Introduction
Primitive neuroectodermal tumors (PNETs) are rare
small round cell neoplasms, first described in 1918 by Stout
(1) as a malignant tumor arising
from major nerve (2). All ages are
affected, but the majority of patients present in their first or
second decades of life, and the tumors are rarely observed in
children <3 years old.
Although peripheral PNET (pPNET; PNET derived from
tissues outside the central and autonomic nervous systems)
frequently occurs in the thoracopulmonary region (Askin’s tumors),
urogenital tract and testis (3–4). pPNET
has also been found in the head and neck region (6). pPNET comprises 1% of all sarcomas, and
is highly malignant (7).
Clinically, the condition presents as a rapidly enlarging, painful
mass, with a high probability of micrometastasis (6).
The symptoms of pPNET in the head and neck are
non-specific. An extensive review of the literature shows only a
few cases of pPNET affecting the parotid gland (8). To the best of our knowledge, the
present study reports the first case of pPNET of the parotid gland
affecting a child <3 years old, which had been previously
misdiagnosed and treated as mumps. Written informed consent was
provided by the patient’s family.
Case report
A 2-year-old male, with no noteworthy medical
history, was brought to the Department of Oral Surgery of Xuzhou
Central Hospital (Xuzhou, China) with a 1-month history of a
painful, progressively enlarging swelling in the left parotid gland
accompanied by cervical lymphadenopathy, which had previously been
treated as mumps by a county hospital. Prior to this referral, the
patient was treated with antibiotics for 1-week. The patient had no
noteworthy medical family history or past history. Upon physical
examination, the patient was afebrile, but anorexic. Upon clinical
examination, a firm non-tender fixed mass (5×5 cm) was found on the
left parotid gland extending to the parapharyngeal space, without
facial paralysis.
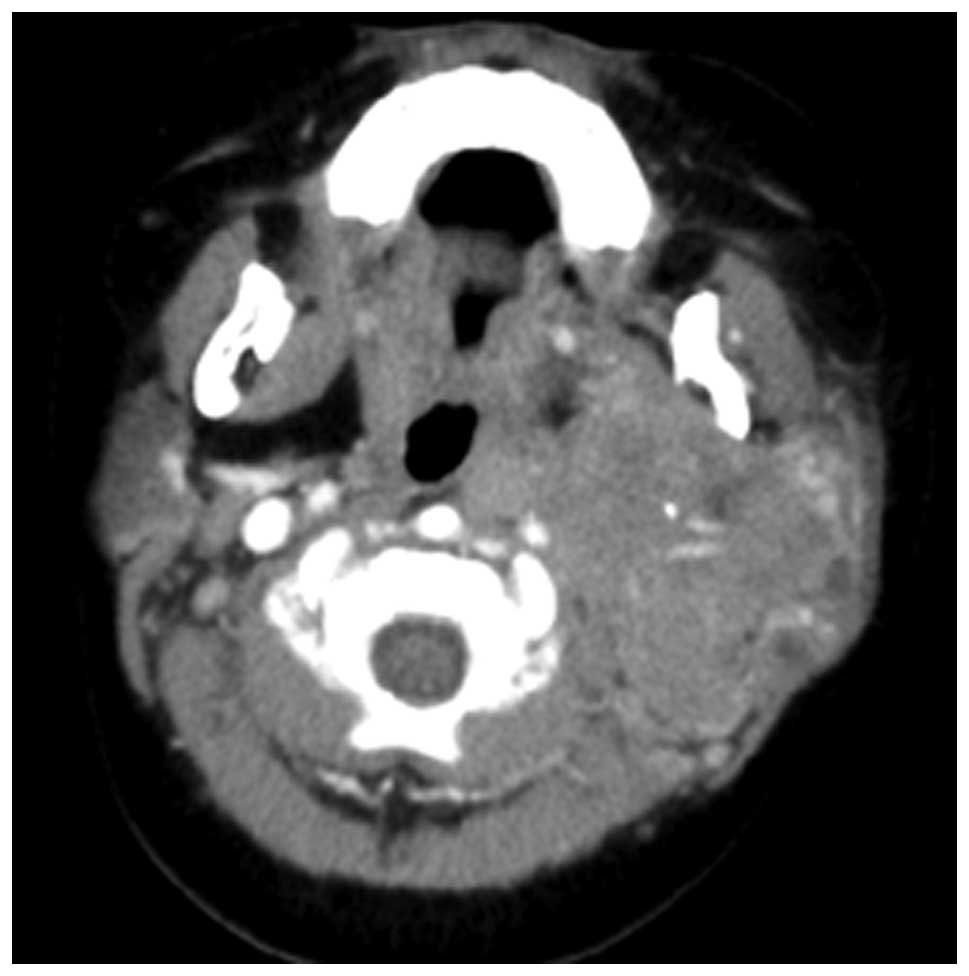
A computed tomography (CT) scan of the head-neck was
indicative of a soft-tissue mass of heterogenous density in the
left side of the parotid gland completely surrounding the carotid
arteries (Fig. 1). The mass was
~5.6×4.8×5.3 cm in size and invaded the left parapharyngeal space
and lateral pterygoid plate without bone erosion. These
observations were confirmatory for a diagnosis of a PNET. A chest
radiograph and abdominal ultrasound were performed to screen for
metastasis, and revealed normal results.
Owing to the requirement for obtaining a
histopathological diagnosis, the patient underwent a needle
aspiration biopsy and excisional biopsy for analysis.
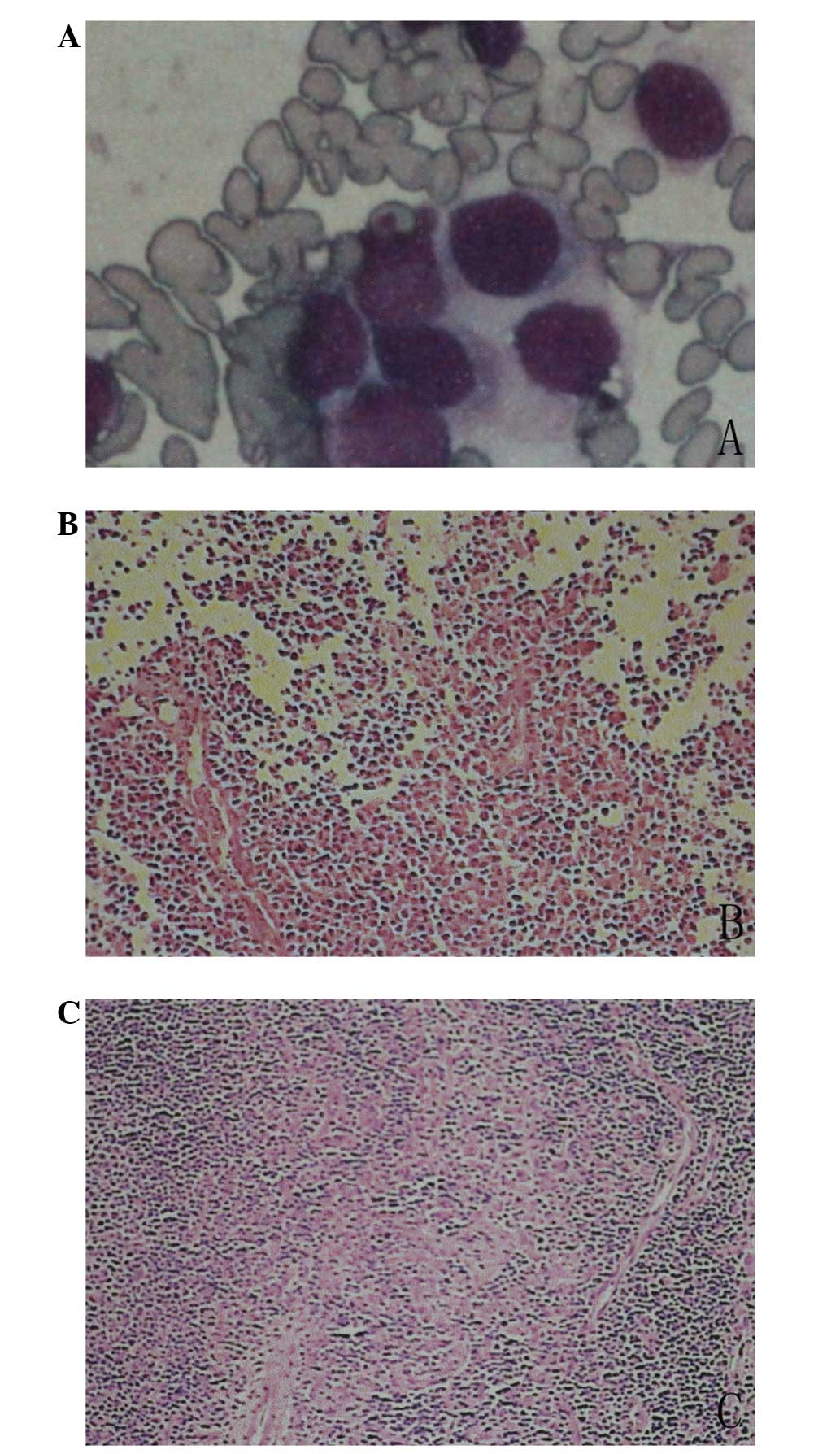
Microscopically, the biopsy specimen exhibited a pleomorphic
cellular infiltrate with hyperchromatic small cells scattered in a
fibrovascular stroma interposed by fibrous septa. Homer-Wright
rosettes consisting of a number of hyperchromatic cells were also
present (Fig. 2). Definitive
management in the form of debulking surgery and parotidectomy was
performed in December 2012. Following the surgery, an
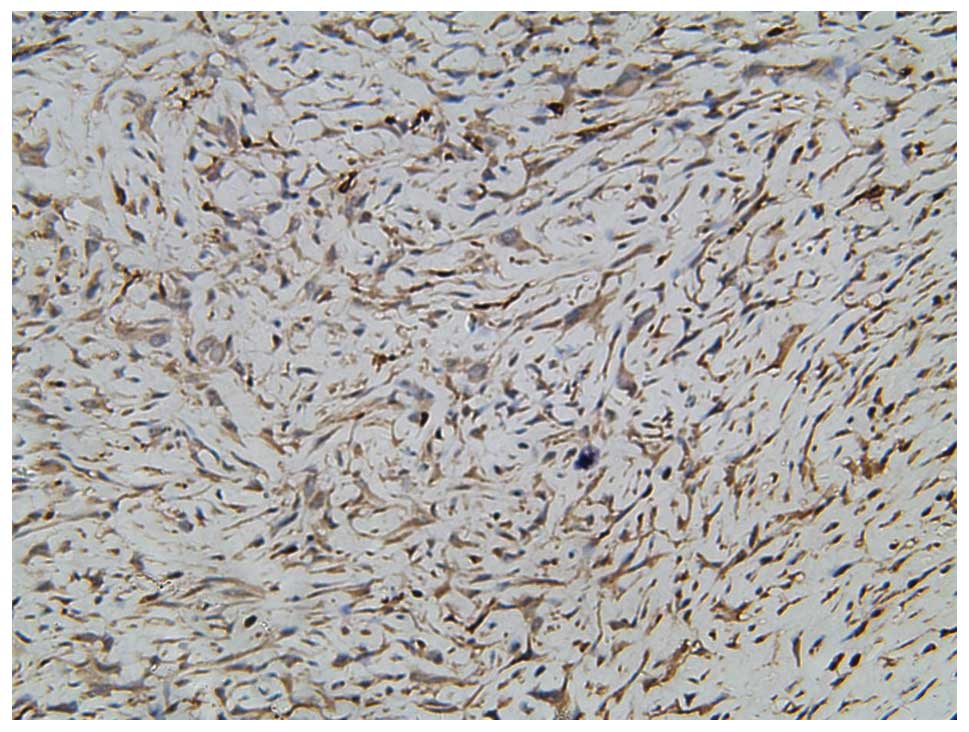
immunohistochemical analysis was performed on the excised material
for a definite diagnosis. The sample was positive for cluster of
differentiation (CD)99 (Fig. 3),
synaptophysin (Syn), vimentin, chromogranin A (CgA) and cytokeratin
(CK). Ki-67 (mib-1) immunochemical staining showed 50% of positive
cells. Other immunomarkers, including desmin, epithelial membrane
antigen and CD38, were all negative. On the basis of these
findings, the lesion was confirmed to be a pPNET of the parotid
gland.
Subsequent to the generation of a definite
diagnosis, the patient was started on radiotherapy (20 Gy)
treatment and multiagent chemotherapy, including cyclophosphamide,
adriamycin and vincristine (CAV). The mass was grossly reduced in
size, but did not completely disappear. Chest CT and abdominal
ultra sonography were performed every 3 months. However, following
the third chemotherapeutic cycle, the patient experienced severe
vomiting and diarrhea, and the parents refused to continue the
treatment. The patient is currently being monitored by follow-up
examinations at 7 months post-surgery.
Discussion
PNET belongs to the Ewing family of tumors. In 1918,
Stout (1) first described the tumor
composed of small-round cells in the ulnar nerve (2). PNET is frequently observed in
adolescents, and can be found anywhere within the body,
particularly in the trunk and extremities. Although pPNET is rarely
noted in the head and neck region, when found, it is often in the
jaw, followed by the mandible and maxilla. pPNET of the parotid
gland is exceedingly unusual. In a review of the English
literature, it was found that pPNET of the parotid gland had only
been reported in adults, aged between 15 and 60 years old (7). Therefore, to the best of our
knowledge, the present case of pPNET of the parotid gland is the
first study in a child.
Clinically, the majority of complaints by patients
at the time of presentation include rapid growth, swelling of the
affected area and pain, all complaints associated with the soft
mass effects of the tumor (6–8).
However, pre-operative diagnosis is difficult, as clinical
presentation may vary greatly between the different sites of
involvement. In children, when parotid gland swelling occurs with
other systemic signs, such as fever, anorexia, fatigue and
respiratory symptoms, the condition may be misdiagnosed and treated
as mumps (9). The patient in the
current case presented with a sudden onset of parotid swelling
accompanied by anorexia and pain. The patient had previously been
misdiagnosed by a county hospital and only treated with
antibiotics. According to the study by Brazão-Silva et al
(10), the complementary
examinations avoid larger areas in the diagnosis, which may affect
the early diagnosis and lead to the risk of more aggressive
approaches. We believe that improved diagnostic techniques and
therapeutic strategies for patients with pPNET could increase the
disease-free survival time.
Imaging studies are essential for the diagnosis and
surgical treatment planning, however, the radiological features of
the tumors are non-specific and frequently cannot be differentiated
from those of other types of bone and soft-tissue tumors (11). Although pPNET most often resemble
large, non-calcified, soft-tissue masses, with cystic or necrotic
areas, this aspect is not a pathognomonic feature, as other lesions
can have the same image pattern. Magnetic resonance imaging also
reveals a non-specific isointensity on T1/T2-weighted images
(11–13). Ultimately, a diagnosis is made by
histopathological and immunohistochemical examination of the
tumor.
Histologically, pPNET consists of a pleomorphic
cellular infiltrate with hyperchromatic small cells, which are
often arranged in lobules separated by fibrous septa (14). In addition, short dendritic
processes lie between the cells in a pPNET, and these are
characteristically absent in Ewing’s sarcoma (10). Homer-Wright-type rosettes with tumor
cells are absent in PNET, which is seen in other round cell tumors.
However, due to their similarity with numerous tumors belonging to
the category of ‘small, round, blue cell tumors’, including
rhabdomyosarcoma, lymphoma and small cell carcinoma, as well as
other members of the PNET family of tumors, particularly
neuroblastoma and Ewing’s sarcoma, which exhibit a similar
histological appearance, the diagnosis of pPNETs can be problematic
on a morphological basis. The use of immunohistochemistry plays a
pivotal role in the diagnosis of this tumor. Normally, the tumor
cells are positive for MIC-2 (CD99) and vimentin, and negative for
desmin and CK, but are not specific markers for pPNET. Schmidt
et al (15) proposed the
following criteria for the pathological diagnosis of PNET: a) Tumor
cells appear round under a light microscope and Homer-Wright-type
rosettes are present; b) immunohistochemistry reveals that the
tumor is positive for 013 (i.e., HBA71, P30/32 and MIC-2), and for
two or more of the NSE, Syn and CGA proteins; and c)
ultrastructural identification of neural elements, especially
neurosecretory granules (14,16).
Due to the rare occurrence of pPNET, optimal therapy
is challenging, particularly if the tumors occur in the head and
neck. Currently, the uniformly accepted strategy is surgery
combined with adjuvant radiotherapy and multiagent chemotherapy
(17,18). However, structural complexity of the
head and neck region leads to difficulty in complete removal. We
believe that close cooperation between surgeons, oncologists and
radiotherapists can improve progression-free survival times. The
patient of the present study underwent maximal tumor reduction
surgery, CAV and low-dose pre-operative radiation therapy for gross
residual disease or microscopic residual disease. The patient has
been under close observation since the treatment (~7 months), and
there have been no signs of recurrence.
In conclusion, the differential diagnosis of pPNET
in a child is extremely important. In the present case, it was
noted that combination therapy can be also be an effective method
for young children with pPNET, while at the same time, attention
must be paid to adverse reactions.
References
|
1
|
Stout AP: A tumor of the ulnar nerve. Proc
NY Pathol Soc. 18:2–11. 1918.
|
|
2
|
Desai SS and Jambhekar NA: Pathology of
Ewing’s sarcoma/PNET: Current opinion and emerging concepts. Indian
J Orthop. 44:363–368. 2010.
|
|
3
|
Tazi I, Zafad S, Madani A, et al: Askin
tumor: a case report with literature review. Cancer Radiother.
13:771–774. 2009.(In French).
|
|
4
|
Song HC, Sun N, Zhang WP and Huang CR:
Primary Ewing’s sarcoma/primitive neuroectodermal tumor of the
urogenital tract in children. Chin Med J (Engl). 125:932–936.
2012.
|
|
5
|
Heikaus S, Schaefer KL, Eucker J, et al:
Primary primitive neuroectodermal tumor/Ewing’s tumor of the testis
in a 46-year-old man-differential diagnosis and review of the
literature. Hum Pathol. 40:893–897. 2009.
|
|
6
|
Hormozi AK, Ghazisaidi MR and Hosseini SN:
Unusual presentation of peripheral primitive neuroectodermal tumor
of the maxilla. J Craniofac Surg. 21:1761–1763. 2010.
|
|
7
|
Nikitakis NG, Salama AR, O’Malley BW Jr,
et al: Malignant peripheral primitive neuroectodermal
tumor-peripheral neuroepithelioma of the head and neck: a
clinicopathologic study of five cases and review of the literature.
Head Neck. 25:488–498. 2003.
|
|
8
|
Helsel JC, Mrak RE, Hanna E, et al:
Peripheral primitive neuroectodermal tumor of the parotid gland
region: report of a case with fine-needle aspiration findings.
Diagn Cytopathol. 22:161–166. 2000.
|
|
9
|
Hviid A, Rubin S and Mühlemann K: Mumps.
Lancet. 371:932–944. 2008.
|
|
10
|
Brazão-Silva MT, Fernandes AV, Faria PR,
et al: Ewing’s sarcoma of the mandible in a young child. Braz Dent
J. 21:74–79. 2010.
|
|
11
|
Ibarburen C, Haberman JJ and Zerhouni EA:
Peripheral primitive neuroectodermal tumors. CT and MRI evaluation.
Eur J Radiol. 21:225–232. 1996.
|
|
12
|
Lawlor ER, Mathers JA, Bainbridge T, et
al: Peripheral primitive neuroectodermal tumors in adults:
documentation by molecular analysis. J Clin Oncol. 16:1150–1157.
1998.
|
|
13
|
Windfuhr JP: Primitive neuroectodermal
tumor of the head and neck: incidence, diagnosis, and management.
Ann Otol Rhinol Laryngol. 113:533–543. 2004.
|
|
14
|
Talesh KT, Motamedi MH and Jeihounian M:
Ewing’s sarcoma of the mandibular condyle: report of a case. J Oral
Maxillofac Surg. 61:1216–1219. 2003.
|
|
15
|
Schmidt D, Harms D and Burdach S:
Malignant peripheral neuroectodermal tumours of childhood and
adolescence. Virchows Arch A Pathol Anat Histopathol. 406:351–365.
1985.
|
|
16
|
Dehner LP: Primitive neuroectodermal tumor
and Ewing’s sarcoma. Am J Surg Pathol. 17:1–13. 1993.
|
|
17
|
Fontaine C, Schots R, Braeckman J, et al:
Long-term survival in an adult metastatic renal peripheral
primitive neuroectodermal tumor (PPNET) with multimodality
treatment including high-dose chemotherapy. Ann Oncol. 8:691–694.
1997.
|
|
18
|
Jairam V, Roberts KB and Yu JB: Historical
trends in the use of radiation therapy for pediatric cancers:
1973–2008. Int J Radiat Oncol Biol Phys. 85:e151–e155. 2013.
|