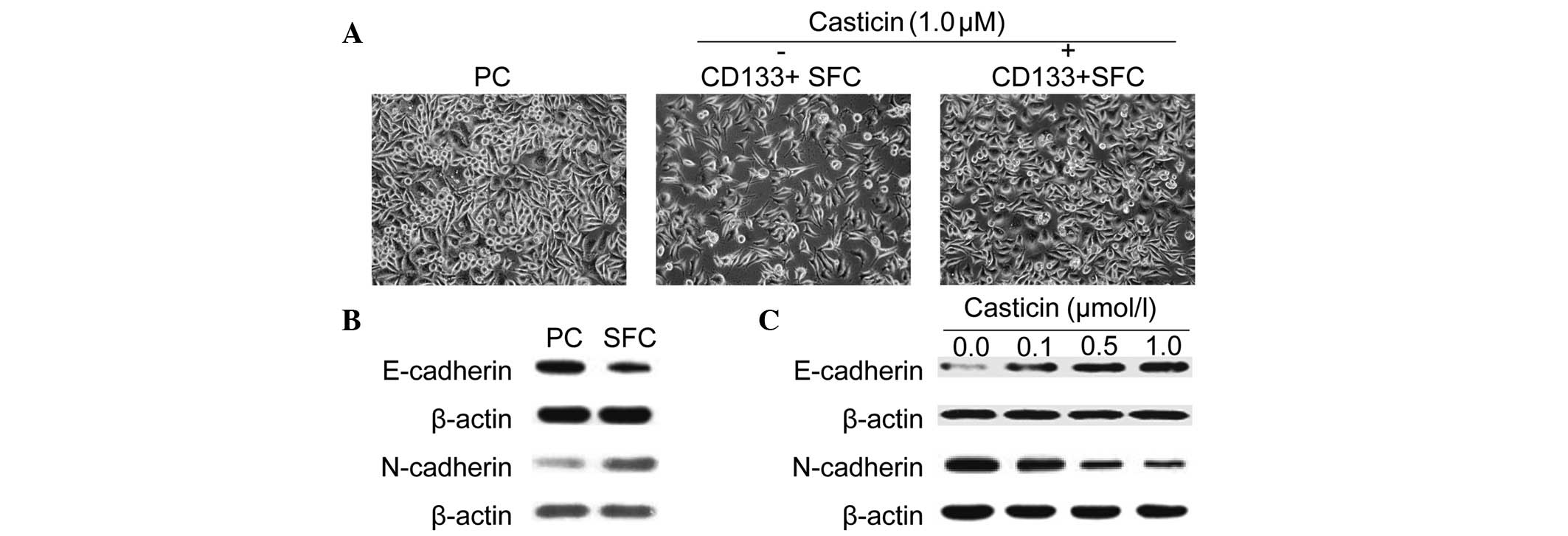
adherently in vitro. As shown in Fig. 2A, LCSCs exhibited a spindle-like
shape, while parental cells displayed a cobble-stone-like
phenotype. However, treatment with 1.0 μmol/l casticin suppressed
EMT in LCSCs as morphological changes from a spindle-like shape to
a cobble-stone-like appearance were observed.
Moreover, similar results were further confirmed by
western blotting using specific antibodies against EMT-relative
markers. As shown in Fig. 2B,
CD133+ SFCs expressed a higher N-cadherin protein level,
which is typically associated with mesenchymal cells and a lower
expression of epithelium-associated E-cadherin protein. Casticin,
at increasing concentrations, induced upregulation of the
epithelial marker, E-cadherin, and downregulation of the
mesenchymal marker, N-cadherin, after treatment for 24 h in
CD133+ SFCs derived from the SMMC-7721 cell line
(Fig. 2C). These results
demonstrated that casticin possessed inhibitory effects of EMT in
LCSCs purified from the SMMC-7721 cell line.
Casticin downregulates the expression of
Twist in LCSCs derived from the HCC SMMC-7721 cell line
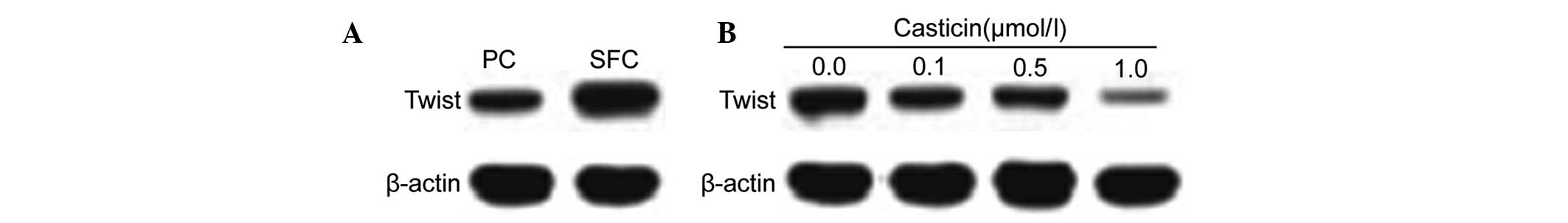
The transcription factor, Twist, was identified as
the critical molecule of EMT (27,28).
Based on our results that casticin inhibited EMT in LCSCs, we
further investigated the effects of casticin on the expression of
Twist. Twist was highly expressed in CD133+ SFCs
(Fig. 3A). Western blot analysis
indicated that the protein levels were downregulated following
treatment with casticin (0.0, 0.1, 0.5 and 1.0 μmol/l) in
CD133+ SFCs (Fig.
3B).
Overexpression of Twist reduced the
inhibition of EMT in LCSCs and self-renewal by casticin
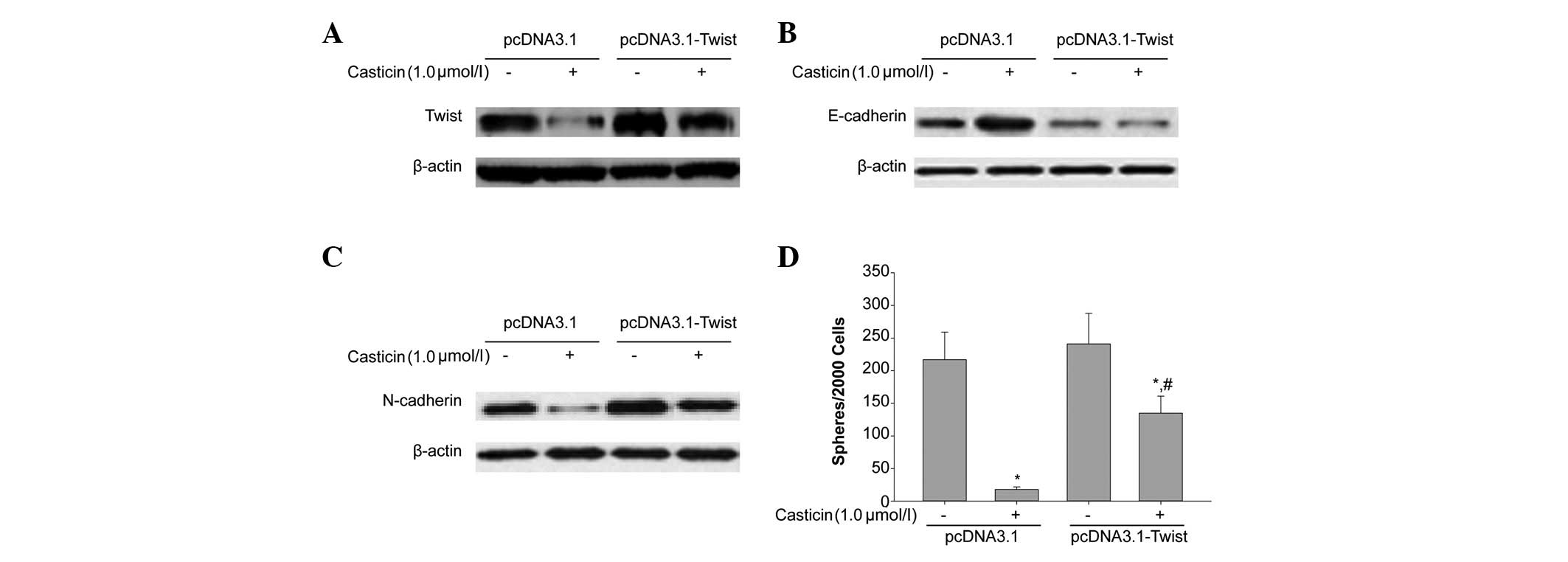
Casticin inhibited EMT of CD133+ SFCs,
while the overexpression of Twist prevented casticin-induced
inhibition of EMT. We transfected the plasmid, pCDNA3.1-Twist, and
the negative control, pCDNA3.1, into CD133+ SFCs derived
from SMMC-7721 cells. As shown in Fig.
4A, compared with the negative control vector, pCDNA3.1,
CD133+ SFCs transfected with pCDNA3.1-Twist
significantly overexpressed Twist. Furthermore, as shown in
Fig. 4B and C, the overexpression
of Twist attenuated casticin-induced upregulation of E-cadherin and
downregulation of N-cadherin, respectively. This tumor
sphere-forming experiment showed that the ectopic expression of
Twist in this cell line promoted tumor sphere formation. We also
treated CD133+ SFCs of SMMC-7721 cells transfected with
pCDNA3.1-Twist with 1.0 μmol/l casticin. As shown in Fig. 4D, after the addition of casticin,
the sphere-forming ability of CD133+ SFCs in this
subpopulation was greater than that of the control group. Taken
together, the overexpression of Twist partly reduced the inhibitory
effect of casticin on EMT of CD133+ SFCs. These results
further supported that the downregulation of Twist expression may
contribute to the inhibitory effects of casticin on EMT of LCSCs
derived from the SMMC-7721 cell line.
Discussion
Targeting LCSCs through repression of the EMT
process is an emerging strategy for the treatment and prevention of
HCC. As these interventions focus on EMT rather than toxicity
induction, they are potentially less toxic than conventional
chemotherapeutic agents. Our study confirmed that CD133+
SFCs sorted by MACS LS column from the SMMC-7721 human liver cancer
cell line showed an enhanced capacity of EMT and had greater
tumorigenicity than the parental cells, which indicated that
CD133+ SFCs enriched LCSCs. Suetsugu et al and
Yin et al demonstrated that CD133+ HCC cells were
cancer stem/progenitor cells (9,10),
which was consistent with our findings. Thus, CD133 could be
regarded as a potential target of stem cell-targeted therapy for
HCC.
EMT is a physiological phenotypic shift that
facilitates invasion and metastasis of liver cancer cells. Motile
and invasive tumor cells have been reported to gain the phenotypic
and molecular characteristics of EMT, including the gene expression
of EMT regulators, such as Twist (30). The ‘mobile CSCs’ were reported to
derive from stationary CSCs in colon cancers that underwent a
transient EMT (30), and the
induction of an EMT by ectopic expression of Twist transcription
factors has been reported to generate CSC properties in human
breast cancer cells (13,14). Our data showed that the protein
levels of the EMT regulator, Twist, were higher in
CD133+ SFCs than that in the parental cells. These
results were consistent with previous reports that Twist was
involved in infiltrative subtypes of HCC and breast cancer
(31). This data may aid in
explaining previous reports in which HCCs expressing CD133 had a
poor survival and high recurrence (32).
E-cadherin, the best-characterized member of
epithelial markers, was lost upon EMT, resulting in migration
(33). Downregulation of E-cadherin
and upregulation of N-cadherin have been reported in various tumors
during EMT (34). In the SMMC-7721
cell line, CD133+ SFCs showed upregulated expression of
N-cadherin and downregulated expression of E-cadherin compared with
that of the parental cells, which suggested that EMT can more
commonly occur in CD133+ SFCs. Thus, finding a natural
agent that targets LCSCs and EMT is considered an emerging strategy
for cancer prevention and recurrence.
In this study, cell morphology of CD133+
SFCs after treatment with casticin (1.0 μmol/l) was assessed.
Consistent with our hypothesis, the CD133+ SFCs showed
typical morphologic phenotypes of EMT, including loss of cell-cell
adhesion, spindle-shaped morphology and increased formation of
pseudopodia. However, following treatment with casticin (1.0
μmol/l) for 48 h, all these features disappeared and were displaced
by epithelial cobble-stone-like cells. This phenomenon suggested
that casticin can reverse the EMT process of CD133+
SFCs, morphologically. For further research, we assessed the
expression of invasion- and EMT-associated proteins, E-cadherin and
N-cadherin, of CD133+ SFCs after treatment with casticin
at various concentrations. Notably, the expression of E-cadherin
increased in a concentration-dependent manner in CD133+
SFCs, while N-cadherin decreased, indicating that casticin can
reverse the EMT process by upregulating E-cadherin and
downregulating N-cadherin. In addition, we also evaluated the EMT
regulator, Twist, using western blot analysis after treatment with
casticin at various concentrations. The results were consistent
with our hypothesis; casticin downregulated the expression of the
transcriptional factor, Twist. We also found that CD133+
SFCs transfected with the pCDNA3.1-Twist plasmid significantly
overexpressed Twist compared with cells with the negative control
vector (pCDNA3.1). Overexpression of Twist attenuated
casticin-induced upregulation of E-cadherin and the downregulation
of N-cadherin. In addition, the suppressive effects of casticin on
EMT of CD133+ SFCs were partially rescued by the
overexpression of Twist. These data demonstrated that casticin
could inhibit the EMT of CD133+ SFCs by downregulating
Twist. Considering the relatively non-toxic nature of casticin,
targeting EMT-type cells and LCSCs by combining it with
conventional chemotherapeutics may be a novel and safe approach for
achieving an improved treatment outcome. However, further in-depth
preclinical and clinical studies are warranted in order to
appreciate its value in reversal of the EMT process on LCSCs.
In conclusion, our study demonstrated that casticin
has the potential to target CD133+ SFCs of the SMMC-7721
cell line, namely LCSCs, and can inhibit the EMT process.
Therefore, we suggest that casticin may be an effective candidate
for the prevention and treatment of recurrence and metastasis as a
cancer therapeutic agent for HCC.
Acknowledgements
The authors would like to thank Dr Jian-Guo Cao
(Medical College, Hunan Normal University, Changsha, Hunan
Province, China) for the critical reading of the manuscript. This
study was supported by the Scientific Research Project of Hunan
Provincial Administration Bureau of Traditional Chinese Medicine
(no. 2010081), the Project of Scientific Research of the Department
of Education of Hunan Province (no. 10C0975), the Major Project
Item of Scientific Research of the Department of Education of Hunan
Province (no. 09A054), the Project of Scientific Research of
Changsha city Bureau of Science and technology (no. K1104060-31)
and the Hunan Provincial Science and Technology Project (no.
2011FJ4144).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
3
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maluccio M and Covey A: Recent progress in
understanding, diagnosing, and treating hepatocellular carcinoma.
CA Cancer J Clin. 62:394–399. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Aguayo A and Patt YZ: Nonsurgical
treatment of hepatocellular carcinoma. Semin Oncol. 28:503–513.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Llovet JM and Bruix J: Systematic review
of randomized trials for unresectable hepatocellular carcinoma:
Chemoembolization improves survival. Hepatology. 37:429–442. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kuvshinoff BW and Ota DM: Radiofrequency
ablation of liver tumors: influence of technique and tumor size.
Surgery. 132:605–611; discussion 611–612. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou BB, Zhang H, Damelin M, et al:
Tumour-initiating cells: challenges and opportunities for
anticancer drug discovery. Nat Rev Drug Discov. 8:806–823. 2009.
View Article : Google Scholar
|
|
9
|
Suetsugu A, Nagaki M, Aoki H, et al:
Characterization of CD133+ hepatocellular carcinoma cells as cancer
stem/progenitor cells. Biochem Biophys Res Commun. 351:820–824.
2006.
|
|
10
|
Yin S, Li J, Hu C, et al: CD133 positive
hepatocellular carcinoma cells possess high capacity for
tumorigenicity. Int J Cancer. 120:1444–1450. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ma S, Chan KW, Hu L, et al: Identification
and characterization of tumorigenic liver cancer stem/progenitor
cells. Gastroenterology. 132:2542–2556. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma S, Lee TK, Zheng BJ, Chan KW and Guan
XY: CD133+ HCC cancer stem cells confer chemoresistance
by preferential expression of the Akt/PKB survival pathway.
Oncogene. 27:1749–1758. 2008.
|
|
13
|
Mani SA, Guo W, Liao MJ, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar
|
|
14
|
Morel AP, Lièvre M, Thomas C, et al:
Generation of breast cancer stem cells through
epithelial-mesenchymal transition. PLoS One. 3:e28882008.
View Article : Google Scholar
|
|
15
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelial-mesenchymal transition: new insights in
signaling, development, and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gavert N and Ben-Ze’ev A:
Epithelial-mesenchymal transition and the invasive potential of
tumors. Trends Mol Med. 14:199–209. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee TK, Poon RT, Yuen AP, et al: Twist
overexpression correlates with hepatocellular carcinoma metastasis
through induction of epithelial-mesenchymal transition. Clin Cancer
Res. 12:5369–5376. 2006. View Article : Google Scholar
|
|
18
|
Li Y, Wicha MS, Schwartz SJ and Sun D:
Implications of cancer stem cell theory for cancer chemoprevention
by natural dietary compounds. J Nutr Biochem. 22:799–806. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li Y, Zhang T, Korkaya H, et al:
Sulforaphane, a dietary component of broccoli/broccoli sprouts,
inhibits breast cancer stem cells. Clin Cancer Res. 16:2580–2590.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rausch V, Liu L, Kallifatidis G, et al:
Synergistic activity of sorafenib and sulforaphane abolishes
pancreatic cancer stem cell characteristics. Cancer Res.
70:5004–5013. 2010. View Article : Google Scholar
|
|
21
|
Haidara K, Zamir L, Shi QW and Batist G:
The flavonoid Casticin has multiple mechanisms of tumor
cytotoxicity action. Cancer Lett. 242:180–190. 2006. View Article : Google Scholar
|
|
22
|
Zhou Y, Peng Y, Mao QQ, et al: Casticin
induces caspase-mediated apoptosis via activation of mitochondrial
pathway and upregulation of DR5 in human lung cancer cells. Asian
Pac J Trop Med. 6:372–378. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kobayakawa J, Sato-Nishimori F, Moriyasu M
and Matsukawa Y: G2-M arrest and antimitotic activity mediated by
casticin, a flavonoid isolated from Viticis Fructus (Vitex
rotundifolia Linne fil.). Cancer Lett. 208:59–64. 2004. View Article : Google Scholar
|
|
24
|
Ko WG, Kang TH, Lee SJ, et al:
Polymethoxyflavonoids from Vitex rotundifolia inhibit proliferation
by inducing apoptosis in human myeloid leukemia cells. Food Chem
Toxicol. 38:861–865. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lin S, Zhang H, Han T, Wu JZ, Rahman K and
Qin LP: In vivo effect of casticin on acute inflammation. Zhong Xi
Yi Jie He Xue Bao. 5:573–576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang J, Yang Y, Tian L, Sheng XF, Liu F
and Cao JG: Casticin-induced apoptosis involves death receptor 5
upregulation in hepatocellular carcinoma cells. World J
Gastroenterol. 17:4298–4307. 2011. View Article : Google Scholar
|
|
27
|
He L, Yang X, Cao X, Liu F, Quan M and Cao
J: Casticin induces growth suppression and cell cycle arrest
through activation of FOXO3a in hepatocellular carcinoma. Oncol
Rep. 29:103–108. 2013.
|
|
28
|
El-Haibi CP, Bell GW, Zhang J, et al:
Critical role for lysyl oxidase in mesenchymal stem cell-driven
breast cancer malignancy. Proc Natl Acad Sci USA. 109:17460–17465.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Barr MP, Gray SG, Hoffmann AC, et al:
Generation and characterisation of cisplatin-resistant non-small
cell lung cancer cell lines displaying a stem-like signature. PLoS
One. 8:e541932013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gavert N and Ben-Ze’ev A:
Epithelial-mesenchymal transition and the invasive potential of
tumors. Trends Mol Med. 14:199–209. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brabletz T, Jung A, Spaderna S, Hlubek F
and Kirchner T: Opinion: migrating cancer stem cells - an
integrated concept of malignant tumour progression. Nat Rev Cancer.
5:744–749. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Song W, Li H, Tao K, et al: Expression and
clinical significance of the stem cell marker CD133 in
hepatocellular carcinoma. Int J Clin Pract. 62:1212–1218. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang J, Mani SA, Donaher JL, et al: Twist,
a master regulator of morphogenesis, plays an essential role in
tumor metastasis. Cell. 117:927–939. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kang Y and Massagué J:
Epithelial-mesenchymal transitions: twist in development and
metastasis. Cell. 118:277–279. 2004. View Article : Google Scholar
|